
Abstract
Traumatic brain injury (TBI) is the leading cause of death and disability for people under 45 years of age. Clinical TBI is often the result of disparate forces resulting in heterogeneous injuries. Preclinical modeling of TBI is a vital tool for studying the complex cascade of metabolic, cellular, and molecular post-TBI events collectively termed secondary injury. Preclinical models also provide an important platform for studying therapeutic interventions. However, modeling TBI in the preclinical setting is challenging, and most models replicate only certain aspects of clinical TBI. This chapter details the most widely used models of preclinical TBI, including the controlled cortical impact, fluid percussion, blast, and closed head models. Each of these models replicates particular critical aspects of clinical TBI. Prior to selecting a preclinical TBI model, it is important to address what aspect of human TBI is being sought to evaluate.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Traumatic brain injury
- Preclinical models
- Controlled cortical impact
- Closed head injury
- Fluid percussion
- Blast injury
1 Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability for people under 45 years of age [1]. Worldwide, in excess of 10 million deaths or hospitalizations are attributable to TBI each year, and more than 57 million people currently are living with the sequelae of TBI [1]. While recent estimates suggest that the disease burden of TBI continues to increase, few targeted therapeutic interventions have proven effective for this common injury. Most therapeutic interventions in TBI are tested in the preclinical setting, using animal models to simulate the pathophysiology of human TBI. Understanding the issues and challenges related to utilizing animal models to study human TBI pathophysiology is a vital first step in translating these models to the clinical setting. This chapter aims to provide a broad overview of some of the most commonly employed animal models of TBI, to identify practical as well as translational issues in both the execution and application of these models.
1.1 Pathophysiology of TBI
TBI is defined as damage to the brain resulting from an external mechanical force, often leading to temporary or permanent impairment of cognitive, physical, and psychosocial functions. The pathophysiology of TBI can be divided into two distinct processes: primary injury and secondary injury. The primary injury is the result of the immediate mechanical force, which can include diverse mechanisms such as blast wave, crush, impact, penetration, or rapid acceleration or deceleration. These diverse mechanisms can manifest in a wide array of primary injuries including contusion, hemorrhage, and axonal shearing. It is important to note that clinical TBI is often the result of a heterogeneous mixture of mechanical forces leading to mixed primary injuries. Interventions targeting primary injury are essentially preventative, and only effective if they preclude or mitigate primary injury. Examples of interventions targeting primary injury would include measures such as mandatory bike helmet laws or improved airbag technology.
In contrast, interventions targeting secondary injury may have a prolonged window in which to act. After the immediate primary injury is sustained, secondary injury develops over minutes to months to possibly years. Secondary injury reflects a complex cascade of metabolic, cellular, and molecular events including glutamate excitotoxicity, disordered cellular calcium homeostasis, mitochondrial dysfunction, inflammation, apoptosis/necroptosis, diffuse axonal injury (DAI), increased free radical generation and lipid peroxidation. In the extreme, secondary injury can lead to cell death, diffuse inflammation, and brain atrophy.
Despite the long window in which mitigation of secondary injury could occur, promising therapeutic interventions in the preclinical setting have failed to translate to clinical trials, with more than 30 failed TBI clinical trials based on successful preclinical studies [2]. While many reasons for failed TBI trials have been cited, including poor central nervous system drug penetration, delayed treatment initiation, heterogeneity across treatment sites, and insensitive outcomes measures [3], one important factor is the heterogeneity of clinical TBI compared to preclinical TBI. Indeed, clinical TBI often reflects a mixture of primary injuries as well as host-specific differences in the secondary injury response. In contrast, preclinical TBI studies rarely account for confounders including location, nature and severity of the primary injury, preexisting medical conditions, genetic background, age, gender, and illicit and prescribed drug use (to name a few). Thus, a promising therapeutic intervention in the preclinical setting may fail in the face of all the clinical confounders for which it was not tested.
Another important caveat to utilizing animal models of TBI is the lack of common terminology around injury severity. In human TBI trials, the Glasgow coma scale (GCS) is the primary means for assessing initial injury severity and the Glasgow outcome scale (GOS), or its extended version (GOSe), is the primary method for assessing outcomes. Whether or not these scales provide the optimal assessment tools is a source of controversy, but irrespective of this controversy, they do provide a common language to describe injury severity and outcomes. No such common language exists in preclinical trials. With no widely adopted common scoring system for injury severity in animal models of TBI, histological changes and functional tests provide the most reliable estimates of severity of TBI. However, subtle changes in injury mechanisms or devices as well as differences in techniques between labs, can sometimes make the comparison of various animal TBI models precarious at best.
Despite these limitations, animal models of TBI are extremely important tools to study the biomechanical, cellular and molecular events after TBI, many of which cannot be feasibly addressed in the clinical setting. The purpose of this chapter is to review some of the most commonly used methods of preclinical TBI both in terms of clinical relevance and potential technical pitfalls. Understanding these models are important prior to developing new models that better recapitulate the spectrum of human TBI.
2 General Experimental Model Approach
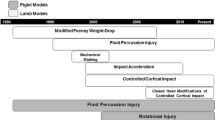
Prior to selecting a preclinical TBI model, it is important to address what aspect of human TBI is being sought to evaluate. For example, an investigator studying sports-related TBI would be wise to avoid a model with high-associated mortality, intraparenchymal hemorrhages, and skull fractures. New model development can take months to years, and translating established models to a new laboratory setting may also require significant effort and time. Understanding sources of variability in the model and establishing baseline functional and histological outcomes are particularly important prior to testing therapeutic interventions. In an attempt to help standardize preclinical TBI studies, the National Institutes of Health (NIH) has recommended the use of common data elements for preclinical TBI models (Table 1).
2.1 Controlled Cortical Impact Injury Model
2.1.1 Introduction
The controlled cortical impact (CCI) model has been utilized mostly to model moderate or severe human TBI. The classic CCI device employs a pneumatic or electromagnetic mechanism to drive a rigid impactor onto the exposed, intact dura, resulting in acute subdural hematoma, axonal injury , blood–brain barrier (BBB) dysfunction, and cortical tissue loss [4, 5]. CCI has been applied to species including mice, rats, ferrets, swine, and monkeys. One of the key advantages of CCI is that injury severity can be carefully calibrated by altering mechanical factors such as time, velocity, and depth of impact. In contrast to weight drop models, CCI lacks a rebound injury and produces a more focal injury compared to FPI. Another advantage of CCI is its use in developing novel therapeutic treatments for brain injury [6–11].
2.1.2 Methods
In rodent CCI models the device is hooked up to an air tank, injury controller, and mounted on a crossbar where the impactor can be adjusted to a range of desired angles of injury. Animals are induced using isoflurane or a comparable anesthetic before the head is secured in a stereotactic frame. A midline incision is made on the top of the head and the periosteum removed to prevent interference with the drill. Depending on desired area of injury, a unilateral [12] or bilateral [13] craniotomy is performed, most commonly, between the bregma and lambda. In both models the diameter of the craniotomy ranges from 4 to 6 mm. Injured mice can receive a mild–severe injury depending on the depth of brain deformation caused by the piston (0.2–1.2 mm) while sham animals receive only a craniotomy. The impact velocity can be adjusted from 0.5 to 10 m/s and impact duration ranges from 25 to 250 ms.
Once the skull flap is removed, using the injury controller the piston is set in the down position and the exposed brain is raised so that it is just touching the piston. The piston is then set back up, the apparatus is unlocked and the dial adjusted to the desired depth. After relocking, the piston is fired onto the brain using the controller. After completion of the injury, the incision is sutured, antibiotic cream is applied, and the animal is placed in a separate cage to wake up (see Notes 1 – 4 ). Common data elements for CCI studies are found in Table 2.
2.1.3 Clinical Relevance
Animals subjected to CCI have a wide range of functional deficits, which are highly related to both the depth of deformation and the velocity of the impact [12, 14]. Functional deficits after CCI include but are not limited to sensorimotor deficits (wire grip, rotarod, wire grip) memory deficits (Morris Water Maze (MWM) performance) and increased depression, anxiety , and impulsivity (forced swim, elevated-plus maze, acoustic startle) [14–18]. Some functional deficits can be persistent up to 1 year post-CCI injury. The most marked histopathological change after CCI is the development of a cavitary lesion. However, CCI can result in a spectrum of anatomic injury including diffuse axonal injury (DAI) [5, 19]. Beta-amyloid precursor protein (APP) positive cells are increased in both the contused cortex and ipsilateral hippocampus as early as 6 h. The highest immunoreactivity levels occur on days 1–3 after injury and the positive cells can be seen for weeks [20]. Neuronal degeneration is also observed in early and late stages after injury [21, 22]. Due of the contusive nature of this injury, CCI causes inflammation and a dramatic increase in cytokines and chemokines [15, 20, 23]. Microglia activation is also accumulated in the lesion [15, 24].
2.1.4 Caveats
Most clinical cases of contusive injuries have a concussive injury as well. However, the CCI model lacks a concussive impact because the animals head is fixed in a stereotactic frame. To overcome this the experimental animals can be subjected to a concussion immediately following the contusive injury [25]. The spectrum of functional deficits after CCI is dependent on the localization of injury. Injuries located in the posterior cortex result in transient motor deficits as well as impaired memory and sensory function [15].
2.2 Fluid Percussion Injury
2.2.1 Introduction
Although it was originally created for use in larger animals, Fluid Percussion Injury (FPI) has become of the most commonly used methods of creating a non-penetrating head injury in rodents [26]. In FPI models, TBI is the result of a fluid pressure pulse transmitted to the intact dura through a craniotomy. The FPI device consists of a cylindrical Plexiglas reservoir of sterile saline with one end attached to a transducer which, through a tube, connects to the cap cemented to the skull. The injury is generated by a pendulum striking the piston, which creates a pressure pulse that travels through the transducer and onto the plastic cap causing a deformation of the intact dura. The fluid pulse produces brief displacement and deformation of brain tissue , and the severity of injury depends on the strength of the pressure pulse. In its most severe forms, FPI can result in intracranial hemorrhage, edema and progressive grey matter damage. FPI models can be categorized by the location of the craniotomy, into midline (centered on the sagittal suture), parasagittal (<3.5 mm lateral to midline), and lateral models (>3.5 mm lateral to midline) [26–29]. Midline and parasagittal FPIs cause bilateral cortical damage, while lateral FPI (LFPI) inflicts primarily unilateral cortical damage, rarely involving the contralateral cortices and brainstem.
2.2.2 Methods
Animals are anesthetized using 3 % halothane via a vaporizer and a vacuum trap or a comparable anesthetic, placed in a prone position on a warming pad and secured in a stereotactic frame. The head is shaved, eye lubricant is applied, and the area of incision is prepped using three alternating treatments of Betadine and ethanol. A 1.5 cm sagittal incision is made at the midline between the ears and extended toward the nose. The periosteum is removed with a cotton swab, then, using a 5 mm drill bit, a craniotomy is performed either centrally over the midline (FPI) or laterally (LFPI) between the bregma and lambda. A cannula is set in the cranial opening so that it is touching the intact dura and fixed it to the skull using glue. The cannula is filled with saline and any bubbles are removed from the apparatus.
To determine the resting position, set the pendulum at 90° perpendicular to the ground and make sure it is touching the piston. Double check and remove any bubbles from all tubing and connections and ensure that the tubing from FPI device is filled with water. Using forceps to hold the cannula and tubing, attach them together by twisting in opposing directions. This prevents the application of excessive torque, which would cause the head cannula to detach from the rat’s skull; Thereby causing leakage and an indeterminable injury. Set the pendulum to the desired height, clear the path from the pendulum to the piston and release the pendulum, allowing it to strike the piston. Severity of the injury depends on the intensity of the pressure pulse, which is controlled by the height of the pendulum drop. There are three levels of injury, mild, moderate, and severe. Mild injury occurs following a percussion of 0.1–1 atm (10.13–101.29 kPa), moderate after 1.5–2.0 atm, and severe after 2.5–3 atm. After the piston strikes, check for any leaking of saline and make sure to catch the pendulum to prevent any inadvertent injury. Remove the cannula, fill the burr hole with bone wax, suture the incision and place the animal on a heating pad to wake up (see Notes 5 and 6 ). Common data elements for FPI studies are found in Table 3.
2.2.3 Clinical Relevance
LFPI, the most commonly utilized FPI procedure in rats, produces a combination of focal cortical contusion and diffuse subcortical neuronal injury (including injury in the hippocampus and thalamus). As in CCI, the contused cortex beneath the injury site becomes a cavitary lesion surrounded by reactive gliosis. Over days to months, progressive degenerative changes may be found in ipsilateral hippocampus, thalamus, medial septum, striatum and amygdala. Widespread β-APP expression, comparable to human DAI , may be seen in more severe FPI models [30–32]. Functional deficits in motor and memory outcomes are similar to those seen after CCI, and can persist for up to 1 year after injury.
2.2.4 Caveats
FPI models are associated with high mortality, probably due to brainstem-mediated apnea. Post-injury seizures are common. The FPI model can be difficult to calibrate, since the height of pendulum is the only adjustable mechanical parameter. Reproducibility has been a challenge of FPI models until recently, when a microprocessor-controlled, pneumatically driven instrument has been developed. Though LFPI is the most commonly employed FPI model, there has been recent interest in midline FPI models, which are believed to better represent sports-related and military TBI.
2.3 Penetrating/Direct Impact TBI
2.3.1 Introduction
A wide variety of experimental penetrating TBI models have been designed to generate cerebral deformation including microinjection [33], cryolesion [34], mechanical suction [35], vacuum pulse [36, 37], and inflation of a balloon [38, 39] and others [40]. However, because of their clinical relevance, the most commonly used are penetrating ballistic like blast injury (PBBI) and fragment penetration models. In PBBI models, the insult is caused by transmission of a high energy projectile that produces a cavitary lesion much larger than the projectile. There may be marked intracerebral hemorrhage, but less diffuse axonal injury than other models. Outcomes are directly related to the anatomical path and energy transfer of the projectile.
2.3.2 Methods
In Carey et al.’s [41–43] model anesthetized animals are placed in a stereotactic frame and the sloping outer wall of the right frontal sinus is removed; thereby allowing the missile like object to penetrate the intact and vertically disposed sinus wall. A 2-mm, 31-mg steel sphere fired from 80 cm at 220 or 280 m/s penetrates the right frontal bone and traverses the right cerebral hemisphere from anterior to posterior. The energy from the projectile can range from 0.9 to 1.4 J. Finnie’s [44] model utilizes a 0.22 caliber firearm to inflict a head wound to restrained sheep. The bullet is fired at the temporal region of the skull, causing a right to left transverse injury to the temporal lobes. Recently several new PBBI models have been developed in rodents [45, 46] and fragment blast models are becoming more common as well [47]. Recently a novel, non fatal, low velocity model for PBBI has been established in rats [48] and is similar to CCI in that a cylindrical carbon fiber rod is accelerated and enters the brain creating a cavitary lesion. However, the pin (2 mm in diameter) is attached to a secondary projectile that is accelerated by a pellet fired from a modified rifle. The secondary projectile and pin, guided by a tube, penetrates the brain at a speed of 90 m/s. The base of the projectile is surrounded by a compressible ring (ferrule) that controls the depth of the penetration. Although the depth is usually set at 5 mm, depth, speed and shape of the pin can be altered to obtain different pathological outcomes . This model has also been modified for use in mice [49].
2.3.3 Clinical Relevance
This model mimics gun-related brain injuries caused by a high-velocity penetrating object. The injury can be induced by multiple mechanisms including the high pressures in front of an object; longitudinal shock wave and pressure waves from kinetic energy transfer [42]. Injury phenotypes are dependent on the injured part of brain and the most common consequences after injury are intracerebral hemorrhages , edema, elevated intracranial pressure, decreased cerebral blood flow, significant neuroinflammation , and notable lesion [38, 50]. Additionally, both apoptosis and necrosis are observed in the lesion [50, 51]. Motor function and cognitive impairments are observed in this model [51–53], including deficits in motor (balance beam and rotarod tasks) and memory (MWM test) that correlate with the degree of injury severity as well as clinical moderate-severe TBI [54, 55].
2.3.4 Caveats
The original model was established by Carey et al. using mongrel cats, however, it is difficult to run behavioral tasks on larger animals, thus rodent models have become increasingly popular [48, 49, 56, 57]. However, rodent PBBI models are relatively new and less standardized than other TBI models. Even though they are underdeveloped, rodent PBBI models provide opportunities to use genetic modification to investigate the mechanisms of injury and therapeutic targets. Due to the high velocity of objects entering the brain, heat damage of the tissue is a potential factor, though this may be also relevant to clinical penetrating TBI.
2.4 Weight Drop Models
2.4.1 Introduction
In weight-drop models, a free falling, guided weight is allowed to impact the skull (with or without a craniotomy). One of the major advantages of weight drop models is their relative inexpensiveness. Injury severity in these models can be altered by adjusting the mass of the weight and the height from which the weight falls. In addition, several modifications of the weight-drop model have been made, which markedly alter both functional and histopathological outcomes .
Models such as Feeney’s weight-drop, are delivered to the exposed dura which results in a cortical contusion [58] and progresses to a cavitary lesion similar to those found in CCI. Shohami’s group later introduced a modified rodent weight drop model, delivering closed head injury (CHI) to the intact but unprotected skull [59–63]. Of Note, in both the Feeney and Shohami methods, there is minimal rotational acceleration of the fixed head. In attempt to add rotational acceleration, a prominent feature in the primary injury of human TBI, Marmarou’s impact acceleration model was developed to recapitulate this important mechanism . In the Marmarou method, a sectioned brass weight is allowed to fall freely from a designated height through a Plexiglas tube, onto the exposed skull of rats placed on a foam bed. Marmarou’s model, which often results in a hight mortality rate in the absence of mechanical ventilation , is characterized by diffuse axonal injury (DAI) and neuronal injury, as well as functional deficits in beam walking and memory [64, 65]. Subsequent modifications of the Marmarou model have been designed to reproduce the frontal impact characteristic of motor vehicle and sports-related injuries [66, 67].
In general, these models do not result in mortality, prolonged apnea, skull fractures or cortical contusions, and are thought to be more representative of the vast majority of human TBI, which is classified as mild. Common data elements for weight drop studies are found in Table 4.
2.4.2 Methodsz
In a recent model of mild TBI [66], animals are induced for 45 s with 4 % isoflurane or alternative anesthetic. Immediately following, a timer is started to initiate the loss of consciousness (LOC) and the animal is placed supine on a Kimwipes. The animal’s tail and each end of the Kimwipes are secured by hand and the animal’s head is placed directly beneath a hollow tube and positioned with the tube opening directly posterior to the eyes. A cylindrical metal rod (54 g) is dropped dorsally on the animal’s head between the coronal and lambdoid sutures. Upon impact the animals head readily penetrates the Kimwipes, causing the head and body to fall in a circular trajectory while the tail remains secured. The severity of the injury can be varied by altering the weight of the object and the height that it is dropped from. Mild injuries are 54 g dropped from 28″, while severe ones are 54 g at 60″. The mouse is then placed on its side and allowed to wake up and the timer is stopped. The (LOC) is recorded as the latency in seconds to spontaneous ambulation. The control group consists of sham animals given anesthesia but no weight drop and all animals are allowed to recover in room air in their cages.
Other commonly used models vary in the level of surgical invasiveness and are aimed to reproduce different clinical aspects of brain injury . The Feeney et al. model involves dropping a weight onto the intact dura through a craniotomy [68], which produces hemorrhages and cell death. In the Marmarou model, which mimics DAI and diffuse TBI, the animals head rests on a foam pad and a guided brass weight impacts a stainless steel disk mounted on the skull [69]. Shohami’s model drops the weight onto one side of an unprotected skull while resting on a hard surface [60] and has been used to investigate BBB disruption. Weight drop models are primarily conducted on mice , rats , and swine (see Notes 7 and 8 ).
2.4.3 Clinical Relevance
Each variation of the weight drop model has distinct clinical features that should be evaluated prior to commencing. Model selection can be based on which clinical features of TBI are desired, for example loss of consciousness may be sought after in modeling TBI in military populations, since 4.9 % of US soldiers returning from Iraq had loss of consciousness [70]. However, the majority of patients with sports-related concussions do not experience loss of consciousness and a model minimizing this outcome might be desirable in modeling sport related TBI [71]. Many of the current weight drop models feature a rapid acceleration and deceleration of head movement [72]. The drastic change in speed and direction of the head is the main cause of concussions and axonal injury [73–75] in closed head traumas. The concussive injury does not induce immediate cell death; however, it does result in axonal injury and DAI at the extreme end of the spectrum. Tau pathology, an area of much clinical investigation in the field of TBI, may or may not be present. Reactive gliosis may also be a prominent feature of these models. The closed head model has been shown to result in impaired neurological and cognitive outcomes including motor, learning , memory , and anxiety [72, 76, 77].
2.4.4 Caveats
While weight-drop models are generally easy and inexpensive to employ, they can result in relatively high variability in injury severity. Another concern is the possibility of rebound injury. However, in general, weight-drop procedures are capable of producing graded axonal injury that is highly relevant to the vast majority of human TBI.
2.5 Non Impact Blast
2.5.1 Introduction
Blast models of TBI have been developed to better understand the effects of primary blast waves on the CNS. Some estimates suggest that nearly one in five veterans of the conflicts in Afghanistan and Iraq were exposed to TBI, much of which was blast-associated TBI. Initial efforts at simulating blast associated TBI were challenged by resultant systemic injuries, but the use of thoracic/abdominal protective strategies have minimized these types of associated injuries, allowing investigators to study the effects of blast associated TBI in isolation. Most commonly, blast models delivery primary injury using a compression-driven shock tube to simulate blast effects.
2.5.2 Methods
The most commonly used blast models have been established in rodents [78–82] and swine [83, 84] and utilize explosives [80, 83, 84] or compressed air [82, 85] to create a shock wave . In these models, anesthetized animals are individually fixed in a cylindrical metal tube in such a way that prevents any movement of the body. However, in some cases, the head and neck are able to freely to flex, extend, and rotate during the injury [86]. Additionally Kevlar vests, which encase the thorax and part of the abdomen, have been used to test acute mortality in rats [79]. The blast is generated by a controlled detonation or by the release of compressed air. Blast parameters can be regulated to generate mild to severe injury. Intensity of the blast and subsequent injury is controlled by altering the amount of explosives or pressure of the compressed air; or by changing the animal’s distance from the blast point. When using compressed air more complex waveforms can be created by adding additional chambers of compressed air. Although reproducible, the blast tube and other models lack the clinical relevance and unpredictable conditions of real life event. In a recent open field study [87], 12 anesthetized animals are loosely fixed in individual compartments using a plastic net and placed at 4 or 7 m from the detonation point. The net allows for their body to be exposed to the blast without being injured by shrapnel or debris. The platforms are then elevated to a height of 1 m to prevent any interference from blast wave reflection off the ground. Pressure meters are placed at each distance to detect the intensity of the blast. After the detonation of 500 g of TNT, the mice are allowed to wake up and the pressure sensors data are recorded (see Notes 9 and 10 ).
2.5.3 Clinical Relevance
Non-impact models of blast injury are characterized by cerebral edema, hyperemia and delayed vasospasm, the degree of which corresponds to injury severity. The most prominent histopathological finding in blast models is DAI, which corresponds to changes in diffuse tractography imaging seen in blast exposed military veterans [88]. Associated with DAI , blast-exposed mice also demonstrate phosphorylated tauopathy, myelinated axonopathy, chronic neuroinflammation and neurodegeneration , consistent with clinical reports of chronic traumatic encephalopathy in military populations. Functional deficits reported after blast models of TBI include deficits in social recognition, spatial memory , and motor coordination [89].
2.5.4 Caveats
Clinically, blast injuries consist of primary, secondary, tertiary, and quaternary mechanisms, of which the blast preclinical models really only recapitulates primary mechanism . In addition, animal placement in relation to the shock tube (that is, inside, outside or near the exit of the tube) can significantly modify injury type and severity. Blast models, while highly clinically relevant, also suffer from the most variability within and between models, making comparisons between studies very difficult. Utilization of common data elements (Table 5) is particularly important in this setting. Additionally shock tube models are incredibly complex and require the use of heavy machinery.
3 Notes
-
1.
Ensuring a snug fit in stereotactic frame is essential before starting as it makes drilling difficult and keeping the head still ensures consistent injuries.
-
2.
Make sure head and stereotactic frame are out of the way when setting the impactor.
-
3.
If the impactor is set at 90 degrees perpendicular to the ground the piston will not be flush with the brain because of the curvature of the brain. Make sure to make the contact between the piston and brain the same between animals.
-
4.
Do not replace skull flap from craniotomy as there will be edema which will cause an increase in ICP.
-
5.
The cannula should fit the burr hole snugly and there should be nothing obscuring visualization of the dura through the center of the cannula. Optionally, a few drops of cresyl violet dye can be added to the saline to facilitate observation of any leaks around the burr hole site during the injury.
-
6.
Bubbles can be removed by disconnecting the distal end of the tubing from the rat and forcing fluid through the reservoir and tubing until all air bubbles have been expelled. The tubing is then reconnected to the rat.
-
7.
When taking mouse and Kimwipes off of the table, hold one end of Kimwipes and tail and place mouse vertical in the air then grab the other end and flatten out. This will stretch the mouse’s body out and make placing the head under the tube easier. Hold the Kimwipes tight but not too tight as to rip it.
-
8.
In order to get a more direct hit on top of the head and not over the cerebellum/spinal cord, tilting the fore body upward towards the pipe.
-
9.
When using compressed air model, it is critical that the mylar membrane is mounted correctly. If the sheet gets folded at all, the membrane doubles in thickness and creates inconsistent blasts.
-
10.
Production of mylar sheets is sometimes inconsistent. Therefore, test each new box before running an experiment by generating and measuring the blast.
References
Langlois JA, Rutland-Brown W, Wald MM (2006) The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil 21:375–378
Loane DJ, Faden AI (2010) Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 31:596–604
Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, Bullock MR, Choi SC, Clifton GL, Contant CF, Coplin WM, Dietrich WD, Ghajar J, Grady SM, Grossman RG, Hall ED, Heetderks W, Hovda DA, Jallo J, Katz RL, Knoller N, Kochanek PM, Maas AI, Majde J, Marion DW, Marmarou A, Marshall LF, McIntosh TK, Miller E, Mohberg N, Muizelaar JP, Pitts LH, Quinn P, Riesenfeld G, Robertson CS, Strauss KI, Teasdale G, Temkin N, Tuma R, Wade C, Walker MD, Weinrich M, Whyte J, Wilberger J, Young AB, Yurkewicz L (2002) Clinical trials in head injury. J Neurotrauma 19:503–557
Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39:253–262
Lighthall JW (1988) Controlled cortical impact: a new experimental brain injury model. J Neurotrauma 5:1–15
Kawamata T, Katayama Y, Maeda T, Mori T, Aoyama N, Kikuchi T, Uwahodo Y (1997) Antioxidant, OPC-14117, attenuates edema formation and behavioral deficits following cortical contusion in rats. Acta Neurochir Suppl 70:191–193
Verweij BH, Muizelaar JP, Vinas FC, Peterson PL, Xiong Y, Lee CP (1997) Mitochondrial dysfunction after experimental and human brain injury and its possible reversal with a selective N-type calcium channel antagonist (SNX-111). Neurol Res 19:334–339
Faden AI, Fox GB, Fan L, Araldi GL, Qiao L, Wang S, Kozikowski AP (1999) Novel TRH analog improves motor and cognitive recovery after traumatic brain injury in rodents. Am J Physiol 277:R1196–R1204
Kroppenstedt SN, Stroop R, Kern M, Thomale UW, Schneider GH, Unterberg AW (1999) Lubeluzole following traumatic brain injury in the rat. J Neurotrauma 16:629–637
Dempsey RJ, Baskaya MK, Dogan A (2000) Attenuation of brain edema, blood-brain barrier breakdown, and injury volume by ifenprodil, a polyamine-site N-methyl-D-aspartate receptor antagonist, after experimental traumatic brain injury in rats. Neurosurgery 47:399–404, discussion 404–406
Sullivan PG, Thompson M, Scheff SW (2000) Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol 161:631–637
Washington PM, Forcelli PA, Wilkins T, Zapple DN, Parsadanian M, Burns MP (2012) The effect of injury severity on behavior: a phenotypic study of cognitive and emotional deficits after mild, moderate, and severe controlled cortical impact injury in mice. J Neurotrauma 29:2283–2296
Geddes RI, Sribnick EA, Sayeed I, Stein DG (2014) Progesterone treatment shows benefit in a pediatric model of moderate to severe bilateral brain injury. PLoS One 9:e87252
Fox GB, Fan L, Levasseur RA, Faden AI (1998) Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma 15:599–614
Bermpohl D, You Z, Lo EH, Kim HH, Whalen MJ (2007) TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J Cereb Blood Flow Metab 27:1806–1818
Mahmood A, Wu H, Qu C, Xiong Y, Chopp M (2013) Effects of treating traumatic brain injury with collagen scaffolds and human bone marrow stromal cells on sprouting of corticospinal tract axons into the denervated side of the spinal cord. J Neurosurg 118:381–389
Wagner AK, Postal BA, Darrah SD, Chen X, Khan AS (2007) Deficits in novelty exploration after controlled cortical impact. J Neurotrauma 24:1308–1320
Ajao DO, Pop V, Kamper JE, Adami A, Rudobeck E, Huang L, Vlkolinsky R, Hartman RE, Ashwal S, Obenaus A, Badaut J (2012) Traumatic brain injury in young rats leads to progressive behavioral deficits coincident with altered tissue properties in adulthood. J Neurotrauma 29:2060–2074
Tran HT, LaFerla FM, Holtzman DM, Brody DL (2011) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31:9513–9525
Ciallella JR, Ikonomovic MD, Paljug WR, Wilbur YI, Dixon CE, Kochanek PM, Marion DW, DeKosky ST (2002) Changes in expression of amyloid precursor protein and interleukin-1beta after experimental traumatic brain injury in rats. J Neurotrauma 19:1555–1567
Colicos MA, Dixon CE, Dash PK (1996) Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res 739:111–119
Pohl D, Bittigau P, Ishimaru MJ, Stadthaus D, Hubner C, Olney JW, Turski L, Ikonomidou C (1999) N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci U S A 96:2508–2513
Dohi K, Kraemer BC, Erickson MA, McMillan PJ, Kovac A, Flachbartova Z, Hansen KM, Shah GN, Sheibani N, Salameh T, Banks WA (2014) Molecular hydrogen in drinking water protects against neurodegenerative changes induced by traumatic brain injury. PLoS One 9:e108034
Kabadi SV, Stoica BA, Byrnes KR, Hanscom M, Loane DJ, Faden AI (2012) Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J Cereb Blood Flow Metab 32:137–149
Dapul HR, Park J, Zhang J, Lee C, DanEshmand A, Lok J, Ayata C, Gray T, Scalzo A, Qiu J, Lo EH, Whalen MJ (2013) Concussive injury before or after controlled cortical impact exacerbates histopathology and functional outcome in a mixed traumatic brain injury model in mice. J Neurotrauma 30:382–391
McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL (1989) Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience 28:233–244
Vink R, Mullins PG, Temple MD, Bao W, Faden AI (2001) Small shifts in craniotomy position in the lateral fluid percussion injury model are associated with differential lesion development. J Neurotrauma 18:839–847
Sanders MJ, Dietrich WD, Green EJ (1999) Cognitive function following traumatic brain injury: effects of injury severity and recovery period in a parasagittal fluid-percussive injury model. J Neurotrauma 16:915–925
Floyd CL, Golden KM, Black RT, Hamm RJ, Lyeth BG (2002) Craniectomy position affects morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. J Neurotrauma 19:303–316
Wang E, Gao J, Yang Q, Parsley MO, Dunn TJ, Zhang L, DeWitt DS, Denner L, Prough DS, Wu P (2012) Molecular mechanisms underlying effects of neural stem cells against traumatic axonal injury. J Neurotrauma 29:295–312
Ekmark-Lewen S, Flygt J, Kiwanuka O, Meyerson BJ, Lewen A, Hillered L, Marklund N (2013) Traumatic axonal injury in the mouse is accompanied by a dynamic inflammatory response, astroglial reactivity and complex behavioral changes. J Neuroinflammation 10:44
Hoshino S, Kobayashi S, Furukawa T, Asakura T, Teramoto A (2003) Multiple immunostaining methods to detect traumatic axonal injury in the rat fluid-percussion brain injury model. Neurol Med Chir (Tokyo) 43:165–173, discussion 174
Fitch MT, Doller C, Combs CK, Landreth GE, Silver J (1999) Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci 19:8182–8198
Sun D, Tani M, Newman TA, Krivacic K, Phillips M, Chernosky A, Gill P, Wei T, Griswold KJ, Ransohoff RM, Weller RO (2000) Role of chemokines, neuronal projections, and the blood-brain barrier in the enhancement of cerebral EAE following focal brain damage. J Neuropathol Exp Neurol 59:1031–1043
Mathew P, Bullock R, Graham DI, Maxwell WL, Teasdale GM, McCulloch J (1996) A new experimental model of contusion in the rat. Histopathological analysis and temporal patterns of cerebral blood flow disturbances. J Neurosurg 85:860–870
Shreiber DI, Bain AC, Ross DT, Smith DH, Gennarelli TA, McIntosh TK, Meaney DF (1999) Experimental investigation of cerebral contusion: histopathological and immunohistochemical evaluation of dynamic cortical deformation. J Neuropathol Exp Neurol 58:153–164
Shreiber DI, Smith DH, Meaney DF (1999) Immediate in vivo response of the cortex and the blood-brain barrier following dynamic cortical deformation in the rat. Neurosci Lett 259:5–8
Williams AJ, Hartings JA, Lu XC, Rolli ML, Dave JR, Tortella FC (2005) Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma 22:313–331
Williams AJ, Wei HH, Dave JR, Tortella FC (2007) Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflammation 4:17
Ghirnikar RS, Lee YL, He TR, Eng LF (1996) Chemokine expression in rat stab wound brain injury. J Neurosci Res 46:727–733
Carey ME (1995) Experimental missile wounding of the brain. Neurosurg Clin N Am 6:629–642
Carey ME, Sarna GS, Farrell JB, Happel LT (1989) Experimental missile wound to the brain. J Neurosurg 71:754–764
Carey ME, Sarna GS, Farrell JB (1990) Brain edema after an experimental missile wound. Adv Neurol 52:301–305
Finnie JW (1993) Brain damage caused by a captive bolt pistol. J Comp Pathol 109:253–258
Shear DA, Lu XC, Bombard MC, Pedersen R, Chen Z, Davis A, Tortella FC (2010) Longitudinal characterization of motor and cognitive deficits in a model of penetrating ballistic-like brain injury. J Neurotrauma 27:1911–1923
Wei G, Lu XC, Yang X, Tortella FC (2010) Intracranial pressure following penetrating ballistic-like brain injury in rats. J Neurotrauma 27:1635–1641
Risling M, Skold M, Larsson I, Angeria M, Davidsson J (2004) Leakage of S-100 protein after high velocity penetration injury to the brain. Restor Neurol Neurosci 23:141, 7th international neurotrauma symposium. Medimond (ed) Adelaide, Australia
Plantman S, Ng KC, Lu J, Davidsson J, Risling M (2012) Characterization of a novel rat model of penetrating traumatic brain injury. J Neurotrauma 29:1219–1232
Cernak I, Wing ID, Davidsson J, Plantman S (2014) A novel mouse model of penetrating brain injury. Front Neurol 5:209
Williams AJ, Hartings JA, Lu XC, Rolli ML, Tortella FC (2006) Penetrating ballistic-like brain injury in the rat: differential time courses of hemorrhage, cell death, inflammation, and remote degeneration. J Neurotrauma 23:1828–1846
Tompkins P, Tesiram Y, Lerner M, Gonzalez LP, Lightfoot S, Rabb CH, Brackett DJ (2013) Brain injury: neuro-inflammation, cognitive deficit, and magnetic resonance imaging in a model of blast induced traumatic brain injury. J Neurotrauma 30:1888–1897
Lu XC, Shear DA, Graham PB, Bridson G, Uttamsingh V, Chen Z, Leung LY, Tortella FC (2015) Dual therapeutic effects of C-10068, a dextromethorphan derivative, against post-traumatic nonconvulsive seizures and neuroinflammation in a rat model of penetrating ballistic-like brain injury. J Neurotrauma 32:1621–1632
Shear DA, Lu XC, Pedersen R, Wei G, Chen Z, Davis A, Yao C, Dave J, Tortella FC (2011) Severity profile of penetrating ballistic-like brain injury on neurofunctional outcome, blood-brain barrier permeability, and brain edema formation. J Neurotrauma 28:2185–2195
Driscoll DM, Dal Monte O, Solomon J, Krueger F, Grafman J (2012) Empathic deficits in combat veterans with traumatic brain injury: a voxel-based lesion-symptom mapping study. Cogn Behav Neurol 25:160–166
Salazar AM, Grafman J, Schlesselman S, Vance SC, Mohr JP, Carpenter M, Pevsner P, Ludlow C, Weingartner H (1986) Penetrating war injuries of the basal forebrain: neurology and cognition. Neurology 36:459–465
Elias PZ, Spector M (2012) Characterization of a bilateral penetrating brain injury in rats and evaluation of a collagen biomaterial for potential treatment. J Neurotrauma 29:2086–2102
Gajavelli S, Kentaro S, Diaz J, Yokobori S, Spurlock M, Diaz D, Jackson C, Wick A, Zhao W, Leung LY, Shear D, Tortella F, Bullock MR (2015) Glucose and oxygen metabolism after penetrating ballistic-like brain injury. J Cereb Blood Flow Metab 35:773–780
Dail WG, Feeney DM, Murray HM, Linn RT, Boyeson MG (1981) Responses to cortical injury: II. Widespread depression of the activity of an enzyme in cortex remote from a focal injury. Brain Res 211:79–89
Shapira Y, Shohami E, Sidi A, Soffer D, Freeman S, Cotev S (1988) Experimental closed head injury in rats: mechanical, pathophysiologic, and neurologic properties. Crit Care Med 16:258–265
Shohami E, Shapira Y, Cotev S (1988) Experimental closed head injury in rats: prostaglandin production in a noninjured zone. Neurosurgery 22:859–863
Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E (1996) An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J Neurotrauma 13:557–568
Stahel PF, Shohami E, Younis FM, Kariya K, Otto VI, Lenzlinger PM, Grosjean MB, Eugster HP, Trentz O, Kossmann T, Morganti-Kossmann MC (2000) Experimental closed head injury: analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J Cereb Blood Flow Metab 20:369–380
Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E (2009) Mouse closed head injury model induced by a weight-drop device. Nat Protoc 4:1328–1337
Heath DL, Vink R (1995) Impact acceleration-induced severe diffuse axonal injury in rats: characterization of phosphate metabolism and neurologic outcome. J Neurotrauma 12:1027–1034
Schmidt RH, Scholten KJ, Maughan PH (2000) Cognitive impairment and synaptosomal choline uptake in rats following impact acceleration injury. J Neurotrauma 17:1129–1139
Khuman J, Meehan WP III, Zhu X, Qiu J, Hoffmann U, Zhang J, Giovannone E, Lo EH, Whalen MJ (2010) Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J Cereb Blood Flow Metab 31:778–789
Kilbourne M, Kuehn R, Tosun C, Caridi J, Keledjian K, Bochicchio G, Scalea T, Gerzanich V, Simard JM (2009) Novel model of frontal impact closed head injury in the rat. J Neurotrauma 26:2233–2243
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG (1981) Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res 211:67–77
Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K (1994) A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J Neurosurg 80:291–300
Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA (2008) Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med 358:453–463
Silverberg ND, Luoto TM, Ohman J, Iverson GL (2014) Assessment of mild traumatic brain injury with the King-Devick Test in an emergency department sample. Brain Inj 28:1590–1593
Meehan WP III, Zhang J, Mannix R, Whalen MJ (2012) Increasing recovery time between injuries improves cognitive outcome after repetitive mild concussive brain injuries in mice. Neurosurgery 71:885–891
White BC, Krause GS (1993) Brain injury and repair mechanisms: the potential for pharmacologic therapy in closed-head trauma. Ann Emerg Med 22:970–979
McAllister TW (1992) Neuropsychiatric sequelae of head injuries. Psychiatr Clin North Am 15:395–413
Krave U, Al-Olama M, Hansson HA (2011) Rotational acceleration closed head flexion trauma generates more extensive diffuse brain injury than extension trauma. J Neurotrauma 28:57–70
Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, Zhang J, Bryant J, Rezaie S, Chung JY, Peters NV, Lee C, Tien LW, Kaplan DL, Feany M, Whalen M (2013) Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol 74:65–75
Mannix R, Berglass J, Berkner J, Moleus P, Qiu J, Andrews N, Gunner G, Berglass L, Jantzie LL, Robinson S, Meehan WP III (2014) Chronic gliosis and behavioral deficits in mice following repetitive mild traumatic brain injury. J Neurosurg 121:1342–1350
Cernak I, Savic J, Malicevic Z, Zunic G, Radosevic P, Ivanovic I, Davidovic L (1996) Involvement of the central nervous system in the general response to pulmonary blast injury. J Trauma 40:S100–S104
Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA (2009) Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma 26:827–840
Clemedson CJ (1956) Shock wave transmission to the central nervous system. Acta Physiol Scand 37:204–214
Cheng J, Gu J, Ma Y, Yang T, Kuang Y, Li B, Kang J (2010) Development of a rat model for studying blast-induced traumatic brain injury. J Neurol Sci 294:23–28
Risling M, Davidsson J (2012) Experimental animal models for studies on the mechanisms of blast-induced neurotrauma. Front Neurol 3:30
Bauman RA, Ling G, Tong L, Januszkiewicz A, Agoston D, Delanerolle N, Kim Y, Ritzel D, Bell R, Ecklund J, Armonda R, Bandak F, Parks S (2009) An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma 26:841–860
de Lanerolle NC, Bandak F, Kang D, Li AY, Du F, Swauger P, Parks S, Ling G, Kim JH (2011) Characteristics of an explosive blast-induced brain injury in an experimental model. J Neuropathol Exp Neurol 70:1046–1057
Wang Y, Wei Y, Oguntayo S, Wilkins W, Arun P, Valiyaveettil M, Song J, Long JB, Nambiar MP (2011) Tightly coupled repetitive blast-induced traumatic brain injury: development and characterization in mice. J Neurotrauma 28:2171–2183
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64
Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, Hoffer BJ, Balaban CD, Schreiber S, Chiu WT, Pick CG (2011) A mouse model of blast-induced mild traumatic brain injury. Exp Neurol 232:280–289
Mac Donald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, Snyder AZ, Raichle ME, Witherow JR, Fang R, Flaherty SF, Brody DL (2011) Detection of blast-related traumatic brain injury in U.S. military personnel. N Engl J Med 364:2091–2100
Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, Eberhart CG, Frangakis CE, Melnikova T, Kim H, Lee D (2011) A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol 70:399–416
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Berkner, J., Mannix, R., Qiu, J. (2016). Clinical Traumatic Brain Injury in the Preclinical Setting. In: Kobeissy, F., Dixon, C., Hayes, R., Mondello, S. (eds) Injury Models of the Central Nervous System. Methods in Molecular Biology, vol 1462. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3816-2_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3816-2_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3814-8
Online ISBN: 978-1-4939-3816-2
eBook Packages: Springer Protocols