
Abstract
The blood–brain barrier (BBB), a specialized interface between the peripheral blood circulation and the central nervous system, specifically regulates molecular and cellular flux between the two. It plays a critical role in the maintenance of brain hemostasis. The BBB restricts the entry of pathogens into the brain, and thus its permeability is a critical factor that determines their central effects. Once the permeability of BBB is compromised, it has serious implications in the etiology of many brain pathologies including West Nile virus (WNV) disease. In this chapter, we describe protocols for preparation, maintenance, infection and permeability measurement of monolayer and bilayer in vitro BBB models to study WNV pathogenesis. We also describe Evans blue dye assay, a well-established method to test vascular permeability in vivo after WNV infection.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
West Nile virus (WNV) , a mosquito -borne flavivirus that causes lethal encephalitis has emerged as a leading cause of arboviral encephalitis in the United States. Since its introduction to North America in 1999, outbreaks of West Nile fever (WNF) and West Nile virus associated encephalitis (WNVE) have occurred in all 48 contiguous states [1]. The fatality rate is approximately 10 % for hospitalized encephalitic cases with increased risk in patients with compromised immune systems, older age and having underlying conditions such as diabetes mellitus [2]. Recent outbreaks of highly virulent WNV strains have also been reported in the Mediterranean basin, southern Europe and Russia [3]. Although the worldwide incidence of WNV infection is increasing, there is no specific treatment or vaccine available for use in humans.
Following peripheral infection, WNV replication is first thought to occur in skin Langerhans dendritic cells. These cells migrate to draining lymph nodes, resulting in primary viremia. By the end of the first week, the virus is largely cleared from the peripheral organs, but in a subset of patients, WNV enters the brain and causes a spectrum of neurological disorders. WNVE is characterized by disruption of the blood–brain barrier (BBB), enhanced infiltration of immune cells into the central nervous system (CNS), microglia activation, inflammation and eventual loss of neurons [2, 4]. Since high viremia is directly correlated with early WNV entry into the CNS [2], it is suggested that WNV in the periphery enters the CNS by crossing the BBB. Increased production of pro-inflammatory mediators facilitate WNV neuroinvasion by compromising the BBB integrity and are associated with high virus titers in the brain and increased mortality in WNV mouse models [5–7]. The BBB, which consists of microvascular endothelial cells, perivascular astrocytes, basement membrane, and pericytes, is a highly regulated interface which separates blood-borne entities from the CNS [8]. The tight junction proteins (TJPs) such as zona occludens, claudins and occludin, the main structural basis of BBB integrity, play a key role in the physiology of the BBB. However, our understanding of the cellular mechanisms associated with WNV-induced BBB disruption, specifically the contribution of BBB-associated cells, is limited. Using an in vitro BBB model comprised of primary human brain microvascular endothelial (HBMVE) cells, we have demonstrated that cell-free WNV can cross the BBB, without compromising the BBB integrity [9]. Further, we have demonstrated that inflammatory mediators secreted from WNV-infected astrocytes degrade key TJPs of HBMVE cells and compromise the integrity of the BBB model thereby contributing to WNV-associated neuropathogenesis [10].
2 Materials
2.1 Cell Culture
Primary human brain microvascular endothelial (HBMVE) cells
-
1.
HBMVE cells are cerebral microvascular endothelium cells from normal human brain cortex tissue (ACBRI 376).
-
2.
Culture medium: For 1 L, add 965 mL Endothelial Cell Media (ECM; Science Cell Research Laboratories, Cat. 1001), 25 mL fetal bovine serum (FBS) (Science Cell Research Laboratories, Cat. 0025), 5 mL 100× Penicillin/Streptomycin (P/S) solution (Science Cell Research Laboratories, Cat. 0503), 5 mL 100× Endothelial Cell Growth Supplement (ECGS; Science Cell Research Laboratories, Cat. 1052).
-
3.
Cell passage reagents: CSC certified Passage Reagent Group [(PRG; Cell Systems Corporation, Cat. 4Z0-800): PRG-1 (EDTA solution), PRG-2 (Trypsin/EDTA solution), PRG-3 (Trypsin Inhibitor solution)], 1× CSC certified attachment factor (Cell Systems Corporation, Cat. 4Z0-210).
-
4.
Culture conditions: HBMVE cells are cultured in a 37 °C and 5 % CO2 incubator . These cells require attachment factor to attach to the bottom of the culture flask (see N ote 1).
Primary human brain cortical astrocyte (HBCA) cells
-
1.
HBCA cells are astrocyte cells from normal human brain cortex tissue (ACBRI 376).
-
2.
Culture Medium: For 1 L, add 980 mL Astrocytic Cell Media (ACM; Science Cell Research Laboratories, Cat. 1801), 10 mL FBS, 5 mL 100× P/S solution, 5 mL 100× Astrocytic Cell Growth Supplement (ACGS; Science Cell Research Laboratories, Cat. 1852).
-
3.
Cell passage reagents: Same as employed for aforementioned HBMVE cells.
-
4.
Culture conditions: HBCA cells are cultured in a 37 °C and 5 % CO2 incubator. These cells require attachment factor to attach to the bottom of the culture flask (see Note 1 ).
2.2 In Vitro BBB Model
-
1.
Inserts: BioCoat® Cell Environment™ Human Fibronectin PET (polyethylene terephthalate) inserts with 3.0 μm pore size (BD Bioscience, Cat. 354543).
-
2.
Instruments: EVOM meter and End Ohm (World Precision Instruments Inc.), Victor™ 1420 fluorescence microplate reader (Perkin Elmer), humidified incubator .
-
3.
Reagents: 1× phosphate buffered saline (PBS), fluorescein isothiocyante (FITC)-dextran (4 kDa molecular weight; Sigma), 24-well plates, 96-well plates, forceps.
2.3 In Vivo BBB Model
-
1.
Animals: 8–12 weeks old C57BL/6 mice.
-
2.
Evans blue dye (see Note 2 ).
-
3.
Reagents: 1× PBS, formamide, isoflurane anesthesia, 70 % ethanol, 29-G 3/8″ needle, syringes (1 mL, 10 mL), needles (26 G), 1.5 mL tubes, dissection board, surgical scissors, forceps.
3 Methods
WNV is a bio-safety level 3 (BSL-3) agent. All procedures involving WNV infection should be restricted to a bio-safety cabinet in an authorized A/BSL-3 laboratory.
3.1 In Vitro Monolayer and Bilayer BBB Model
Cell culture (3–7 days prior to BBB assembly) and maintenance
-
1.
Coat the tissue culture flasks with 1× CSC certified attachment factor (2 mL for T-75 flasks) before adding any media or plating HBMVE and HBCA cells (see Note 1 ).
-
2.
Maintain low passage (2–8) HBMVE and HBCA cells in ECM and ACM complete media (described in Subheading 2), respectively, at 37 °C, 5 % CO2 and 100 % humidity.
-
3.
Split confluent HBMVE and HBCA cells at a ratio of 1:3–1:6. Wash the cells with 2 mL of PRG-1. Add 2 mL of PRG-2 and incubate for 2 min. Add 2 mL of PRG-3 to neutralize the trypsin.
-
4.
Collect HBMVE and HBCA cells by centrifugation and count with 1:2 dilution.
-
5.
Resuspend the cell pellet in ECM and ACM complete media to get 1 × 105 HBMVE cells/mL and 1 × 106 HBCA cells/mL, respectively.
Monolayer HBMVE BBB model
-
1.
Place tissue culture inserts in the wells of a 24-well plate (Fig. 1) and hydrate in ECM at 37 °C, 5 % CO2 and 100 % humidity for 1 h.
Fig. 1 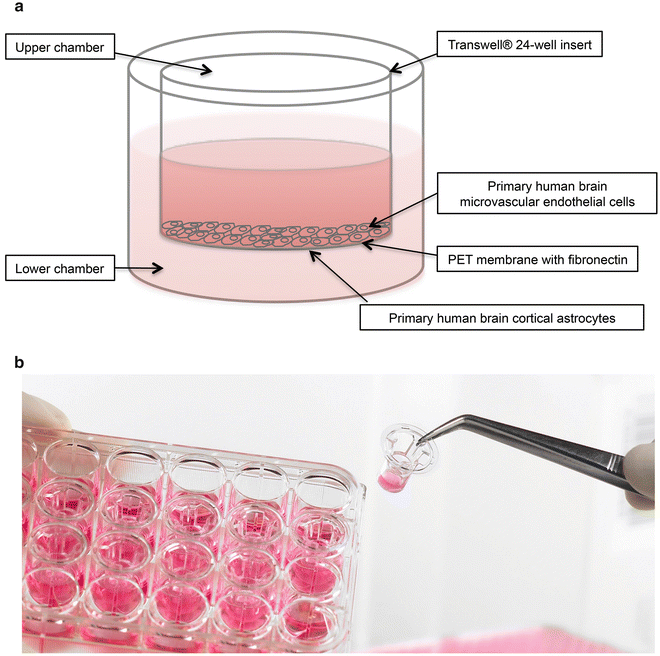
In vitro BBB model. (a) A BBB insert and (b) demonstration of how the inserts are situated in the plate containing wells
-
2.
Wash the inserts once with 1× PBS to remove the residual media .
-
3.
Add 100 μL 1× CSC certified attachment factor in each insert (see Note 1 ).
-
4.
To bottom of the well add 500 μL of ECM complete media.
-
5.
To top of inserts (luminal membrane surface) add 500 μL of ECM complete media with cells (5 × 104 HBMVE cells). In 1–2 inserts, add only ECM complete media to be used as controls (see Note 3 ).
-
6.
Incubate the inserts at 37 °C, 5 % CO2 and 100 % humidity.
-
7.
Change fresh media every 2–3 days.

Bilayer HBMVE-HBCA BBB model
-
1.
Place tissue culture inserts in the wells of a 24-well plate and hydrate in ECM or ACM at 37 °C, 5 % CO2 and 100 % humidity for 1 h.
-
2.
Wash the inserts once with 1× PBS to remove the residual media .
-
3.
Add 100 μL 1× CSC certified attachment factor to each insert from the abluminal and luminal sides (see Note 1 ).
-
4.
Invert the inserts and to bottom of inserts (abluminal membrane surface) add 100 μL of ACM complete media with cells (1 × 105 HBCA cells). In 1–2 inserts, add only ACM complete media to be used as controls.
-
5.
Incubate the inserts at 37 °C, 5 % CO2 and 100 % humidity for 4 h. Rehydrate the inserts with ACM complete media periodically to keep the cells wet.
-
6.
To bottom of the well add 500 μL of ECM complete media.
-
7.
After 4 h incubation, transfer the inserts to a plate and to the top of inserts (luminal membrane surface) add 500 μL of ECM complete media with 5 × 104 HBMVE cells. In controls, add only ECM complete media.
-
8.
Incubate the inserts at 37 °C, 5 % CO2 and 100 % humidity.
-
9.
Change fresh media every 2 days.
3.2 Integrity of the BBB Model
It generally takes between 5 and 6 days to form a tight BBB model (see Note 4 ). Check tightness by measuring transendothelial electric resistance (TEER) and FITC-dextran transmigration starting day 3 after putting cells onto inserts [9–12].
TEER measurement
-
1.
Remove the old ECM complete media from the upper chamber.
-
2.
Add 500 μL of fresh warmed ECM complete media to upper chamber of cell insert.
-
3.
Add 1.5 mL fresh warmed media to End Ohm connected to the EVOM meter (TEER measuring instrument) (Fig. 2). For monolayer BBB, add ECM media . For bilayer BBB, add ECM or ACM media.
Fig. 2 
EVOM meter and End Ohm for the measurement of TEER. (a) EVOM meter. (b) End Ohm TEER measurement chamber with BBB insert
-
4.
Transfer insert (with media) carefully into End Ohm using forceps.
-
5.
Turn machine on after putting measuring tip of End Ohm into the insert and toggle the switch to measure resistance (see Notes 5 and 6 ). Make sure machine reads 0 before toggling measuring switch.
-
6.
After taking reading, transfer insert (with media) back to the well in the 24-well plate.

FITC-dextran transmigration assay (All steps must be done in the dark)
-
1.
Add 750 μL ECM to bottom chamber of fresh 24-well plate.
-
2.
Decant media from upper chamber of cell inserts.
-
3.
Transfer cell inserts into new 24-well plate with 750 μL ECM in corresponding wells.
-
4.
Add 150 μL of FITC-dextran (with ECM at 100 μg/mL concentration) to upper chamber of the insert.
-
5.
Incubate plate for 2 h in dark at 37 °C.
-
6.
Remove 150 μL samples from lower chamber of each cell insert.
-
7.
Place samples into 96-well plate (add samples in the second row).
-
8.
Make standards in serial dilution from the stock of FITC-dextran (100–0.098 μg/mL) in a 96-well plate.
-
9.
Read 96-well plate at 485 nm excitation and 535 nm emission wave lengths using the plate reader (Victor™ 1420 fluorescence microplate reader, Perkin-Elmer Wallance Inc).
-
10.
The FITC-dextran transmigration across the inserts is calculated as percentage of the total amount added in the upper well.
3.3 Infection of In Vitro BBB Models
-
1.
Decant media from upper chamber of cell inserts.
-
2.
Make WNV working dilution in serum -free ECM from the stock. Bring the titer to 50,000 plaque-forming units per 100 μL of serum-free ECM to infect the inserts with multiplicity of infection 1.
-
3.
Add this 100 μL of serum-free ECM containing WNV to each insert.
-
4.
Add 100 μL of serum-free ECM without virus for the controls.
-
5.
After adsorption for 1 h at 37 °C, aspirate the media and wash the inserts two times with 1× PBS and then add 500 μL of fresh ECM.
-
6.
Incubate the inserts at 37 °C, 5 % CO2 and 100 % humidity.
-
7.
At 6, 12, 24, 48, and 72 h after incubation, remove the media from the upper chamber of the inserts and wash the inserts twice with 1× PBS and assay the BBB integrity by TEER and FITC-dextran transmigration assay as described above.
3.4 In Vivo BBB Model
West Nile virus inoculation
-
1.
Dilute the WNV NY99 stock to 100 (LD50 dose) or 1000 (LD100 dose) plaque-forming units (PFU) per 20 μL in 1× PBS and keep it on ice (see Note 7 ). Appropriately diluted WNV should be drawn into the syringe and kept ready for inoculation.
-
2.
Anesthetize mice in an induction chamber using 2–5 % Isoflurane at a rate of 1–2 L/min of O2. After 30–40 s, observe the mice to ensure complete anesthesia. Carefully monitor the following symptoms during this time: (a) mouse is still but breathing and (b) ears are still pink.
-
3.
Once anesthetized, place the mouse on a warming table and hold the hind limb with a flat end forceps.
-
4.
Use the forceps to extend the left foot and wipe the foot with 70 % alcohol to remove debris before injection.
-
5.
Inject 20 μL of WNV subcutaneously into the center of hind foot forming a small bleb at the injection site (see Note 8 ).
-
6.
Wipe the foot with paper towel wet with 70 % ethanol.
-
7.
Discard the needle and syringe in a puncture resistant sharps container.
-
8.
In control mice, inject 20 μL of PBS by the same route.
-
9.
Return the mice to their cage.
-
10.
Make sure that the mice are awake before leaving the room.
Intraperitonial injection of Evans blue dye (at specific days after inoculation )
-
1.
Prepare a 1 % sterile solution of Evans blue dye in PBS. If necessary, filter-sterilize the solution to remove any particulate matter that has not dissolved.
-
2.
Aspirate 1 mL of 1 % Evans blue dye solution into a syringe. Avoid all air bubbles that might have escaped into the syringe.
-
3.
Scruff the mouse with the right hind limb immobilized and the head and body tilted downward. Hold onto the tail with the nondominant hand between the thumb and the forefinger.
-
4.
Disinfect the right lower abdominal wall with gauze dampened with alcohol.
-
5.
Insert the needle (small gauge, 26) at a 10–15° angle into the peritoneal cavity in the caudal right abdominal quadrant through the skin and the abdominal wall, thereby avoiding injection into the cecum or the stomach on the left side.
-
6.
Lift the needle tip slightly and slowly inject 1 mL of 1 % Evans blue dye solution.
-
7.
Put the mouse back into its cage and observe it for 1 h.
Organ collection and extraction of Evans blue dye from the brain
-
1.
Anesthetize the mice using Isoflurane. Keep the mice anesthetized during the whole procedure.
-
2.
Place the mice on their backs and pin their feet on a dissection board.
-
3.
Spray external area of abdominal and thoracic cavity with 70 % ethanol. Open the abdominal and thoracic cavity using surgical scissors to expose thoracic and abdominal organs.
-
4.
Flush the heart slowly with 10–15 mL of 1× PBS using small gauge (26 G) needle (see Note 9 ).
-
5.
After perfusion, harvest brains using surgical scissors and forceps.
-
6.
Take representative pictures to show differences in Evans blue dye extravasation (Fig. 3) (see Note 10 ) [13]. Include all brains in the same field in order to have identical lighting conditions.
Fig. 3 
In vivo BBB model. Mice were inoculated with PBS or 100 PFU of WNV via footpad and BBB permeability was measured using Evans blue dye at day 6 after infection. Mice were injected i.p. with 1 mL Evans blue dye (1 % w/v) and after 1 h were cardiac perfused with PBS. The extravasation of the dye was evident in the WNV-infected mice brain
-
7.
Collect brains in 1.5 mL tubes.
-
8.
Weigh an empty tube and bring the balance value to zero.
-
9.
Transfer the brain and weigh it. Repeat for all brain samples.
-
10.
Add 500 μL formamide to each tube.
-
11.
Transfer all tubes to a 55 °C water bath. Incubate for 24–48 h to extract Evans blue dye from the brain.
-
12.
Centrifuge the formamide/Evans blue dye mixture to pellet any remaining tissue fragments.
-
13.
Measure absorbance at 610 nm. Use 500 μL-formamide as blank.
-
14.
Calculate nanograms of Evans blue dye extravasated per mg brain tissue (see Note 11 ).

4 Notes
-
1.
HBCA and HBMVE cells require attachment factor to attach to the bottom of the culture flask and inserts. Coat the tissue culture flasks and inserts with 1× CSC certified attachment factor (2 mL for T-75 flasks, 100 μL for inserts) at room temperature. Wait at least 10 min before adding media or plating HBMVE and HBCA cells.
-
2.
Evans blue dye is a dye that binds albumin. Under physiologic conditions the BBB endothelium is impermeable to albumin. Therefore, Evans blue dye-bound albumin remains restricted within the blood vessels. In WNV infection, there is increased vascular permeability and the BBB becomes permeable to small proteins such as albumin. This condition allows for extravasation of Evans blue dye in the brain tissue .
-
3.
Always have blank inserts with no cells as controls.
-
4.
It generally takes between 5 and 6 days to form a tight BBB model. Start checking tightness via permeability and TEER methods on day 3 after plating cells onto the insert.
-
5.
TEER is expressed as Ω/cm2. TEER of 450 Ω/cm2 is considered good/tight for BBB.
-
6.
High TEER is equivalent to higher resistance (tighter BBB).
-
7.
Dose is based upon virus titration studies using WNV NY99 strain.
-
8.
Use 29-G 3/8″ needle for inoculation of WNV via footpad route.
-
9.
Perfusion of the heart with 10–15 mL of PBS is done slowly over 10–12 min.
-
10.
In case of increased BBB permeability, brains will show significantly increased blue coloration compared to brains isolated from the mice with intact endothelium.
-
11.
By using a standard curve for Evans blue dye, optical density measurements can be converted into milligram dye captured per milligram of tissue.
References
Beasley DW, Barrett AD, Tesh RB (2013) Resurgence of West Nile neurologic disease in the United States in 2012: what happened? What needs to be done? Antiviral Res 99:1–5
Lim SM, Koraka P, Osterhaus AD, Martina BE (2011) West Nile virus: immunity and pathogenesis. Viruses 3:811–828
Calistri P, Giovannini A, Hubalek Z, Ionescu A, Monaco F, Savini G, Lelli R (2010) Epidemiology of west nile in europe and in the mediterranean basin. Open Virol J 4:29–37
Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM (2005) Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med 202:1087–1098
Wang P, Dai J, Bai F, Kong KF, Wong SJ, Montgomery RR, Madri JA, Fikrig E (2008) Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J Virol 82:8978–8985
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA (2004) Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10:1366–1373
Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, Wong SJ, Montgomery RR, Fikrig E, Bucala R (2007) Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest 117:3059–3066
Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD (2006) Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmun Pharmacol 1:223–236
Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR (2009) West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology 385:425–433
Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR (2010) Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 397:130–138
Kelley JF, Kaufusi PH, Nerurkar VR (2012) Dengue hemorrhagic fever-associated immunomediators induced via maturation of dengue virus nonstructural 4B protein in monocytes modulate endothelial cell adhesion molecules and human microvascular endothelial cells permeability. Virology 422:326–337
Roe K, Orillo B, Verma S (2014) West Nile virus-induced cell adhesion molecules on human brain microvascular endothelial cells regulate leukocyte adhesion and modulate permeability of the in vitro blood-brain barrier model. PLoS One 9, e102598
Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S (2012) West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J Gen Virol 93:1193–1203
Acknowledgements
This study was partially supported by grants (P30GM114737) from the Centers of Biomedical Research Excellence (COBRE), National Institute of General Medical Sciences, National Institutes of Health (NIH), and Institutional funds.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Kumar, M., Nerurkar, V.R. (2016). In Vitro and In Vivo Blood–Brain Barrier Models to Study West Nile Virus Pathogenesis. In: Colpitts, T. (eds) West Nile Virus. Methods in Molecular Biology, vol 1435. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3670-0_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3670-0_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3668-7
Online ISBN: 978-1-4939-3670-0
eBook Packages: Springer Protocols