
Abstract
Shoot-tip meristem cryopreservation methodologies are reported for the complementary cryoprotective strategies of vitrification and equilibrium freezing using traditional controlled-rate freezing and chemical additive cryoprotection. Pregrowth, pretreatment, and cold acclimation approaches for the improvement of tolerance to liquid nitrogen are also presented. The chapter concludes by reporting an analytical protocol that profiles volatile hydrocarbon stress markers (for ethylene, hydroxyl radicals, and lipid peroxidation products) during cryopreservation. This method uses noninvasive headspace sampling and gas chromatography, and it is widely applicable across cryogenic systems.
Access provided by Autonomous University of Puebla. Download protocol PDF
Similar content being viewed by others


Key Words
1 Introduction
Recent advances in shoot-tip cryopreservation have been significant, this is largely because of: (1) development of vitrification-based cryoprotection protocols, (2) refinements in tissue culture practices, (3) identification of critical points in cryopreservation technology transfer, and (4) the wider uptake and validation of cryostorage technologies in international germplasm repositories. There still remain some genotypes intractable to cryogenic storage, and fundamental research is progressively facilitating the identification of decisive factors in recalcitrance, with a view to aiding storage protocol development. With these issues in mind, this chapter will report the routine storage and investigative methodologies currently applied to shoot cryopreservation. Generic cryopreservation protocols will be described and Ribes (currants) are used as an exemplar, as this genus has been studied in detail with regard to critical point thermal analysis, protocol validation, and technology transfer to germplasm repositories. An analytical gas chromatogram (GC)-head space sampling technique will also be described. This is a noninvasive tool that has been optimized to pinpoint stress components in shoot-tip cryopreservation protocols with a view to assisting method development.
Anatomically, the shoot meristem contains the apical dome and the youngest, unexpanded primordial leaf. For cryopreservation, selection of tissues that comprise the meristem apex, subjacent tissue, and several larger leaf primordia may be also necessary. Shoot-tip size and origin (apical, lateral, axillary) influences poststorage survival and it is also possible to cryopreserve nodal stem cuttings containing an axillary meristem. Physiological status is critical and storage responses contrast between temperate and tropical species. In temperate genotypes, cold hardiness, acclimation, and dormancy responses are important factors and are used in cryoprotective treatments. Cryopreservation of cold-hardened winter buds of woody perennial species is therefore an alternative approach to in vitro cryopreservation. In contrast, tropical and warm temperate species are more sensitive to chilling and desiccation stresses, and their cryopreservation protocols have to be developed accordingly.
As described in Chapter 3, there are two main approaches to cryopreservation based on cryoprotective modality. The first is termed traditional, controlled rate, or two-step freezing and involves the application of single or combined mixtures of colligative cryoprotectants, sometimes in conjunction with osmotic additives. Controlled cooling of tissues to an intermediate freezing temperature causes a water vapor deficit to be created between the inside and outside of the cell initiated by the preferential formation (termed “seeding” or ice nucleation) of extracellular ice. Thus, intracellular water moves across the plasmalemma and cellular dehydration results; this process is called equilibrium freezing. Under optimum freezing rates this is a cryoprotective process, as the amount of water available for intracellular ice formation is reduced. However, excessive dehydration can lead to colligative damage owing to the harmful concentration of solutes. Critical cryogenic factors in controlled-rate freezing are:
-
1.
Cryoprotectant composition, the components of which must include a penetrating colligative cryoprotectant.
-
2.
Cooling rate.
-
3.
The control of, and point at which, ice nucleation occurs.
-
4.
A “hold step” programmed as a fixed time at a fixed terminal transfer temperature. Usually at, or around, the point of homogeneous ice formation, which is −40°C.
-
5.
Transfer to liquid nitrogen.
The second approach to plant cryopreservation is vitrification which usually comprises different permutations of chemical additive as well as vitrification, and encapsulation, osmotic-, evaporative-, and chemical (silica gel)-dehydration. The cryoprotective basis is the concentration of solutes to such an extent that cell viscosity becomes so high that on exposure to cryogenic temperatures, a glass, rather than ice, is formed. The process, termed vitrification, involves the creation of a metastable amorphous glassy state, characterized by the glass transition temperature, Tg, the thermal point at which a glass is formed. However, glasses can be thermally unstable and it is possible that devitrification occurs on cooling and rewarming (as this risks the formation of ice it is critical that glasses are stabilized). This can be achieved by the manipulation of dehydration and cryoprotective additives, particularly the inclusion of sugars and careful control of rewarming. Vitrification is a useful alternative to controlled-rate cooling and freezing, as it has the major advantage that tissues are directly immersed into liquid nitrogen, circumventing the need for expensive programmable freezing equipment. In addition to cryogenic factors, the development of successful cryopreservation protocols also depends on the physiological status of the tissues, and pre- and post-treatments are applied to maximize the ability of shoot tips to survive cryopreservation.
To assist the reader in accessing information as to the theory and applications of plant cryopreservation, a bibliography is recommended. For fundamental theory see Mazur (1) and Chapter 3 of this volume; for cryophysics and instrumentation see Benson et al. (2); for historical perspectives and wider areas of in vitro plant conservation see Benson (3,4); and for a comprehensive overview of the contemporary aspects of fundamental and applied cryobiology, see Fuller et al. (5).
2 Materials
Cryogenic safety protocols and protective clothing should be used throughout all liquid nitrogen handling procedures. In vitro materials should be sterile and manipulations performed in a laminar-flow cabinet. Shoot cultures should be checked before cryopreservation for health, vigor, and the presence of contaminants, particularly incipient systemic infections that are manifested as opaque white-cream haloes in the culture medium surrounding explants.
2.1 Shoot-Tip Dissection
-
1.
Binocular, dissecting microscope (magnification ×20).
-
2.
Two 10-mL syringes fitted with hypodermic needles.
-
3.
Liquid growth medium (see Note 1 ).
-
4.
50-mm Petri dish lined with a sterile filter paper.
-
5.
Scissors, scalpel, forceps, Pasteur pipets.
2.2 Common Cryopreservation Materials
-
1.
Liquid nitrogen.
-
2.
Small bench top Dewar with 1-L capacity.
-
3.
Cryovials, canes, Pasteur pipets, 90- and 50-mm Petri dishes, sterile filter papers, forceps, scissors, scalpels, heated magnetic stirrer, and binocular dissecting microscope.
-
4.
Long-term storage Dewar and inventory system.
-
5.
Recovery medium (see Note 2 ).
-
6.
Safety equipment.
2.3 Controlled-Rate Freezing
-
1.
Two representative cryoprotectants have been selected for controlled-rate freezing, both use dimethyl sulfoxide (DMSO), one of the most penetrating colligative cryoprotectants known (see Notes 3 – 5 ). The second incorporates a mixture of additives and includes the dehydrating osmotic agent polyethylene glycol and glucose.
-
2.
Programmable freezer.
-
3.
Water bath at 45°C.
2.4 Cryoprotective Dehydration
-
1.
One or more of a series of pregrowth media containing 0.5–1 M of a dehydrating agent selected from sucrose, mannitol, or sorbitol (see Note 6 ).
-
2.
An airtight vessel (e.g., a Parafilm-sealed 9.0-cm glass Petri dish) containing a known loading (30–50 g) of sterile heat-activated silica gel.
-
3.
An open sterile Petri dish located in sterile, horizontal laminar airflow.
2.5 Vitrification Using Cryoprotective Additives
-
1.
100 mL of plant vitrification solution 2 (PVS2 [8–11]) made up in standard liquid growth medium to which is added 30% (w/v) glycerol, 15% (w/v) ethylene glycol, 15% (w/v) DMSO, and sucrose to a final concentration of 0.4 M (see Notes 3 – 5 ).
-
2.
A dilution series range (50, 60, 70, 80% [v/v]) of PVS2 solution made in liquid medium (see Note 6 ).
-
3.
Ice and ice bucket.
-
4.
100 mL of “unloading” solution comprising 1.2 M sucrose made up in liquid culture medium.
-
5.
Water bath at 45°C.
2.6 Encapsulation and Dehydration
-
1.
A 5-mL Gilson Inc., Middleton “Pipetteman” fitted with a tip or a 3-mL plastic Pastet (see Note 7 ).
-
2.
Liquid culture medium containing 0.75 M sucrose (see Note 6 ) aliquoted (20 mL) into 100-mL conical flasks.
-
3.
Calcium-free, liquid culture medium containing 3% (w/v) sodium alginate (SIGMA [SIGMA-ALDRICH, Gillingham, Dorset, UK] low viscosity, sodium salt derived from sea kelp) dispensed into small bottles as 20-mL aliquots (see Notes 8 and 9 ).
-
4.
Liquid culture medium containing 100 mM CaCl2 dispensed as 30-mL aliquots into conical flasks or beakers.
-
5.
A 250–500-µm mesh sieve.
-
6.
Sterile tissues or pieces of filter paper.
-
7.
Reciprocal shaker.
2.7 Encapsulation and Vitrification
-
1.
PVS2 solution (see Subheading 2.5. ).
-
2.
Calcium-free, liquid culture medium (12) containing 2 or 3% (w/v) sodium alginate (SIGMA low viscosity, sodium salt derived from sea kelp) and incorporating 0.4 M sucrose (12) dispensed into small bottles as 20-mL aliquots (see Note 8 ).
-
3.
Liquid culture medium containing 100 mM CaCl2 (12) dispensed as 30-mL aliquots into conical flasks or beakers.
-
4.
Liquid culture medium containing 0.75 or 0.8 M sucrose (see Note 6 ) aliquoted (20 mL) into 100-mL conical flasks.
-
5.
A 250–500-µm mesh sieve.
-
6.
Sterile tissues or pieces of filter paper.
2.8 Droplet Freezing
-
1.
Sterile pieces of aluminium foil (of 2–3 cm length × 0.5–1.0 cm width × 0.003 to 0.005 cm depth dimensions).
-
2.
Sterile liquid culture medium.
2.9 Volatile Headspace Analysis of Cryopreservation Injury in Shoot Tips
-
1.
GC equipped with an injector at 150°C, Porapak Q- or Haysep Q-packed column (2-m long, 1/8-in. ID stainless steel tubing, 80–100 mesh), oven temperature initially at 60°C for 3 min (increase to 160°C at 20°C/min, and hold at 160°C until all compounds have eluted from the column [see Note 10 ]), carrier gas of oxygenfree nitrogen set to a flow-rate of 25–30 mL/min, flame ionization detector at 225°C, supply of hydrogen (30 mL/min) and air (300 mL/min) for the flame ionization detector, and an output linked computer, integrator, or chart recorder.
-
2.
Gas calibration standards (Supelco, Scotty I-analyzed gases, cat. no. 2-2566).
-
3.
4 mL autoclave-resistant glass vials with open-top caps and inert silicon/Teflon septum (Supelco, SIGMA-ALDRICH cat. no. 27209-U).
-
4.
1-mL glass, gas-tight syringe fitted with closable inlet valve (see Note 11 ).
-
5.
5% (v/v) DMSO in distilled H2O (filter-sterilized).
-
6.
Preculture and recovery medium (see Note 12 ).
-
7.
100-µL and 5-mL automatic pipets and sterile tips.
3 Methods
3.1 Pregrowth
Pregrowth of cultures, shoot nodes, and excised shoot-tips can be undertaken 24–72 h before cryopreservation (see Notes 6 and 13 ). The procedure involves the application of an osmotica (sucrose, sorbitol, mannitol), DMSO, a plant growth regulator (abscisic acid), and a stress-ameliorating factor (proline) at predetermined optimized concentrations.
3.2 Acclimation
Cold acclimation may be simulated for chill-sensitive species, as well as for naturally acclimating genotypes. This can be achieved by the application of a 0.75 M sucrose pregrowth treatment (13) to shoot nodes supported on semisolid growth medium 3–7 d before meristem excision (see Note 13 ). Cold-hardened dormant buds, which have undergone natural seasonal acclimation, may also be used (10,11). Alternatively, it is possible to induce cold hardiness in rejuvenated buds or in vitro plants using temperature-controlled environments (14; see Notes 14 and 15 ). It may be necessary to optimize low temperatures (−5 to 10°C), treatment duration (days to months), and temperature cycling (diurnal alternation of growth and low-temperature treatments) to achieve enhanced shoot-tip survival after cryopreservation (see Note 15 ).
3.3 Shoot-Tip Dissection
-
1.
With the aid of a dissecting microscope (×20–40) and two hypodermic needles, remove the larger expanded leaves from the shoot tips, while retaining several non- or partly expanded leaf primordia and a small amount of subjacent tissue.
-
2.
Dissection is best achieved on a medium-moistened, sterile filter paper platform in a Petri dish placed on the microscope-viewing platform.
-
3.
Trim the shoot tip to an approximate size of 1–5 mm (see Note 15 ).
-
4.
Immediately after dissection transfer shoot tips to filter paper bridges (placed in a 50-mm Petri dish or similar) soaked with pregrowth medium (see Note 16 ).
-
5.
For some species and genotypes a short recovery period after dissection may be required to overcome wounding injury. Thus, transfer dissected shoot tips from filter paper bridges to standard solid or liquid medium (see Note 17 ).
-
6.
For some germplasm the incorporation of 5% (v/v) DMSO in the recovery medium can enhance survival and help ameliorate the effects of wounding.
3.4 Chemical Cryoprotection and Controlled Freezing
This method is adapted from that originally developed for potatoes (6) and Ribes (7).
-
1.
Transfer 10–20 shoot tips to a cryovial containing an appropriate cryoprotective additive (see Subheading 2.3. ) that can be applied all at once, gradually at ambient temperature, or chilled/ice temperatures.
-
2.
Incubate at room temperature for 1 h (see Note 6 ).
-
3.
Transfer to a programmable freezer and set a freezing cycle (see Note 18 ) with parameters in the following ranges:
-
a.
Cooling rate (−0.25 to −5°C/min).
-
b.
Terminal transfer temperature (0 to −40°C).
-
c.
Holding time at temperature of transfer (30–45 min).
-
a.
-
4.
An optional “seeding” stage can also be incorporated within a range normally between −5 and −15°C, which includes a hold for 5–15 min to allow ice nucleation. This is achieved manually by touching the cryovials with an ultracold instrument (e.g., a spatula that has been briefly held in liquid nitrogen), or by initiating the automatic seeding facility on a programmable freezer (see Note 18 ).
-
5.
On completing the controlled temperature program, rapidly transfer the vials to a small liquid nitrogen-filled Dewar located by the side of the freezer (see Note 19 ).
-
6.
Subsequently transfer the vials to a large storage Dewar.
-
7.
For thawing, transfer the vials to a 45°C water bath until all the ice has melted, then place in a flow bench and wipe the outside of the vial with a tissue soaked in a sterilizing solution to remove excess and potentially contaminating water.
-
8.
Expel the vial contents onto filter papers contained in a Petri dish; the cryoprotectant is removed by capillary action.
-
9.
Transfer the shoot tips to recovery medium (see Note 2 ).
3.5 Cryoprotective Dehydration
This method is adapted from those of Niino et al. (8), Uragami (9), Bagniol and Engelmann (15), and Sherlock et al. (16).
-
1.
Remove nodal segments or shoot tips (2–5 mm) from plantlets and detach expanded leaves using a scalpel (see Note 20 ).
-
2.
Culture the prepared explants in a series of media containing 0.5–0.75 M of osmotic agent selected from sucrose, mannitol, or sorbitol prepared on standard solid medium for 1–5 days (see Notes 13 and 17 ).
-
3.
Transfer plant tissues to standard medium in order to optimize step 2 and select the protocol that maximizes dehydration with minimal loss of viability (see Note 18 ).
-
4.
After step 3 has been optimized, transfer the osmotically dehydrated tissues to airtight vessels containing activated silica gel and desiccate for 2–16 h. Alternatively, the germplasm may be desiccated in an evaporative airflow.
-
5.
Place shoot tissue on recovery medium to determine which treatment is optimal for maximum dehydration and minimal loss of viability.
-
6.
Choose the optimal pregrowth treatment and test its effectiveness in supporting postfreeze recovery after ultrarapid freezing (see Notes 13 and 17 ).
-
7.
Transfer the dehydrated shoot tips to cryovials and plunge directly into liquid nitrogen.
-
8.
Rewarm the vials at ambient temperatures.
-
9.
Transfer tissues to recovery medium (see Note 2 ).
3.6 Chemical Additive Vitrification
These methods are largely based on those originally developed by Uragami (9) and Sakai (11) and has since been adapted extensively by Kim et al. (17) and Thin et al. (18) to produce different derivative methodologies. These use different combinations of cryoprotectants applied at higher concentrations than would be the case for standard cryoprotection. They also include protocol permutations that incorporate pregrowth and dehydration treatments that assist the recovery of more sensitive genotypes (11). The basic protocols are presented with some alternative options highlighted.
3.6.1 PVS2
-
1.
Prepare PVS2 (see Subheading 2.5. ; see Note 22 ).
-
2.
Prepare a range of diluted PVS2 solutions (e.g., 10–80% [v/v] PVS2 solutions in standard liquid medium).
-
3.
Prepare a solution of standard liquid medium containing 1.2 M sucrose (see Note 23 ).
PVS3 is an alternative solution comprising 50% (w/v) sucrose and 50% (w/v) glycerol prepared in standard liquid culture medium (8).
3.6.2 Basic Vitrification Procedure
Prepare shoot tips (see Subheading 3.3. ) and construct a toxicity test (see Notes 23 and 24 ) for the PVS2 solutions as follows:
-
1.
Add on ice, chilled 1-mL aliquots of PVS2 within the range of 50–80% (v/v) to a specific concentration for 1–10 min and remove almost all the vitrification solution (see Note 25 ).
-
2.
Replace the intermediate concentrations with a stepwise, higher concentration of PVS2 solution (see Note 6 ).
-
3.
Gradually increase the concentration of PVS2 until the tissues are in the 100% solution (see Note 24 ).
-
4.
Remove the 100% PVS2 and replace with “unloading” solution containing 1.2 M sucrose in standard liquid medium. Perform two to three washes in fresh unloading solution and maintain in this solution for up to 30 min.
-
5.
Expel vial contents onto a filter paper; the paper will soak away the liquid excess.
-
6.
Transfer the shoots to recovery medium and select the treatment that permits maximum survival and cryoprotection.
-
7.
After the stepwise delivery of PVS2 has been optimized, transfer the vials directly to liquid nitrogen.
-
8.
For rewarming, place vials in a water bath at 45°C and recover shoot tips as described in steps 4 – 5 of this protocol.
3.7 Encapsulation and Dehydration
Fabre and Dereuddre (19) developed this cryoprotective approach and applied it for the first time to Solanum phureja. The following method has been adapted for Ribes spp. (20–22) and as such, has been used to assist in cryopreservation technology transfer and training programs that also incorporate PVS2 and controlled-rate freezing methodologies. It may be applied with appropriate modification and optimization to other shoot-tip systems.
-
1.
Transfer shoot-tip meristems to 3% (w/v) alginate solution (see Subheading 2.6. ) and gently swirl the vial to assist tissue immersion, taking care not to form air bubbles.
-
2.
Fill a 3-mL plastic pastette with 2 mL of alginate containing ca. five meristems, care is again required to ensure the alginate contains no air bubbles.
-
3.
Hold the pastette vertical to ensure that spherical drops are formed, and deliver droplets at ca. 1-s intervals into 0.1 M CaCl2 containing liquid culture medium to form uniform spherical beads (45–50 µL in size).
-
4.
Allow the beads to polymerize in the calcium solution for ca. 15 min.
-
5.
Pour the calcium solution and beads into a 90-mm sterile Petrie dish, and retrieve the beads containing shoot tips.
-
6.
Transfer the encapsulated germplasm to a 100-mL conical flask containing ca. 25 mL 0.75 M sucrose liquid pretreatment medium.
-
7.
Place flasks on an orbital shaker (e.g., a Gertomat M, 125 revolutions per min) for 16–22 h under standard culture conditions.
-
8.
Pour the beads from the flask into a 90-mm sterile Petri dish or through a sieve, and blot dry on sterile filter paper to remove excess surface moisture.
-
9.
Transfer the beads to the base of a sterile, empty 90-mm sterile Petri dish. Ensure the beads do not touch each other and that they are evenly distributed across the base of the dish.
-
10.
Place the dish to the back of a horizontal air stream of a laminar flow hood for 4 h, or as optimized for the germplasm type (see Notes 26 – 28 ).
-
11.
Record the temperature and relative humidity (RH) of the laminar air-flow cabinet at the onset and end point of the 4-h period to ensure quality control (see Note 29 ) between different cryopreservation experiments. RH and ambient temperature can critically influence the bead desiccation rate and final moisture content (this should be 20–25% on a fresh weight basis in order to ensure that a stable glass is form during cryopreservation).
-
12.
Place desiccated beads in a cryovial and immerse in liquid nitrogen until required.
-
13.
Warm at ambient temperature in the laminar flow hood for 20–30 min.
-
14.
Wipe the exterior of the cryovials with a sterilant solution.
-
15.
Rehydrate beads in liquid medium for 20 min to remove sucrose (see Note 30 ).
-
16.
Transfer to recovery medium (see Note 31 ).
3.8 Encapsulation-Vitrification
This method is a combination of the encapsulation technique created by Fabre and Dereuddre (19) and the vitrification protocol developed by Sakai (11). It involves encapsulation followed by treatment with PVS2 solutions. Use the basic preparative and bead-making procedures for alginate and PVS2 (see Subheadings 3.6. and 3.7. ).
-
1.
Suspend shoot-tips meristems following an appropriate pregrowth or hardening treatment (see Subheadings 3.2. and 3.3. ), in Ca2+-free 2 or 3% (w/v) Na-alginate loaded with a final concentration of 0.4 M sucrose, or a mixture of 2 M glycerol and 0.4 M sucrose.
-
2.
Dispense as droplets into 0.1 M CaCl2 solution made up in liquid culture medium containing a final concentration of 0.4 M sucrose, or a mixture of 2 M glycerol and 0.4 M sucrose.
-
3.
Incubate in this solution for 1 h at 25°C.
-
4.
Remove the encapsulated shoot-tip meristems and place in a 50- to 100-mL flask containing 100% PVS2 solution ( Subheading 2.5. ).
-
5.
Place on a reciprocal shaker for an optimized incubation time (e.g., 60 rpm up to 1 h).
-
6.
Remove beads and place in a 1.8- to 2.0-mL cryovial at 5–15 beads/vial and add 1 mL of PVS2 solution.
-
7.
Directly immerse the beads into liquid nitrogen.
-
8.
On retrieval from storage, rapidly rewarm the vials in a water bath at 35–45°C for approx 1 min.
-
9.
Drain the PVS2 solution away and replace with 1.2 M sucrose unloading solution for 10 min.
-
10.
Blot the beads dry of excess moisture and place on recovery medium.
-
11.
Hirai et al. (12) also report a variation on this method (see Notes 26 – 31 ), which involves an additional evaporative drying step.
-
a.
Prepare encapsulated shoot tips in 2% (w/v) alginate as described in steps 1 – 2 .
-
b.
Transfer to 0.8 M sucrose prepared in liquid culture medium and incubate for 16–20 h on a rotary shaker at approx 60 rpm.
-
c.
Remove, blot dry on sterile filter papers, and transfer to sterile glass Petri dishes containing sterile heat-activated 50 g silica gel and sealed with Parafilm.
-
d.
Desiccate for 2–5 h optimized to a moisture content of approx 25% on a fresh weight basis.
-
e.
Proceed as in steps 6 – 10 , but omitting the application of PVS2 and sucrose unloading stages.
-
a.
3.9 Droplet Freezing
This method is based on that originally developed for cassava and potato (23); see ref. 2 for a review of droplet freezing. It uses DMSO as the cryoprotectant. More recently it has been applied in combination with vitrification solutions (24). Shoot tips may require pregrowth treatments before proceeding with the protocol ( Subheadings 3.2. and 3.3. ).
-
1.
Prepare 10% (v/v) DMSO in liquid medium ( Subheading 2.3. ).
-
2.
Dispense using an analytical 2.5–20 µL Gilson Pipetteman or equivalent 2.5-µL droplets of cryoprotectant (5–10 droplets/strip dependent on loading size) onto the surface of sterile, aluminium foil strips ( Subheading 2.8. ).
-
3.
Transfer one shoot-tip meristem to each droplet.
-
4.
Using liquid nitrogen-tolerant sterile forceps drop the foils directly into liquid nitrogen containing a small (5–10 mL) Dewar located in a laminar flow cabinet.
-
5.
Transfer the foils to cryovials at two foils per vial.
-
6.
Store in liquid nitrogen storage vessels.
-
7.
For rewarming, remove the foils from the vials and place on liquid medium at ambient room temperatures (~25°C). The shoots will dislodge on rewarming and can be retrieved and plated onto the recovery medium.
3.10 Postfreeze Recovery Assessments
It is not always possible to perform viability assays based on microscopy (e.g., vital staining) on shoot tissues that are often optically too dense. Recovery can be assessed initially on survival and then regeneration, and it is the latter that is the definitive assessment of protocol success.
-
1.
Within 7 d of thawing, examine tissues under a binocular microscope.
-
2.
Record the number of shoot tips, which are green and have expanded leaf primordia (see Note 32 ).
-
3.
Continue to undertake weekly assessments and record a time-course of recovery events as leaf expansion, callus formation, shoot and plantlet regeneration (see Note 33 ).
-
4.
Evaluate recovery as (1) normal events (plantlet formation in the absence of callogenesis and adventitious shooting), and (2) abnormal events (callus and adventitious shoot development) (see Note 33 ).
3.11 Volatile Headspace Analysis of Cryopreservation Injury in Shoot Tips
The survival and regeneration of shoot tips following cryopreservation often varies between genotypes and this can require lengthy optimization steps to determine the best cryopreservation protocol. This approach does not identify the underlying physical and physiological causes of cryopreservation injury in recalcitrant species, making it difficult to develop protocols that match the physiological requirements (e.g., desiccation and cold sensitivities) of each species. A complementary approach is to use analytical techniques to measure stress responses in tissues at each stage of a cryopreservation protocol so that critical points for recovery can be identified. This approach has been successfully applied to dedifferentiated cultures of Daucus carota (25) and algae (26) using volatile markers of oxidative stress and were used to create improved cryopreservation protocols. The method has now, and as follows, been optimized for the profiling of cryoinjury in shoot-tip meristems. The small mass of the shoot tips and the time-consuming nature of meristem excision manipulations often restricts the production of sufficient tissue mass to detect stress markers using analytical procedures based on calorimetry. The nondestructive and sensitive nature of headspace volatile analysis eliminates this problem as it reduces the need for large numbers of meristems to be surveyed. Moreover, the issue of compound detection from tissues with low mass can be circumvented by increasing the time shoot tips are sealed in air-tight vials, allowing more time for volatile accumulation before sampling. The nondestructive attribute of this method also allows time-course experiments to be conducted on the same batch of vials, and assuming sterile conditions are maintained, the same tissues can be assessed for recovery. This means direct comparisons can be made between volatile markers and shoot-tip recovery.
The following headspace protocol describes the detection of ethane, ethylene, pentane, and methane. Ethane, ethylene, and pentane are secondary products of lipid peroxidation, and the detection of these compounds during cryopreservation may indicate membrane damage (25,26). Ethylene and ethane are produced from the peroxidation of linolenic acid, and pentane from linoleic acid (27,28). A second, and usually more abundant, source of ethylene in plants is produced enzymatically (29) from 1-aminocyclopropanecarboxylic acid. An intact membrane is considered essential for 1-aminocyclopropanecarboxylic acid-dependant ethylene biosynthesis, and a reduction in ethylene production during in vitro plant cryopreservation intimates that membrane damage has occurred (25). The growth-regulating effects of ethylene also influence growth and development, such as inhibiting in vitro shoot growth and promoting senescence (29,32), and these could impact postcryopreservation regeneration. Ethylene may also affect the oxidative stress status of the tissue as it can induce lipid peroxidation, deplete ascorbate and α-tocopherol levels, and induce antioxidant enzyme activity (30,31).
Methane is produced during DMSO scavenging of hydroxyl radicals (·OH), one of the most reactive and damaging free radicals known (25). The ·OH reacts with DMSO to produce methane sulfinic acid and a methyl radical (see Fig. 1 ), which in turn produces either methane or methanol and formaldehyde (33).

Reaction between dimethylsulfoxide and the hydroxyl radical (·OH) to produce methane. ·CH3, methyl radical; FA, formaldehyde; MSA, methane sulfinic acid.
The following protocol has been adapted to measure methane, ethane, ethylene, and pentane in cryopreserved shoot-tips cultures. It may also detect other small-chained products of lipid peroxidation, such as alcohols and aldehydes (28), allowing further insight into the stress responses during cryopreservation.
-
1.
Measure the empty weight of each capped vial.
-
2.
Submerge each vial in H2O, cap, and record the full weight.
-
3.
Calculate the empty headspace volume of each vial by subtracting the empty weight from the full weight (normally ~5.1 mL for 4-mL vials).
-
4.
Autoclave the vials, lids, and septa in the absence of plastic. Then immediately vent each vial in a laminar flow bench for 1–2 h to remove any contaminating volatiles.
-
5.
To each vial add a known volume of medium using a 5-mL automatic pipet. Once the medium is solidified, ventilate the vials for a further 10 min (see Note 34 ).
-
6.
Add 75 µL of 5% (v/v) DMSO to each vial, cap, and record the weight.
-
7.
Add five alginate-encapsulated meristems or 10 nonencapsulated meristems to each vial, ensuring the bead or tissue is in contact with the medium and DMSO layer. Seal the vials, note the time, and record the weight. Also incorporate control vials (see Note 35 ).
-
8.
Place vials in culture room under normal temperature and light conditions.
-
9.
Calculate the headspace volume (; see Note 36 ): where: vol. = volume (L) and DMSO = dimethylsulfoxide.
-
10.
Turn on the GC, turn on the flow of carrier gas, and set the temperature for the injector, column, and detector. Leave to equilibrate overnight.
-
11.
On the day of headspace sampling (see Note 37 ), turn detector on and equilibrate for 30 min.
-
12.
Inject 1 mL of standard into the GC and calibrate the integrator. Repeat at least three times (see Note 38 ).
-
13.
Flush the syringe in the flow bench, take a 1-mL sample from a vial, and record the time (see Note 39 ).
-
14.
Once all the vials have been sampled, open the vials and allow them to vent for 10 min.
-
15.
If required, reseal each vial and record the time. Repeat steps 11 – 15 as needed (see Note 40 ).
-
16.
Calculate volatile production (): where: r volatile = rate of volatile production; s conc = concentration in a 1-mL sample (µL/L); b conc = blank concentration in a 1-mL sample (µL/L); s vol = headspace volume (L); b vol = blank headspace volume (L); t2 − t1 = time between sealing and removal of a 1-mL sample(s); FW = sample tissue weight (g); T = temperature during sampling (K); 22.4 = L of space occupied by 1 mol of gas at 273.15 K and 1 atm; 106 = conversion factor from L to µL.
4 Notes
-
1.
Tissue culture growth optimization must precede cryopreservation applications.
-
2.
Recovery medium composition is species specific and may require the incorporation of growth regulators for shoot regeneration.
-
3.
Cryoprotectants and pregrowth additives must be of high purity; spectroscopically pure DMSO is recommended.
-
4.
Cryoprotectants and vitrification solutions are usually prepared in standard liquid culture medium; it is advisable to check medium pH after incorporating the additives.
-
5.
Where possible, cryoprotectant mixtures should be filter sterilized, this may be technically difficult for high-viscosity solutions for which autoclaving may be the only practical option.
-
6.
Concentration, duration, and temperature (ambient or on ice) of exposure to PVS2, sugars, and other cryoprotectants is genotype specific, this range is only a guide.
-
7.
Uniform bead size is critical for reproducible bead dehydration and desiccation so it is important to use an alginate dispenser of a constant size. Adjusting the pipet tip diameter with a hot scalpel can accommodate larger structures. The authors’ use Fisherbrand (Fisher Scientific, Loughborough, UK) 5 mL plastic pastettes. However, any pastette of similar type will suffice, but it is important to standardize the pastette used, as different manufacturers products have different apertures. If undertaking technology transfer with other laboratories ensure that an identical or equivalent type of Pastette is used. Size difference in beads will lead to nonuniform desiccation profiles on air and silica gel drying.
-
8.
Prepare calcium-free standard liquid medium (Ca2+ salts of vitamins accepted) and add 3% (w/v) sodium alginate (SIGMA low-viscosity kelp alginate as the sodium salt). It is very difficult to solubilize alginate and this is best achieved by: (1) adding the alginate to liquid medium on a magnetic stirrer and then heating to boiling while agitating vigorously, dispense small amounts of alginate, step by step; (2) the alginate solution may be vigorously agitated by shaking and then autoclaved. Discard flocculated solutions.
-
9.
Knowledge of cold-hardiness status is especially important for woody perennial species and seasonal screening for freeze tolerance is recommended to determine at which time in the growth cycle buds are best able to survive cryopreservation. Laboratory-induced cold hardiness is frequently used to enhance poststorage recovery. A suggested procedure for plants normally maintained at 20–25°C in vitro is cold acclimation for 7–10 d using 8 h days at 15–22°C and 16 h nights at −1 to 4°C. If specialist growth room facilities are not available for cold hardening it is possible to improvise and use manual transfers from a refrigerator set at the lowest setting to a standard growth room.
-
10.
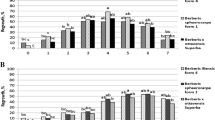
A shorter isothermal (constant temperature) program can be used when measuring C1 to C3 compounds. Gradient separation of C1 to C6 compounds requires 25–30 min ( Fig. 2 ), whereas isothermal separation of C1 to C3 compounds requires 2–10 min ( Fig. 3 ). A temperature of 60°C is suggested for isothermal separation.
-
11.
If a glass syringe is unavailable, a plastic syringe can be used as an alternative as long as samples are injected into the GC immediately to minimize syringe compound adsorption.
-
12.
To determine volatile production during each phase of cryopreservation, vials will need to contain the corresponding medium.
-
13.
The duration of exposure and loading of sucrose and other compounds (e.g., polyols, mannitol, proline, abscisic acid) used as a pregrowth additive or to simulate cold acclimation will require genotype-specific optimization for concentration.
-
14.
Tissues harvested ex situ may exhibit different natural cold-hardiness responses to freezing. For cold acclimation in vitro it will be necessary to optimize the cold-hardening regimes on a species and sometimes genotype basis.
-
15.
It is important to standardize and optimize shoot tip and nodal cutting size.
-
16.
Shoot tips rapidly dehydrate after dissection and they should be transferred to a suitable medium immediately after removal. Ensure that this has the appropriate osmotic balance relative to the pregrowth medium from which the shoots were originally removed.
-
17.
Newly dissected tissues are frequently sensitive to wounding, which is exacerbated by freezing. A short period of pregrowth (e.g., 16–48 h) in the presence of chemical additives such as (at 1–5% [v/v]) DMSO can significantly enhance recovery. Dehydrating pregrowth treatments applied as osmotica do not provide adequate protection against cryogenic treatments and additional cryoprotection is required. Comparing the efficacy of several treatments leads to the development of a pregrowth strategy. Pregrowth additives are incorporated into solid media or in filter papers moistened with additive-containing liquid media. The most commonly used osmotic pregrowth additives are, sorbitol, mannitol, and sucrose.
-
18.
Modern programmable freezers consist of a freezing chamber cooled by liquid nitrogen. With the use of temperature probes (connected to the sample and the chamber) and precise computer programming it is possible to investigate a wide range of cooling/freezing parameters. With an appropriate output device (e.g., chart recorder/PC) the temperature at which extracellular ice is formed (latent heat of crystallization) may be determined with reasonable accuracy. This event, termed nucleation, can mark the onset of intracellular dehydration and it can be an important factor in developing a controlled cooling method for shoot tips. Extracellular ice formation causes a vapor pressure deficit, which is compensated by the movement of intracellular water to the outside of the cell. The effect is “cryoprotective” as the amount of intracellular water available for ice formation is reduced. In the absence of external intervention, it is possible for extracellular ice nucleation to occur randomly; however, this may be problematic and lead to variable freezing responses. The control of nucleation is thus an important consideration in developing slow-freezing methods for shoot tips. Some programmable freezers are fitted with a device that initiates nucleation by mechanically agitating the cryogenic samples. It is also possible to induce nucleation by touching the outside of the tube with a liquid nitrogen-chilled instrument.
-
19.
It is important that vials are plunged immediately into liquid nitrogen after they are removed from the programmable freezer. Rewarming of the samples during this transition is detrimental to the tissues. For convenience a small Dewar (e.g., of 1-L capacity) can be located near the programmable freezer and used to transport vials to the long-term storage Dewar site.
-
20.
Nodal segment stem size may be difficult to standardize and optimization is required.
-
21.
Determination of water loss (using controlled oven drying) aids in the development of cryoprotective-dehydration protocols. However, it is important to use an appropriate oven temperature (e.g., 105°C) and duration time for accurate moisturecontent determinations. Moisture-content status should be accurately determined on a fresh and dry weight basis as appropriate. In humid environments, transfers to and from the drying ovens should be undertaken over activated silica gel desiccators.
-
22.
The mixture will be extremely viscous and takes some time to completely dissolve in solution. Gradual addition of the cryoprotectants during vigorous agitation on a magnetic stirrer is recommended.
-
23.
To reduce osmotic shock, thawed tissues may be sequentially transferred to a range of media containing decreasing concentrations of a nontoxic osmoticum such as sucrose.
-
24.
Shoot tips may not survive direct exposure to 100% PVS2 and stepwise addition of PVS2 in an increasing concentration series is necessary. Following vitrification and exposure to liquid nitrogen the shoots are initially recovered in an “unloading” solution (1.2 M sucrose). This prevents osmotic shock while replacing the toxic vitrification solution with a less damaging sucrose solution.
-
25.
Gradually add increasing concentrations of PVS2 solution, e.g., in a range of 50–100% (v/v), exposure duration is short (5–30 min), and requires species optimization. Adding chilled solutions on ice may reduce the toxic effects of vitrification cocktails. It may be possible to expose tissues to higher concentrations of PVS2 if applied at 0°C.
-
26.
This is a critical point (see also Note 21 ) as it is essential that the beads desiccate uniformly and reproducibly between different cryopreservation experiments. Desiccation rates can be determined by assessing the changes in moisture content of a batch of empty (do not include meristems) beads before and after 4 h of desiccation. This can be determined from bead fresh and dry weights as follows:
-
27.
New desiccation profiles should be determined when working in different laminar flow benches and/or laboratories to ensure the bead residual water content is optimum after desiccation. Note that alginate beads become extremely hard when airdehydrated, and are sometimes difficult to manipulate. It is important that bead dry weights are determined at the correct temperature (105°C) as lower temperatures will underestimate their water content.
-
28.
Failure to take note of environmental quality control parameters will result in nonuniform bead desiccation.
-
29.
Another option in high RH environments is to desiccate beads over activated silica gel.
-
30.
Encapsulated shoot tips may be rewarmed at ambient temperatures and placed on culture medium. Beads rehydrate within 1 h of placing on solid medium and can be rehydrated by placing in liquid culture medium for ca. 30 min following rewarming and before placing on recovery medium.
-
31.
Regenerating shoots usually grow out of the bead, although this is not the case for all species. Removal from the alginate may be necessary for regeneration to proceed.
-
32.
This is usually the first postfreeze recovery response.
-
33.
Shoot-tip cryopreservation is successful if whole plants are regenerated from the cryopreserved meristem. Recovery proceeds via several morphological patterns of development. Leaf expansion, callus, and root formation indicate viability but not regeneration, which should proceed via an original meristem (primary or axillary) and not an indirect adventitious route (e.g., organogenesis from a callus). This criterion is important to ensure the genetic stability of plants regenerated from cryopreserved tissues. Quantitatively acceptable levels of recovery are of obvious importance. In the first stages of method development, survival levels approaching 50% of the total number of shoot tips frozen may be considered encouraging. The application of the technique on a reproducible and routine basis demands high levels of regeneration (e.g., approx 80% of the total number of shoots frozen).
-
34.
For 4-mL vials, a medium volume of 3.5 mL is recommended as this allows sufficient space for five alginate-encapsulated meristems; volume will need modification for meristem size. A large headspace volume will make volatile detection more difficult.
-
35.
Control vials should be prepared with medium alone, medium + DMSO, and tissue without DMSO. This will measure background volatiles in the laboratory (medium alone), medium methane production (difference between medium ± DMSO), tissue methane production not produced by the reaction between DMSO, and the hydroxyl radical (difference between tissue and medium-only vials without DMSO). All vials should be sealed at the same time to prevent background differences in volatiles.
-
36.
It is often difficult to accurately measure the volume of small pieces of plant tissues. For nonencapsulated meristems, determine the average mass of a meristem and use this number to estimate the collective meristem mass in each vial (assume 1 g of tissue equates to a volume of 1 mL [density of water]). For encapsulated tissues, weigh the vial before and after the addition of beads. Use this weight to estimate bead volume, again assuming 1 g = 1 mL.
-
37.
The sealing time before headspace sampling will depend on the tissue mass. For some species, the meristems will need sealing for up to 7 d to allow the volatiles to accumulate to detectable concentrations. It is equally important not to seal the meristems for too long, as this may induce anaerobiosis (detected as ethanol and acetaldehyde peaks during gradient separation; Fig. 2 ).
-
38.
Blocked needles and leaky syringes cause considerable error. To check the syringe is gas tight, fill it with air, block the needle outlet by pushing it gently into a rubber bung or by closing the needle valve, submerge the syringe under H2O, and gently press the syringe plunger. If air bubbles escape from any part of the syringe other than the needle, then the syringe is no longer gas tight. A blocked needle often occurs if the GC septum is too tight. A blockage can also occur while puncturing a vial septum. A blocked needle is difficult to detect during injection if the syringe plunger does not move freely in the barrel. To check if the needle is blocked, fill the syringe with air, submerge the needle tip under water, and gently press the syringe plunger. The absence of air bubbles indicates a needle blockage.
-
39.
If the sample volume to be withdrawn is greater than 10% of the headspace volume, a negative pressure will develop inside the vial and syringe needle, which upon withdrawal of the needle from the vial, will cause air to move uncontrollably into the syringe diluting the sample. This problem is solved by closing the valve on the syringe needle prior to withdrawal from the vial, or by adding a known volume of sterile air to the vial before sample withdrawal (added volume has to be included in headspace volume calculations). A disadvantage of adding sterile air is that the sample will be diluted, which may make peak detection more difficult. Solid phase microextraction provides another viable sampling option.
-
40.
After a number of injections high molecular weight compounds may accumulate in the column and cause a noisy baseline. These compounds can be removed by increasing the column temperature to 150–170°C for 1–4 h. It is recommended that this column cleaning procedure is routinely undertaken before and after each experiment.

Gradient separation of headspace volatiles from Ribes sanguineum (cv King Edward VII) shoot tips on Ribes (RIB) medium containing 0.75 M sucrose.

Isothermal separation of headspace volatiles from Ribes ciliatum shoot tips and gas standard (15 µL/L for methane and ethane, 30 µL/L for ethylene).
References
Mazur, P. (2004) Principles of cryobiology. In: Life in the Frozen State, (Fuller, B., Lane, N., and Benson, E. E., eds.), CRC Press, Boca Raton, FL, pp. 3–66.
Benson, E. E., Johnston, J., Muthusamy, J., and Harding, K. (2005) Physical and engineering perspectives of in vitro plant cryopreservation. In: Plant Tissue Culture Engineering, (Dutta & Gupta, S. and Ibaraki, Y., eds.), Springer, Netherlands, pp. 441–476.
Benson, E. E. (1999) Plant Conservation Biotechnology. Taylor and Francis, London, UK.
Benson, E. E. (2004) Cryo-conserving plants and algae: historical perspectives and future challenges. In: Life in the Frozen State, (Fuller, B., Lane, N. and Benson, E. E., eds.), CRC Press, Boca Raton, FL, pp. 299–328.
Fuller, B., Lane, N., and Benson, E. E. (2004) Life in the Frozen State. CRC Press, Boca Raton, FL.
Harding, K., Benson, E. E., and Smith, H. (1991) The effects of tissue culture duration on post-freeze survival of shoot-tips of Solanum tuberosum. CryoLetters 12, 17–22.
Reed, B. A., Dumet, D. J., Denoma, J. M., and Benson, E. E. (2001) Validation of cryopreservation protocols for plant germplasm conservation: A Pilot study using Ribes L. Biodiversity and Conservation 10, 939–949.
Niino, T., Sakai, A., and Yakuwa, H. (1992) Cryopreservation of dried shoot tips of Mulberry winter buds and subsequent plant regeneration. CryoLetters 13, 51–58.
Uragami, U. (1991) Cryopreservation of Asparagus (Asparagus officinalis L.) cultured in vitro. Res. Bull. Hooaido, Natl. Agric. Exp. Stn. 156, 1–37.
Sakai, A. (2000) Development of cryopreservation techniques. In: Cryopreservation of Tropical Plant Germplasm: Current Research Progress and Application, (Engelmann, F. and Takagi, H., eds.), JIRCAS International Agriculture Series No 8, IPGRI, Rome, Italy, pp. 1–7.
Sakai, A. (2004) Plant cryopreservation. In: Life in the Frozen State, (Fuller, B., Lane, N. and Benson, E. E., eds.), CRC Press, Boca Raton, FL, pp. 329–346.
Hirai, D., Shirai, S., and Sakai, A. (1998) Cryopreservation of in vitro-grown meristems of strawberry (Fragaria X ananassa Duch.) by encapsulation-vitrification. Euphytica 101, 109–115.
Dumet, D., Chang, Y., Reed, B. M., and Benson, E. E. (2000) Replacement of cold acclimatization with high sucrose pre-treatment in blackcurrant cryopreservation. In: Cryopreservation of Tropical Plant Germplasm: Current Research Progress and Application, (Engelmann, F. and Takagi, H., eds.), JIRCAS International Agriculture Series No 8, IPGRI, Rome, Italy, pp. 385–387.
Reed, B. M. (1988) Cold acclimation as a method to improve survival of cryopreserved Rubus meristems. CryoLetters 9, 166–171.
Bagniol, S. and Engelmann, F. (1991) Effects of pregrowth and freezing conditions on the resistance of meristems of date palm (Phoenix dactylifera L. var. Bou Sthammi Noir) to freezing in liquid nitrogen. CryoLetters 12, 279–286.
Sherlock, G., Block, W., and Benson, E. E. (2005) Thermal analysis of the plant encapsulation/dehydration protocol using silica gel as the desiccant. CryoLetters 26, 45–54.
Kim, J., Kim, H.-H., Baek, H.-Y., Cho, E.-G., Kim, Y.-H., and Engelmann, F. (2005) Changes in sucrose and glycerol content in garlic shoot-tips during freezing in PVS3 solution. CryoLetters 26, 103–112.
Thienh, N. T., Takagi, H., and Sakai, A. (2000) Cryopreservation of in vitro-grown apical meristems of some vegetatively propagated tropical monocots by vitrification. In: Cryopreservation of Tropical Plant Germplasm: Current Research Progress and Application, (Engelmann, F. and Takagi, H., eds.), JIRCAS International Agriculture Series No 8, IPGRI, Rome, Italy, pp. 227–232.
Fabre, J. and Dereuddre, J. (1990) Encapsulation-dehydration a new approach to cryopreservation of Solanum shoot-tips. CryoLetters 11, 413–426.
Reed, B. A., Dumet, D. J., DeNoma, J. M., and Benson, E. E. (2001) Validation of cryopreservation protocols for plant germplasm conservation: A Pilot study using Ribes L. Biodiversity and Conservation 10, 939–949.
Reed, B. M., Schumacher, L., Dumet, D., and Benson, E. E. (2005) Evaluation of a modified encapsulation-dehydration cryopreservation procedure incorporating high sucrose pretreatments for the conservation of Ribes germplasm. In Vitro Plant 41, 431–436.
Reed, B. M., Kovalchuk, I., Kushnarenko, S., et al. (2005) Evaluation of critical points in technology transfer of cryopreservation protocols to international plant conservation laboratories. CryoLetters 25, 341–352.
Menuhr-Schaffer, A., Schumacher, H. M., and Mix-Wagner, G. (1997) Long-term storage of old potato varieties by cryopreservation of shoot-tips in liquid nitrogen. Plant Genetic Resources Newsletter 111, 19–24.
Leunufna, M. and Keller, E. R. J. (2005) Cryopreservation of yams using a vitrification modified by including droplet method: effects of cold acclimation and sucrose. CryoLetters 26, 93–102.
Benson, E. E. and Withers, L. A. (1987) Gas chromatographic analysis of volatile hydrocarbon production by cryopreserved plant tissue cultures—a nondestructive method for assessing stability. CryoLetters 8, 35–46.
Fleck, R. A., Benson, E. E., Bremner, D. H., and Day, J. G. (2000) Studies of free radical-mediated cryoinjury in the unicellular green alga Euglena gracilis using a non-destructive hydroxyl radical assay: a novel approach for developing protistan cryopreservation strategies. Free Radic. Res. 32, 157–170.
Frankel, E. N. (1982) Volatile lipid oxidation products. Prog. Lipid Res. 22, 1–33.
Frankel, E. N. (1999) Antioxidant activity by headspace gas chromatography of volatile oxidation products of ω-6 and ω-3 polyunsaturated lipids. Methods Enzymol. 299, 190–201.
Yang, S. F. and Hoffman, N. E. (1984) Ethylene biosynthesis and its regulation in higher plants. Ann. Rev. Plant Physiol. Plant Mol. Biol. 35, 155–189.
Biddington, N. L. (1992) The influence of ethylene in plant tissue culture. Plant Growth Regul. 11, 173–187.
Ievinsh, G., Kruzmane, D., Rusite, E., Arente, G., and Gertnere, D. (2000) Modulation of Solanum tuberosum L. morphogenesis and antioxidant status in a stem explant culture by limitation of gas exchange: putative effects of ethylene. J. Plant Physiol. 156, 717–723.
Munné-Bosch, S., Peáuelas, J., Asensio, D., and Llusià, J. (2004) Airborne ethylene may alter antioxidant protection and reduce tolerance of holm oak to heat and drought stress. Plant Physiol. 136, 2937–2947.
Babbs, C. F. (1989) Scatchard analysis of methane sulfinic acid production from dimethyl sulfoxide: a method to quantify hydroxyl radical formation in physiologic systems. Free Radic. Biol. Med. 6, 493–503.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2007 Humana Press Inc., Totowa, NJ
About this protocol
Cite this protocol
Benson, E.E., Harding, K., Johnston, J.W. (2007). Cryopreservation of Shoot Tips and Meristems. In: Day, J.G., Stacey, G.N. (eds) Cryopreservation and Freeze-Drying Protocols. Methods in Molecular Biology™, vol 368. Humana Press. https://doi.org/10.1007/978-1-59745-362-2_12
Download citation
DOI: https://doi.org/10.1007/978-1-59745-362-2_12
Publisher Name: Humana Press
Print ISBN: 978-1-58829-377-0
Online ISBN: 978-1-59745-362-2
eBook Packages: Springer Protocols