
Abstract
Angiogenesis is an important determinant of tissue function, from delivery of oxygen and other substrates to removal of waste products, in health and disease (e.g., adaptive or pathological remodelling). The phenotype and functional responses of endothelial cells are conditioned by systemic humoral signals and local environmental factors, including the haemodynamic forces that act upon them. Here we describe some interventions that have been helpful in unraveling the integrative nature of the complex in vivo response, and quantitative assessment of angiogenesis in muscle.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
In principle, endothelial cell (EC) cultures can be used to measure responses relevant to angiogenesis (e.g., migration and proliferation ), to determine gene expression in response to putative signals, and to quantify intracellular, surface or secreted proteins (e.g., adhesion receptors, cytokines, proteases). In the circulation ECs are continually exposed to shear stress (luminal viscous drag applied by flowing blood), to compression by blood pressure (pulsatile distortion), and to tension from strain in the extracellular matrix (e.g., cyclic deformation during muscle activity). Intracellular signalling pathways and gene expression respond to such changes and modify EC functions including proliferation, apoptosis , adhesion, motility, and matrix deposition or degradation [1]. However, modifications of angiogenic pathways have not been systematically analysed, although responses to cytokines or growth factors will be modified if applied to EC in the presence of mechanical stress rather than to undisturbed cells [3]. In contrast to the majority of studies on chemical factors in pathological angiogenesis and in vitro models, the physical environment is known to play an important role in controlling capillary growth in normal cardiac and skeletal muscle in vivo [2, 4]. The control of in vivo angiogenesis is complex and a reductionist approach to dissect potential mechanisms is tempting, but although in vitro models are often described as ‘angiogenesis assays’ they can at best only partially represent the phenomenon. For example, use of Matrigel as a substrate may be better than flat monocultures but the subsequent tube formation is not a unique property of endothelium, and the mechanism of lumen formation is different to that found in tissues [2]. While there has been considerable progress in this approach, e.g., flow-based studies are a more physiologically relevant modelling of angiogenesis, there is a long way to go before in vitro studies can reproduce the situation in vivo [3]. In addition, each step of the angiogenic cascade described in reviews based primarily on tissue culture experiments is known to have exceptions in observed capillary growth [4].
In vivo, ECs continually experience haemodynamic forces: shear stress due to flowing blood, compressive pressure, and circumferential tension from the cardiac cycle, a pressure wave traveling along the wall. In addition to circulating humoral factors and local metabolites, in striated muscle capillaries are also subjected to stretch and compression during a duty cycle. ECs are sensitive to the shear stress applied [5] with rapid responses, e.g., production of reactive oxygen species and/or nitric oxide, and prolonged adaptations involving changes in gene expression. The physical environment is therefore recognized as an important modifier of a range of endothelial functions including proliferation , apoptosis, motility, adhesion, and matrix deposition or degradation; changes in which may directly induce angiogenesis. In animal models, skeletal muscle capillaries exposed to hyperaemia and hence increased wall shear stress, or to a sustained strain, showed similar angiogenic responses [6, 7]. Metabolic consequences of systemic hypoxaemia or local hypoxia can stimulate capillary growth, but is dependent on regional phenotype and/or requires an additional stimulus such as muscle activity [8, 9]. The responses are reliant on key chemical mediators, both pro- and anti-angiogenic growth factors, but with different time courses [10, 11].
Angiogenesis is a complex, multifactorial process that is regulated in different ways among vascular beds and according to the stimulus, often involving proliferation and migration of EC to form new capillaries from pre-existing vessels. Angiogenesis is essential for development of the vasculature and in wound repair, and is highly regulated allowing capillary growth when required or capillary rarefaction when not. Many diseases are associated with excessive angiogenesis (e.g., rheumatoid arthritis, diabetic retinopathy, gastric ulcers), or insufficient angiogenesis (e.g., cardiac hypertrophy, peripheral vascular disease), making it a major target for therapeutic intervention [12, 13]. However, there are relatively few data on angiogenesis in normal adult organs where, apart from the female reproductive tract, capillary growth is very rare [14]. Irrespective of the cause of angiogenesis, it is assumed there is a common sequence of events initiated by proteolytic breakage of the basement membrane surrounding a capillary, followed by migration of normally quiescent EC into the interstitium, accompanied by modification of the extracellular matrix and mitosis at the base of the column of migrating EC. These abluminal sprouts elongate and develop lumen, eventually fusing with existing vessels or other sprouts to create functional anastomoses. However, despite repetition in most reviews, this paradigm is not universal [15, 16].
Endothelial cell proliferation and/or migration seen in vitro are not necessarily synonymous with angiogenesis in vivo, and care in interpretation is needed if the ability to isolate putative mechanisms is not to be achieved at the expense of contextual complexity. For example, different patterns of angiogenesis may occur in vivo when the physical environment, both inside and outside vessels, changes. During development, angiogenesis by intussusceptive growth has been described, where extracellular material penetrates and divides vessels [17], perhaps driven as much by growth and remodelling of surrounding tissue as by endothelial cell-specific stimuli. Muscle activity represents a complex mix of cellular, chemical, and physical influences on angiogenesis. However, some reductionism is possible: ECs are sensitive to mechanical strain imposed by the surrounding tissue, and stretch of skeletal muscle during the early phases of overload provides a uniquely sprouting phenotype, whereas intravascular mechanical stimuli such as high shear stress as a result of hyperaemia produce a unique phenotype of longitudinal splitting [4]; the latter two forms of capillary growth exhibit both common and differential signalling responses [10, 18], with the distinct growth patterns involving perivascular cells in different ways [16]. In this chapter three in vivo models of muscle angiogenesis are described, along with techniques to assess various parameters.
Animal models of exercise usually involve treadmill or wheel running, with apparent differences in outcomes between approaches and species. For example, control of duration/intensity is difficult to normalise using running wheels, whereas the treadmill is imposed rather than voluntary so may include a stress component. Angiogenesis appears to be easier to induce in rats than mice on treadmill training, but direct comparisons are difficult due to likely different workloads imposed. Here an alternative method of muscle activity, that of indirect electrical stimulation by implanting electrodes in the vicinity of a motor nerve, is described [19, 20]. This allows more targetted recruitment of muscle and better-defined recruitment parameters.
Endothelial sprouting can also be induced by unilateral muscle overload [7]. This procedure involves the removal (surgical ablation, or extirpation) of the majority of the m. tibialis anterior (TA) from the hindlimb of the animal, which leads to a compensatory overload of the synergist m. extensor digitorum longus (EDL) and m. extensor hallucis proprius (EHP). The consequence is stretch of the EDL leading to an increase in sarcomere length of ~20 % [21]. Longitudinal splitting angiogenesis can be induced by oral ad libitum administration of prazosin [6]. Prazosin is an α1 adrenoreceptor antagonist and acts as a vasodilator by blocking tonic sympathetic tonus on the resistance vessels, predominantly within skeletal muscle, mainly affecting the arteriolar side of the vasculature. It has little or no effect on capillary diameter and therefore increases blood flow and shear stress in capillaries [14].
2 Materials
2.1 Animals
All work must be performed in accordance with local and national policies and guidelines governing animal work.
-
1.
Animals should be housed at ~21 °C with a 12 h:12 h light:dark cycle, kept with littermates in an enriched environment, and given standard laboratory feed pellets and tap water ad libitum. Mice: Male C57Bl/6 or C57Bl/10 mice should be 6–8 weeks old, and weigh ~25 g for optimal results (see Note 1 ).
-
2.
Rats: Male Sprague-Dawley or Wistar rats should be used when they are 200–250 g (see Notes 1 and 2 ).
2.2 Surgery
-
1.
Isoflurane: For induction of anesthesia use 5 % Isofluorane (Novartis) delivered at 5 L/min O2 flow rate, and maintain with 2.5 % at 2 L/min O2 (see Note 3 ).
-
2.
Buprenorphine analgesic: 2.5 mL/kg buprenorphine (Temgesic; National Veterinary Services) delivered subcutaneously.
-
3.
Duplocillin LA antibiotic: Dilute Duplocillin LA antibiotic (NVS) 1:10 with sterile water.
-
4.
Scalpels: No. 10 blades for surgical procedures.
-
5.
Monofilament sutures: Use 6-0 Vicryl (Ethicon) for mice; 4-0 Vicryl for rats (see Note 4 ).
-
6.
Teflon-coated multi-stranded stainless steel wires (e.g., Clark or AM Systems).
-
7.
Neurotech Multichannel Stimulator (Bio-Medical Research).
-
8.
Food-safe silicone sealant (e.g., Dow Corning 744).
2.3 Vasodilator Treatment
-
1.
Prazosin solution: Dissolve 50 mg/L Prazosin (see Note 5 ) in tap water, and gently warm while stirring overnight (solubility is very low). Add a small amount (suggested 1–2 g/L) of sugar to the solution to mask the bitter taste.
2.4 Tissue Sampling
-
1.
Liquid nitrogen.
-
2.
2-Methylbutane (isopentane).
-
3.
Optimal Cutting Temperature embedding matrix (OCT).
-
4.
Cork discs, 20 mm diameter.
-
5.
Poly-l-lysine-coated slides (e.g., polysine™; VWR International).
-
6.
Cryostat microtome.
2.5 Biochemical Assays
-
1.
Lysis buffer: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (975 μL), containing 10 μL Protease Inhibitor Cocktail (Sigma), 10 μL phenylmethylsulfonyl fluoride (PMSF), 5 μL sodium vanadate solution.
-
2.
ELISA kits and reagents as required, e.g., mouse and human VEGF protein and PGF1α (Cambridge Biosciences, UK).
-
3.
Bradford or similar protein assay reagents.
2.6 Alkaline Phosphatase
-
1.
Stock buffer: Add 1.7 g magnesium sulfate (MgSO4∙7H2O) and 3.8 g sodium borate (NaBO2∙4H2O) to distilled water and make up to a total volume of 1 L. Store at 4 °C (see Note 6 ).
-
2.
Working buffer: Add 7–10 mg nitroblue tetrazolium salt (NBT), 2 mg 5-bromo-4-chloro-3-indoxyl phosphate, toluidine salt (BCIP) to the stock buffer and make up to 10 mL. Sonicate to dissolve and adjust to pH 9.3 with boric acid (3.09 g/L).
2.7 Lectin Staining
-
1.
Paraffin wax pen.
-
2.
Electron microscopy grade 4 % (w/v) formaldehyde (e.g., TAAB, Berkshire, UK).
-
3.
Lectin solution: Griffonia simplicifolia Lectin I labelled with 5 μL/mL fluorescein- or rhodamine-labelled Griffonia simplicifolia Lectin I.
-
4.
Phosphate buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10 mM, Na2HPO4, 2 mM KH2PO4, and adjust pH to 7.4 with HCl.
-
5.
Fluorochrome-safe mountant (Vectashield; Vector Labs, UK) with and without 4′,6-diamidino-2-phenylindole (DAPI).
2.8 α-Smooth Muscle Actin Staining
-
1.
Formalin fixative: 10 % buffered neutral formalin solution. Add 100 mL of formaldehyde solution (37–40 %), to 900 mL distilled water containing 4.0 g NaH2PO4, 6.5 g Na2HPO4 (anhydrous).
-
2.
Tris-Buffered Saline (TBS): Prepare 10× stock TBS solution containing 0.5 M Tris Base and 9 % (w/v) NaCl at pH 7.6. Store at room temperature. Dilute the stock 1:10 with distilled water before use and adjust pH to 7.6 if necessary.
-
3.
Blocking solution: Add 10 % (v/v) normal goat serum to TBS containing 1 % (w/v) bovine serum albumin (BSA). Three milliliters is sufficient for ten slides.
-
4.
Anti-alpha smooth muscle actin antibody (e.g., rabbit monoclonal, Abcam): Dilute to 1:500 in TBS with 1 % BSA—6 μL primary antibody solution in 3 mL TBS with 1 % BSA is enough for ten slides.
-
5.
Horse radish peroxidase (HRP) labelled secondary antibody (e.g., goat anti-rabbit; Abcam); diluted to 1:1000 on the slides in TBS with 1 % BSA.
-
6.
3,3-Diaminobenzidine (DAB): Add two DAB tablets (10 mg) to 5 mL of water, vortex and filter.
2.9 BrdU Pulse Labelling
-
1.
In Situ Cell Proliferation Kit, FLUOS (Roche, Mannheim, Germany).
2.10 Proliferating Cell Nuclear Antigen (PCNA) Staining
-
1.
Formalin fixative (see Subheading 2.8, item 1).
-
2.
Washing buffer: Add 500 μL of 6 % (v/v) Triton X-100, 666 μL 7.5 % (w/v) BSA in water to 3.83 mL PBS.
-
3.
Rabbit anti-PCNA antibody (Santa Cruz); 1:100 (5 μL antibody in 500 μL washing buffer).
-
4.
Donkey anti-rabbit antibody: CY2 conjugated Donkey anti-rabbit antibody (Jackson ImmunoResearch Laboratories) dilute 1:50 with rhodamine conjugated GSL-1 lectin (1:100; Vector) (10 μL antibody, 5 μL lectin in 500 μL washing buffer).
-
5.
Aquamount/glycerol jelly.
2.11 TUNEL Staining
-
1.
Formalin fixative (see Subheading 2.8, item 1).
-
2.
Proteinase K solution: Add 1 μL TUNEL staining kit solution (Trevigen 2 TdT-DAB in situ apoptosis detection kit—Trevigen Inc., Gaithersburg, MD, USA) in 50 μL of water per slide.
-
3.
Quenching solution: Add 10 mL of 30 % hydrogen peroxide solution to 90 mL of methanol.
-
4.
Labelling reaction mix: 1 μL TdT-dNTP kit solution, 1 μL Mg2+ kit solution, 1 μL TdT enzyme kit solution, 50 μL TdT labeling buffer (kit solution diluted 1:9 in dH2O).
-
5.
DAB solution: Add 50 mL PBS, 250 μL DAB stock, 50 μL 30 % hydrogen peroxide.
-
6.
Nuclear counter stain: Add 0.5 g of methyl green (ethyl violet free, Sigma) to 100 mL 0.1 M sodium acetate buffer, pH 4.2.
-
7.
Histomount mounting solution.
2.12 Histological Quantification
-
1.
Standard light microscope system equipped with a digital camera (see Note 7 ).
-
2.
Image analysis software (see Note 8 ).
2.13 Fibre Type Composition
-
1.
Acid preincubation buffer: 0.2 M sodium acetate adjusted to pH 4.5 containing 50 % (v/v) acetic acid.
-
2.
Alkali preincubation buffer: Dissolve 3.445 g sodium metaborate and 2.36 g calcium nitrate (Ca(NO3)2) in 500 mL of distilled water and adjust the pH to 10.3 using 0.1 M sodium hydroxide.
-
3.
Glycine/calcium chloride buffer: Add 3 g glycine and 2.94 g calcium chloride in distilled water, adjust the pH to 9.4 using 0.1 M potassium hydroxide and make up to 1 L.
-
4.
Incubation buffer: Add 160 mg of adenosine 5-triphosphate (ATP) to 100 mL of glycine/calcium chloride buffer and adjust to pH 9.4 with 0.1 M KOH.
-
5.
1 % cobalt chloride (or nitrate) and 1 % ammonium sulphide in distilled water.
3 Methods
3.1 Muscle Stimulation
-
1.
Surgery should be conducted under aseptic conditions with the use of inhalation anesthetic (see Note 3 ). Five to seven animals are required in each treatment groups. Control groups: Animals without any intervention are used as controls. The untreated contralateral limbs of animals receiving surgery may also serve as controls (see Note 9 ). Sham control animals should have the electrodes implanted, but not receive stimulation (see Note 10 ).
-
2.
In the test group of animals implant seven-stranded stainless steel wires close to the lateral popliteal nerve to indirectly stimulate ankle flexors. Shave the site prior to making the incision (see Note 11 ).
-
3.
Strip the insulation from the end of the electrodes and wrap them around a 25 G needle to produce a coil, which aids inter-muscle location (located deep between the vastus lateralis and the tibialis anterior, which requires minimal dissection of covering muscle), and fix in place with sutures.
-
4.
Feed the electrodes under the skin onto the back of the animals and exteriorise through a piece of Velcro® attached to the skin at the nape of the neck. Attach the electrodes to an external stimulator. When not in use the electrodes should be coiled up inside a Velcro® pocket to prevent them being damaged by the animals.
-
5.
The animals should be recovered within 20–30 min of anesthetic removal and given systemic analgesic and topical antibiotic twice a day for the first 2 days post-surgery.
-
6.
Stimulate the extensor digitorum longus and tibialis anterior muscles for 2–28 days, starting ~24 h after surgery at 10 Hz, pulse width 0.3 ms, and sufficient voltage (3–6 V) to produce palpable contractions using an external stimulator [19].
-
7.
Alternatively, in-house constructed miniaturised implantable stimulators may be used when located subcutaneously on the back of the animal. Home Office guidelines suggest these should be <15% of the animals body mass (easy to achieve <10% with rats, less so with mice due to battery size). These are either encased in heat-shrink plastic and sealed with silicone sealant, or coated with hypo-allergenic beeswax. The stimulator is operated through a superficial magnetic reed-switch allowing it to be activated 24 h following surgery. A similar approach has been used by Greene and colleagues [20]. The limitation for their use is battery weight versus life; lithium coin batteries give ~1 week life and allow stimulators to be implanted at ~10 % body weight in mice.
3.2 Muscle Overload
-
1.
Anaesthetise animals using isoflurane delivered in oxygen to maintain blood oxygen saturation during cardiorespiratory depression, and maintain anaesthesia using a lower dose throughout the duration of the procedure (see Subheading 2.2, item 1).
-
2.
Systemic buprenorphine analgesic 2.5 mL/kg is administered subcutaneously, usually by scruffing the skin at the neck (see Subheading 2.2, item 2).
-
3.
Shave the hindlimb (see Note 11 ) and disinfect the site with 70 % ethanol spray. After a few seconds wipe away alcohol with a sterile cotton bud or tissue; surface evaporation causes cutaneous vasoconstriction that minimises bleeding on incision.
-
4.
Locate the TA tendon, usually visible under the skin at the instep. It can usually be found by feeling for the femur with the back of the scalpel blade, and running down the anterolateral surface. The outline of the TA can be seen as a slight depression on leg extension.
-
5.
Using moderate pressure, with the scalpel make an incision from the tendon to about midway up the TA. Make a single incision of 1–2 cm through the skin. Free the skin and carefully clear away any fascia overlaying the muscle with blunt dissection using strabismus scissors.
-
6.
Free up the tendon from surrounding connective tissue (ensure the surface is glistening white), being careful to avoid damage to any blood vessels in close proximity (the ankle is well vascularised).
-
7.
Slide a pair of curved forceps under the tendon and cut with scissors or scalpel, leaving a generous portion still attached to the muscle (see Note 12 ).
-
8.
Holding the cut tendon, lift the muscle free from the underlying tissue, blunt dissect the inner surface from the underlying EDL, and free the lateral edges. With one clean cut slice through the muscle with a scalpel 2/3–3/4 along its length.
-
9.
Replace the cut end of the muscle and apply pressure to the site for a few moments in order to stop any bleeding.
-
10.
Apply one drop of topical antibiotic, leaving for about 10 s before blotting away excess.
-
11.
Using monofilament sutures close the wound with 4–5 interrupted stitches (see Note 13 ). Clean the wound carefully, as any dried blood invites the animal to nibble at sutures; an anaesthetic cream may dampen irritation but is not usually necessary.
-
12.
Allow the animal to recover in a warmed cage, with jelly feed to aid rehydration. Check on them regularly—the only issue noted is occasional biting of sutures that require repair within the first 12 h. Thereafter they feed and drink normally, with minor gait impairment lasting 36–48 h. Normal climbing activity returns earlier.
3.3 Chronic Vasodilatation
-
1.
Replace normal drinking water of animals with the prazosin solution (see Note 14 ) and allow access ad libitum (see Note 15 ).
-
2.
Replace with fresh Prazosin solution every 2–3 days, as the drug will precipitate (evident as a cloudy appearance of the water).
3.4 Tissue Sampling
-
1.
For immunohistochemistry and protein and mRNA quantification kill the animals by stunning and cervical dislocation.
-
2.
Immediately dissect the muscles and divide into two. Process as described below for biochemical and histological analysis in items 3 and 4 respectively. For the EDL aim to sample from the central half of the muscle.
-
3.
Snap-freeze one half of the muscle in liquid nitrogen for biochemical analysis.
-
4.
Coat the other half of muscle sample with OCT for structural support and cryoprotection. Orientate the muscle perpendicularly on a cork disc by leaning the muscle against a pin or soft tissue (e.g., piece of liver). Carefully invert and drop the disc into liquid nitrogen-cooled isopentane bath (see Note 16 ).
-
5.
Keep discs in liquid nitrogen for short-term storage before sectioning; for long-term storage wrap in aluminum foil packets to aid sorting and place at −80 °C.
-
6.
Cut ~ 10 μm sections (8 μm is better, 12 μm is acceptable) on a cryostat microtome at −20 °C, depending on the rigidity of the sections obtained this may be varied by ±2 °C only.
-
7.
Pick up sections on poly-l-lysine-coated slides for better adhesion during subsequent immunohistochemistry and allow to air-dry for 30–60 min before staining or re-freezing and storage at −20 °C (see Note 17 ).
3.5 Protein Quantification
-
1.
Powder frozen tissue using a chilled pestle and mortar under liquid nitrogen and suspended in ice-cold lysis buffer.
-
2.
Centrifuge at 5000 × g for 20 min at 4 °C.
-
3.
Assay samples for the protein of interest in duplicate by ELISA, according to manufacturer’s instructions (5–6 animals/group).
-
4.
Determine total tissue protein concentration by Bradford assay performed in duplicate at the same time as the ELISA.
3.6 Alkaline Phosphatase Staining
Alkaline phosphatase is an enzyme found at high levels in capillary endothelium, but may also stain terminal arterioles and motor nerves; it is not suitable for all tissues or species (e.g., not found in human muscle, lower staining in mouse muscle). This staining protocol uses the phosphatase activity to cleave BCIP, which becomes a strong reducing agent, then acts on NBT to form an insoluble dye which deposits on the capillary wall.
-
1.
Air-dry cryostat sections (30 min if fresh or taken from freezer).
-
2.
Fix sections in precooled acetone (4 °C or less) for 5 s.
-
3.
Air dry (5–10 min).
-
4.
Stain the slides with working buffer for 45 min to 1 h at room temperature.
-
5.
Wash in distilled water.
-
6.
Mount with aquamount and a cover slip. Seal with glue or nail varnish if sections are to be kept for long periods.
3.7 Lectin Staining
Lectins are carbohydrate-binding proteins from plant extracts; those isolated from Griffonia (Bandeiraea) simplicifolia seeds (GSL-1/BSL-1) identify ECs in mouse and rat tissues. They bind to galactose residues, which are found at high concentration on the proteoglycans in the glycocalyx lining blood vessels and laminin glycoproteins, which are major components of the basal lamina that surround the vasculature. The choice of which lectin to use is species-dependent [22], and other variables, e.g., use Ulex europaeus lectin, UEA-I, for humans but staining may be influenced by blood group.
-
1.
Air-dry cryostat sections fully (30 min if fresh or taken from freezer).
-
2.
Draw around sections with a paraffin wax pen, and allow to dry completely (~15 min). This produces a shallow well for incubating tissue, and helps to reduce problems with evaporation while minimizing incubation volume.
-
3.
Fix in ice-cold acetone (4 °C) for 15 s. Alternatively, fix with formaldehyde, followed by 3 × 5 min PBS washes.
-
4.
Incubate for 1 h at room temperature using fluorescein- or rhodamine-labelled lectin.
-
5.
Wash 3 × 5 min in PBS.
-
6.
Wash in distilled water.
-
7.
Mount using 1:10 Vectashield plus DAPI:Vectashield without DAPI (to normalise fluorescent intensity of different structures).
3.8 α- Smooth Muscle Actin
During vascular remodelling, haemodynamic control needs to be preserved and so estimating the size of microvascular units (number of capillaries fed by individual arterioles) is an important readout. The process of arteriolisation has been shown to accompany, or even precede angiogenesis . Alpha actins are used to label vascular smooth muscle cells , and identify arterioles and venules on the basis of size and layer thickness.
-
1.
Day 1: Fix in formalin fixative for 10 min.
-
2.
Wash the slides 4 × 5 min in TBS + 0.025 % Triton X-100 with gentle agitation.
-
3.
Incubate in blocking solution for 2 h.
-
4.
Drain slides for a few seconds.
-
5.
Incubate with antibody to α-smooth muscle actin overnight at 4 °C.
-
6.
Day 2: Wash 2 × 5 min in TBS + 0.025 % Triton X-100 with gentle agitation.
-
7.
Wash 2 × 5 min TBS + 0.025 % Triton X-100 with gentle agitation.
-
8.
Incubate the slides in 0.3 % hydrogen peroxide in TBS for 15 min.
-
9.
Wash 2 × 5 min TBS + 0.025 % Triton X-100 with gentle agitation.
-
10.
Apply HRP secondary antibody and incubate for 1 h at RT.
-
11.
Wash 3 × 5 min TBS.
-
12.
Develop with DAB for 10 min at RT.
-
13.
Wash in running tap water for 5 min.
-
14.
Mount in glycerol.
3.9 Bromodeoxyuridine (BrdU) Pulse Labelling
Understanding the relationship between cellular proliferation and angiogenesis is important; e.g., the longitudinal splitting form is efficient in that it requires little mitotic activity, while proliferative activity in perivascular or mural cell s gives insight into local regulatory mechanisms. Incorporation of an identifiable nucleoside during mitosis has long been used to label the site of proliferation, with BrdU (a synthetic thymidine analog) often preferred. Co-localisation of BrdU with capillaries can provide a measure of cell proliferation associated with the capillaries (though not necessarily ECs), and that associated with interstitial cells or myonuclei.
-
1.
Animals are injected i.p. with BrdU labeling reagent 16 h prior to tissue sampling. Other time points can be used to assess the kinetics of cell turnover.
-
2.
Cryosections are obtained as above (see Subheading 3.4).
-
3.
Immunostaining is also performed using primary anti-BrdU antibodies as described above (see Subheading 3.8) with the addition of 1:100 rhodamine Griffonia simplicifolia lectin-1 to allow co-localisation of BrdU-labelled cells with vascular structures.
-
4.
Slides are mounted using 1:10 Vectashield + DAPI:Vectashield to permit confirmation of nuclear localisation of BrdU labelling.
3.10 PCNA Staining
For archived material, an alternative approach to BrdU labeling is to retrospectively stain for factors upregulated during the cell cycle. PCNA (a.k.a. cyclin) is a DNA polymerase co-factor expressed by cells undergoing mitosis, and so can act as a measure of cell proliferation .
Air-dry cryostat sections (30 min).
-
1.
Fix in formalin fixative for 5 min.
-
2.
Wash in PBS for 3 × 5 min.
-
3.
Block with washing buffer at RT for 30 min.
-
4.
Incubate with primary antibody for 1 h (usually 1 % (see Subheading 2.10, item 3).
-
5.
Wash in PBS 3 × 5 min.
-
6.
Incubate with secondary antibody (usually 1/200) and lectin for 1 h. Keep sections covered and out of direct light.
-
7.
Wash in PBS 3 × 5 min.
-
8.
Wash in distilled water.
-
9.
Mount with Aquamount/glycerol jelly if required.
3.11 TUNEL Staining
-
1.
An important balance to angiogenesis during remodelling is vascular pruning. During apoptosis , endonucleases digest DNA to form short double-stranded DNA fragments that can be detected by attaching biotinylated nucleotides to the 3′-OH ends using a terminal deoxynucleotidyl transferase (TdT) enzyme, visualised using a streptavidin-horseradish peroxidase conjugate followed by the substrate DAB which forms a brown precipitate when broken down by peroxidase. Apoptotic cells must be distinguished from necrotic cells morphologically. Air-dry cryostat sections fully (30 min if fresh or taken from freezer).
-
2.
Draw round sections with a paraffin wax pen, and allow to dry (~15 min).
-
3.
Fix for 10 min in formalin fixative.
-
4.
Wash slides for 2 × 5 min in PBS.
-
5.
Blot slides dry, then immediately add 50 μL of proteinase K solution and cover.
-
6.
Incubate for 15 min and then wash 2 × 2 min in dH2O.
-
7.
Place slides into 50 mL of quenching solution for 5 min.
-
8.
Wash 2 × 1 min in PBS.
-
9.
Incubate slides in 50 mL TdT labeling buffer for 5 min.
-
10.
Blot slides dry, then immediately add 50 μL labelling reaction, mix and cover.
-
11.
Incubate slides in a 37 °C incubator in a humidified chamber for 2 h.
-
12.
Transfer slides into 50 mL TdT stop buffer (kit solution diluted 1:9 in dH2O) and incubate for 5 min.
-
13.
Wash slides 2 × 2 min in PBS.
-
14.
Blot slides dry and then add 50 μL streptavidin-HRP detection solution onto each sample (1 μL kit solution in 50 μL dH2O per slide) and cover. Incubate at RT for 10 min.
-
15.
Wash slides 2 × 2 min in PBS.
-
16.
Incubate slides in DAB solution for 5 min.
-
17.
Wash slides in dH2O and transfer to fresh water.
-
18.
Counterstain nuclei by transferring slides to 50 mL methyl green solution for 5 min.
-
19.
Wash slides by sequentially dipping in 1-butanol until the sample turns from blue to mainly green, dip once more in fresh 1-butanol, then dip ten more times in two changes of xylene.
-
20.
Dry xylene from the back of the slide. Leave xylene on the surface of the slide to aid the mounting process.
-
21.
Place two drops of Histomount on the samples and gently lower the coverslip.
-
22.
Leave to harden overnight. Store out of direct light.
3.12 Quantification of Capillary Supply
-
1.
Once capillary visualisation is complete, three to four images per section are photographed and analysed (see Note 18 ).
-
2.
Photomicrographs can be taken using ×10 or ×20 objectives, depending on the field of view required (e.g., 0.14–0.20 mm2); ×40 is better for co-localisation studies.
-
3.
Some pre- or post-processing may be required to compensate for the dynamic range of capillary staining intensities, or to adequately visualise fibre boundaries (try equalisation or logarithmic filters).
-
4.
For each image place one to four sampling squares (regions of interest, ROI) on the field of view (Fig. 1), and count the number of capillaries and fibers using an unbiased counting rule (Fig. 2).
Fig. 1 
Sampling ROI for individual muscle cross sections. Arrows show the gradation in oxidative capacity, decreasing in the medial-lateral axis (see Ref. 8)
Fig. 2 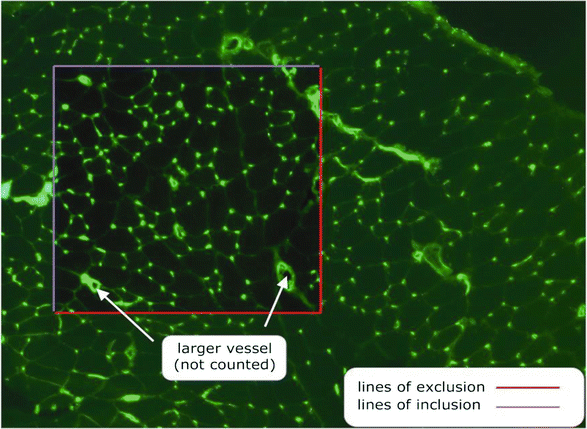
Image showing a cross section of muscle stained with FITC-labelled Griffonia simplicifolia (GSL-1) illustrating the protocol for unbiased sampling for estimating capillary supply in skeletal muscle
-
5.
The top and left sides of the sampling square are designated ‘inclusion lines’ with the bottom and right sides designated ‘exclusion lines.’ Any capillary or fibre exclusively touching a line of inclusion is counted in the analysis; any touching a line of exclusion is excluded from the analysis.
-
6.
It is best if the counting frame is systematically oriented on the same circumscribed regions, relative to the major axis and the section boundaries, to provide a systematic-random sampling. This is statistically preferable to simple random sampling, which will increase data variance and tend to obscure any localised response of specific muscle regions. Location is especially important in the outer cortex of the TA muscle, where apparent capillarisation/fibre composition is very sensitive to the sample position.
-
7.
A fibre is defined as an isolated nonstained, convex area surrounded by collagen IV or laminin .
-
8.
A capillary is defined as an ALP/lectin/CD31 -positive structure <8 μm in diameter. They are seen as either a circle in cross section or elongated structures when sectioned obliquely, in which case individual branches are counted as single capillaries.
-
9.
The size of the ROI is important in limiting ‘edge effect’ errors, the most common being too few fibres (ideally >50) or capillaries (>100) included.
-
10.
There are a large number of indices used to describe the adaptive nature of the microcirculation, most of which have severe limitations in their utility (for detailed explanation, see Ref. 23).
-
11.
An unbiased counting rule [24] should be used to estimate the capillary density (CD or NA(c,f) in stereological notation; expressed as number of capillaries per mm2 of section (ideally fibre area), the capillary to fiber ratio (C:F, NN(c,f)) is the number of capillaries/total number of fibers), and the average fiber cross-sectional area (FCSA; total area of fibres/number of fibres) (see Note 19 ).


3.13 Fibre Type Composition
Muscle fibre types may be detected using enzymatic methods, exploiting differential pH stability of different myosin ATPases [25]. For example, in sections preincubated at pH 4.55 dark staining indicates Type I (slow oxidative), light staining indicates Type IIa (fast oxidative glycolytic), and intermediate staining intensity indicates Type IIb (fast glycolytic) fibre types. Intrinsic mATPase activity may be revealed by omitting the preincubation step. It does not easily allow discrimination between Type IIb and Type IId/x/c fibres whose oxidative capacity is between Type IIa and IIb, which may be assessed by staining for succinic dehydrogenase activity (note differences in notation between rodent and human fibre types). This approach is increasingly replaced by monoclonal anti-MHC antibodies allowing for targeted multiple fibre staining (see Ref. 26).
-
1.
Preincubate sections: 2–4 min in either acid or alkali preincubation solution.
-
2.
Wash in distilled water (dH2O).
-
3.
Wash in glycine/CaCl2 buffer.
-
4.
Incubate sections: 15 min in incubation solution at room temperature.
-
5.
Wash three times in dH2O.
-
6.
Incubate in 2 % CaCl2 three times for 30 s each.
-
7.
Wash three times in dH2O.
-
8.
Incubate in 1 % CoCl2 for 3 min.
-
9.
Wash five times in dH2O.
-
10.
Incubate in 1 % ammonium sulphide for 3 min in a fume cupboard.
-
11.
Wash five times in dH2O.
-
12.
Mount with Aquamount/glycerol jelly and coverslips.
-
13.
The muscle cross-sectional area occupied by the three main fibre types may be quantified using standard stereological point-counting methodology [24]. Type I or Type IIa fibres from fast or mixed muscles are about half the size of Type IIb fibres in adult rats, and consequently their total oxidative capacity is overestimated if only numerical frequency is quantified. In addition, muscle tension development is proportional to FCSA; thus a quantitative estimate of their relative area may be a better index of functional capacity.
3.14 Power Calculations
Based on previously published data, significant differences can be seen in capillary supply indices and most ultrastructural data between two experimental groups using n = 4 animals; however if submaximal responses are induced this will require a larger sample size. Our previous data has a median average standard deviation (SD) of 0.12 for C:F which requires n = 6 to give 80 % power to means separated by 0.2, or roughly half the response to our mechanical stimuli. The most subtle ultrastructural change we would expect to see is changes in pericyte coverage. Our previously published data has a median average SD of 2.1, which therefore requires n = 7 to have 80 % power in a mean difference of 3, again half the response in our previous model. Power calculation for microarray analysis is a rather contentious topic, but based on a power of 85 % and P < 0.05, previous experience gives a pooled variance term S = 0.15, then using D/S × N(2/n) gives a requirement for 6–8 animals/experimental group. Data are usually presented as mean ± SEM, with statistical analysis by one-way ANOVA using PLSD or Bonferroni post-hoc analysis.
4 Notes
-
1.
Males are almost always used in order to avoid interference in vascular remodeling that may occur during the estrous cycle in females.
-
2.
Sprague-Dawley rats are fast growing with relatively aerobic muscles, whereas Wistar rats tend to be smaller at similar age and have slightly less aerobic muscles.
-
3.
We originally used halothane (Fluothane®, ICI), but now use Isoflurane (IsoFlo®) routinely.
-
4.
It is preferable to use absorbable sutures for internally and nonabsorbable for cutaneous sutures.
-
5.
Prazosin was originally a gift from Pfizer. It is now commercially available from Fluka, Tocris, and Sigma-Aldrich.
-
6.
The stock buffer can be stored for several months at 4 °C.
-
7.
Any standard microscope system is adequate for acquisition of photomicrographs. We used a Zeiss Axioskop 2 plus fluorescent microscope, with image capture using an Axiocam MRc digital camera and Axiovision software (Karl Zeiss).
-
8.
Analysis of digital images can be performed using commercial image processing packages, but we routinely use ImageJ (NIH).
-
9.
In animals receiving surgery there is some gait adjustment following surgery that means the untreated contralateral limb muscles are not exactly the same as those from naïve control animals, but serve to normalise inter-animal variation.
-
10.
Sham controls serve to normalize for surgical trauma, but are probably only important for the earliest time points as for example edema resolves within 2–3 days of surgery.
-
11.
Removing the fur avoids allergic reaction to fine hair, and allows clean wound closure with minimal ingress of bacteria etc.
-
12.
Leaving a small stump of the TA may be preferable to complete removal as it can cause less bleeding, surgical trauma, and/or inflammation (the latter consequences may affect angiogenesis), but there is no noticeable difference in extent of final capillary supply.
-
13.
Continuous sutures may unravel if the animal chews them.
-
14.
Prazosin has been the vasodilator drug of choice for our group, although other compounds may have similar effects.
-
15.
Each mouse receives approximately 175 μg per day, based on the average water consumption monitored throughout the experiment. The rat dosage should be scaled according to body mass and drinking rates.
-
16.
Note this avoids prolonged freezing due to volatilisation of liquid nitrogen alone, but the wetting agent must therefore be treated carefully. To avoid freezing artifacts the isopentane should be close to its freezing point, which can be ascertained through an increase in viscosity and clouding whilst stirring, and preferably has solid bits floating in it; for larger tissue samples this is essential.
-
17.
Avoid condensation on removal from the freezer as freezing artifacts can also occur on temperature reversal and re-storage.
-
18.
More sections may be analyzed provided they are adequately separated to avoid the same microvascular units being sampled twice (e.g., 100–200 μm apart), but this rarely provides additional accuracy in the absence of gross structural heterogeneity.
-
19.
I would recommend these three indices as a minimum: CD offers insight into the functional capacity of the microcirculation, C:F is a robust index of angiogenic activity under most conditions, while FCSA is a direct determinant of CD so cannot be ignored. Numerical or areal fiber type composition can offer additional insights (see Subheading 3.13).
References
Lekes PI (1999) Endothelium and mechanical forces. Harwood Academic Publishers, London
Egginton S, Gerritsen M (2003) Lumen formation: in vivo versus in vitro observations. Microcirculation 10:45–61
Nash GB, Egginton S (2005) Modelling the effects of the haemodynamic environment on endothelial cell responses relevant to angiogenesis, in angiogenesis assays: a critical appraisal of current techniques. In: Staton CA, Lewis C, Bicknell R (eds) Angiogenesis assays—a critical appraisal of current techniques. John Wiley & Sons, London
Egginton S, Zhou A-L, Brown MD et al (2001) Unorthodox angiogenesis in skeletal muscle. Cardiovasc Res 49:634–646
Barakat A, Lieu D (2003) Differential responsiveness of vascular endothelial cells to different types of fluid mechanical shear stress. Cell Biochem Biophys 38:323–343
Zhou A, Egginton S, Hudlicka O et al (1998) Internal division of capillaries in rat skeletal muscle in response to chronic vasodilator treatment with alpha1-antagonist prazosin. Cell Tissue Res 293:293–303
Zhou AL, Egginton S, Brown MD et al (1998) Capillary growth in overloaded, hypertrophic adult rat skeletal muscle: an ultrastructural study. Anat Rec 252:49–63
Deveci D, Marshall JM, Egginton S (2001) Relationship between capillary angiogenesis, fiber type, and fiber size in chronic systemic hypoxia. Am J Physiol Heart Circ Physiol 281:H241–H252
Milkiewicz M, Hudlicka O, Verhaeg J et al (2003) Differential expression of Flk-1 and Flt-1 in rat skeletal muscle in response to chronic ischaemia: favourable effect of muscle activity. Clin Sci (Lond) 105:473–482
Williams JL, Weichert A, Zakrzewicz A et al (2006) Differential gene and protein expression in abluminal sprouting and intraluminal splitting forms of angiogenesis. Clin Sci (Lond) 110:587–595
Olfert IM, Birot O (2011) Importance of anti-angiogenic factors in the regulation of skeletal muscle angiogenesis. Microcirculation 18:316–330
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27–31
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307
Hudlická O, Brown M, Egginton S (1992) Angiogenesis in skeletal and cardiac muscle. Physiol Rev 72:369–417
Egginton S (2009) Invited review: activity-induced angiogenesis. Pflügers Arch 457:963–977
Egginton S (2011) Physiological factors influencing capillary growth. Acta Physiol (Oxf) 202:225–239
Djonov V, Baum O, Burri PH (2003) Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res 314:107–117
Williams JL, Cartland D, Hussain A et al (2006) A differential role for nitric oxide in two forms of physiological angiogenesis in mouse. J Physiol 570:445–454
Hudlicka O, Egginton S, Brown MD et al (1994) Effect of long-term electrical stimulation on vascular supply and fatigue in chronically ischemic muscles. J Appl Physiol 77:1317–1324
Linderman JR, Kloehn MR, Greene AS (2000) Development of an implantable muscle stimulator: measurement of stimulated angiogenesis and poststimulus vessel regression. Microcirculation 7:119–128
Egginton S, Hulicka O, Brown MD et al (1985) Capillary growth in relation to blood flow and performance in overloaded rat skeletal muscle. J Appl Physiol 85:2025–2032
Kirkeby S, Mandel U, Vedtofte P (1993) Identification of capillaries in sections from skeletal muscle by use of lectins and monoclonal antibodies reacting with histo-blood group ABH antigens. Glycoconj J 10:181–188
Egginton S (1990) Morphometric analysis of tissue capillary supply. Adv Comp Environ Physiol 6:73–141
Egginton S (1990) Numerical and areal density estimates of fibre type composition in a skeletal muscle (rat extensor digitorum longus). J Anat 168:73–80
Brooke MH, Kaiser KK (1970) Muscle fiber types: how many and what kind? Arch Neurol 23:369–379
Bloemberg D, Quadrilatero J (2012) Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One 7:e35273
Acknowledgements
Development of the ideas in this chapter has been supported by the BBSRC, BHF, MRC, and Wellcome Trust; discussions with many colleagues and students have been extremely helpful.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Egginton, S. (2016). In Vivo Models of Muscle Angiogenesis. In: Martin, S., Hewett, P. (eds) Angiogenesis Protocols. Methods in Molecular Biology, vol 1430. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3628-1_25
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3628-1_25
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3626-7
Online ISBN: 978-1-4939-3628-1
eBook Packages: Springer Protocols