
Abstract
Angiogenesis refers to the growth of new capillaries from a pre-existing capillary bed which can occur during normal physiological and pathological conditions by sprouting and non-sprouting processes, which are activated by different stimuli. Various studies have demonstrated that exercise increases the expression of several growth factors for both sprouting and non-sprouting angiogenesis, including vascular endothelial growth factor and other cytokines in skeletal and cardiac muscle, which are associated with an increase in the number of capillaries in the heart and skeletal muscle. Exercise is known to stimulate the release of several pro- and anti-angiogenic proteins and transcription factors and it appears that hypoxia and/or ischemia play a major role in the growth and expansion of new capillaries and has also been suggested that mechanical forces, such as shear stress or muscle overload, stimulate exercise-induced angiogenesis. More importantly, an in-depth understanding of the factors that influence exercise-induced angiogenesis may contribute to the development of potential therapeutic strategies for the treatment of different diseases including hypertension and ischemic heart disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Angiogenesis
- Physical activity
- Exercise training
- Vascular endothelial growth factor
- Matrix metalloproteinase
- Angiopoietins
- Endostatin
- Thrombospondin
- Tissue inhibitors of matrix metalloproteinases
- Endothelial progenitor cells
- Chronic disease
1 Introduction
Blood vessels and capillaries play a vital role in supplying oxygen and nutrients to metabolic tissues as well as play a functional role in the endocrine and immune systems [1]. Much of the current understanding of the anatomy and physiology of capillaries stems from the early work of Krogh [2–5], who examined the distribution and number of capillaries in organs and tissues, the structure of the capillary wall, and the exchange of substances through the capillary wall [5]. His work has also eluded to the fact that blood vessels have the capacity to grow (i.e., arteriogenesis) as well as to form new blood vessels from a pre-existing capillary bed, which is known as angiogenesis. The term angiogenesis was first used to describe the formation of new blood vessels during placental growth [6] and was first observed by implanting a transparent chamber into the ear of a rabbit [7]. Angiogenesis is important for normal physiological growth, such as in the female reproductive tract [8], wound healing, and muscle remodeling [9]. Deviations from normal vessel growth and maintenance can lead to several chronic diseases and angiogenesis plays an important role in the healing process after conditions such as stroke [10], myocardial infarction [11], ulcers [12], and neurodegeneration [13]. On the other hand, abnormal blood vessel growth or remodeling is associated with cancer [14], inflammatory disorders [15], pulmonary hypertension [16], and in eye diseases [17].
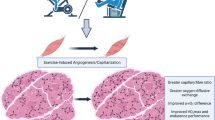
Physical activity has been shown to be a powerful stimulus of cardiac and skeletal muscle adaptation and physiological remodeling. For example, exercise training plays a major role in the remodeling process to increase capillary density of muscle [18] and mitochondrial enzyme activity after a single bout of exercise [19] as well as after exercise training [20]. These changes may occur because skeletal and cardiac muscle are metabolically active tissues that can increase their metabolic requirements during exercise 30–50-fold compared to basal conditions [21]. Therefore, adequate circulation to supply oxygen and nutrients to these tissues and to remove “metabolic waste” from these tissues is essential for their optimal performance and health. Since its discovery, angiogenesis has been studied extensively and several investigators have attempted to elucidate the mechanisms to explain how exercise stimulates angiogenesis. Hudlicka et al. [22] have provided data on some of the possible stimuli for exercise-induced angiogenesis, including increases in blood flow (hyperemia), shear stress, and muscle stretch. Other suggested stimuli include hypoxia and metabolic disturbance [23] (Fig. 11.1).

Simplified pathway of exercise angiogenesis and the suggested mechanisms that exercise affects, including hypoxia, shear stress, muscle activity/stretch, and metabolic disturbance. VEGF vascular endothelial growth factor; MMPs matrix metalloproteinases; Ang angiopoietin; EPCs endothelial progenitor cells; miRNAs microRNAs; TSP-1 thrombospondin-1; TIMPs tissue inhibitors of matrix metalloproteinases
Angiogenesis is a highly regulated process and is controlled by several pro- and anti-angiogenic factors which can be turned on when needed and completely shut down [22, 24]. Over 30 years ago, Dvorak et al. [25] discovered a new molecule involved in tumor growth they termed vascular permeability factor, which was later renamed as vascular endothelial growth factor (VEGF) [26]. VEGF is thought to be the key mediator in the angiogenic process and there is a growing body of evidence indicating that exercise increases the transcription and proteins of VEGF in cardiac and skeletal muscle [27–34]. Two other pro-angiogenic factors that increase with exercise are matrix metalloproteinases (MMPs) and angiopoietins (Ang-1, Ang-2). The three most investigated anti-angiogenic factors that are upregulated with exercise include endostatin, thrombospondin-1 (TSP-1), and tissue inhibitors of matrix metalloproteinases (TIMPs). Accordingly, evidence for the role of acute and chronic exercise training for upregulating these factors will be reviewed in this article. We will discuss a few other pro- and anti-angiogenic factors to determine if these proteins change in response to acute exercise or chronic exercise training. Since endothelial progenitor cells (EPCs) as well as microRNAs (miRNAs) also increase with acute exercise and chronic exercise training, their role in angiogenesis will be reviewed.
2 Sprouting and Non-sprouting Angiogenesis Pathways
Animal studies have revealed at least two forms of angiogenesis to occur, at least in skeletal muscle: sprouting angiogenesis and non-sprouting angiogenesis [35]. Irrespective of the cause of angiogenesis, it is assumed that there is a common sequence of events; the activation of endothelial cells followed by the degradation of the basement membrane and the extracellular matrix allowing endothelial cells to migrate to sites where capillaries are needed. Sprouting angiogenesis forms new capillaries via stalk cells and tip cells, whereas non-sprouting angiogenesis occurs when capillaries split into two via intussusception. Non-sprouting angiogenesis is suggested to be a more efficient form of angiogenesis as it permits a more rapid expansion of capillaries and does not require an initial proliferation of endothelial cells [36]. Several reviews have described the details for both sprouting [1, 9, 21, 22, 24, 37–44] and non-sprouting angiogenesis [21, 35, 45–49].
Sprouting angiogenesis is considered to be the major type of angiogenic growth induced by exercise [21] and may be induced by stimuli such as hypoxia, metabolic disturbance, and muscle stretch [35]. It involves the activation of normally quiescent endothelial cells [50], which branch out from an existing capillary and extend through a surrounding matrix to form a cord-like structure. Initiation of sprouting angiogenesis requires the proteolytic degradation of the basement membrane [51], formed by inactive endothelial cells and pericytes. Normally quiescent blood vessels are built with angiogenic sensors, and following the angiogenic signal, pericytes detach from the vessel wall [52] and are removed from the basement membrane [53]. Endothelial cells begin to loosen their junctions from the vessel and the vessel begins to dilate [54]. Permeability of the endothelial cells’ layer forces plasma proteins to enter the extracellular matrix and stored angiogenic molecules begin to remodel the extracellular matrix [54]. An endothelial “tip cell” leads the migration of endothelial cells and neighboring stalk cells have filopodia to guide their elongation [55]. In order for the abluminal sprouting capillary to become functional, it must fuse with a neighboring blood vessel and become mature [54]. Endothelial cells return to their inactive state. A myeloid bridge aids the elongation of the stalk as well as the fusion with another blood vessel and allows the initiation of blood flow [54]. Pericytes cover the newly formed capillary and protease inhibitors deposit a basement membrane. Junctions are re-established to ensure optimal blood flow. Regression occurs in the absence of angiogenic stimuli [56, 57].
New capillaries can also form by non-sprouting angiogenesis by intussusception and this was first observed in the rat lung [58]. Opposing capillary walls protrude towards the vessel lumen, and once contact has been established between the capillary walls, the inter-endothelial cell junctions are reorganized and the endothelial bilayer of the capillary becomes centrally perforated [58]. An interstitial pillar-like structure is formed, which is invaded by pericytes and myofibroblasts and these lay down collagen fibrils [59]. The final phase of intussusception is when the pillars increase in girth without any other change in structure [58]. When comparing sprouting vs. non-sprouting angiogenesis, it would appear that non-sprouting angiogenesis is a more efficient way of expanding the capillary network as there is much less endothelial cell proliferation and capillary maturation involved [60].
3 Factors That Stimulate Angiogenesis in Response to Exercise
Angiogenesis is a complex, multi-step process and is thought to involve multiple signaling pathways which are coordinated by pro- and anti-angiogenic mediators to generate effective angiogenesis. The following are various pro-angiogenic factors which are considered to be the key mediators in the angiogenic process specifically relevant to exercise-induced angiogenesis.
3.1 Vascular Endothelial Growth Factor in Acute Exercise and Chronic Exercise Training
VEGF has been shown to play a crucial role in exercise-induced angiogenesis [61–63]. VEGF is a 35–45 kDa peptide growth factor and includes a number of isoforms (VEGF-A to VEGF-D), with the most relevant to exercise-induced angiogenesis being VEGF-A (commonly known as just VEGF). Sources of VEGF include skeletal muscle fibers, endothelial cells, fibroblasts, macrophages, pericytes, as well as mast cells and smooth muscle cells [33, 63]. VEGF plays a major role in endothelial cell stimulation, survival and differentiation, vascular smooth muscle proliferation and migration, and induces capillary permeability and dilation of arterioles [21]. An increase in VEGF transcription is thought to be regulated by several factors. One of these factors includes hypoxia-inducible factor-α (HIF-1α) during hypoxia [64]. VEGF contains a hypoxic-response element [65] and binds to HIF-1α during hypoxic states. This results in an increase in VEGF transcription when there is reduced oxygen availability and it would seem that HIF-1α would also regulate VEGF protein during exercise. For example, in response to moderate to heavy exercise (knee extension exercise at 75–100 % maximum work rate), intracellular PO2 decreases from 30 mm Hg at rest [66] to approximately 3–4 mmHg during exercise in human quadriceps muscle [67]. However, the exercise-induced increase in VEGF is more complex than by activation of HIF-1α alone. It is pointed out that VEGF expression was still observed in HIF-1α knockout mice following exercise training [68]. Moreover, there is a reduced VEGF transcription response with restricted blood flow; however, there was still a similar activation in HIF-1α between the restricted and non-restricted exercising skeletal muscle [69]. Therefore, VEGF must be regulated by some factors other than HIF-1α.
More recently, metabolic activity has been highlighted as playing a role in VEGF regulation [70]. For example, AMP-activated protein kinase (AMPK) has been suggested to play a role in exercise-induced angiogenesis. AMPK is involved in the regulation of the stress response as well as metabolic homeostasis and is associated with increased glucose uptake in muscle [71]. It also leads to the promotion of angiogenesis by increasing the expression of VEGF in skeletal muscle cell culture (i.e., C2C12 cell line) through activation of the AMPK-p38 MAPK-dependant pathway [72]. Skeletal muscle cell culture has also indicated that adenosine enhances mRNA expression of VEGF and this may be mediated by the A2B receptor [73]. Several studies in animal models have revealed that peroxisome-proliferator activated receptor γ co-activator-1α (PGC-1α) through the activation of estrogen-related receptor α (ERR-α) serves as a principal regulator of increased expression of VEGF through exercise [72–74].
There is a growing body of evidence that VEGF and its receptors VEGFR1, VEGFR2, and neuropilin 1 (NRP-1), which is also a receptor of VEGF, increase in response to a single bout of exercise [27–34]. Moreover, an acute bout of exercise in untrained subjects releases higher amounts of VEGF mRNA and protein response compared with trained subjects [75]. Several studies have shown that an acute bout of single-leg knee extension exercise lasting for 45 min at approximately 25% maximum workload increases mRNA expression of VEGF by at least twofold and VEGF protein expression ~1.5-fold [27–29]. Splice variants VEGF121 and VEGF165 increase initially after an acute bout of exercise by 3- and 3.5-fold, respectively, after 45 min of one-leg knee extension exercise at 25% of maximum workload, followed by a 3.5-fold increase in VEGF189 [33]. The relative change in expression after exercise may be represented by endothelial activation with VEGF121 and VEGF165 as they are diffusible [76], whereas VEGF189 contains a domain encoded by exon 6 and is bound to cell-surface heparin sulfate proteoglycans which represent a later chemoattractant and differentiation role in the VEGF system [76].
Prolonged moderate intensity exercise training is known to upregulate VEGF mRNA expression and protein content. For example, 60 min of cycling at 50% of maximal aerobic capacity (i.e., VO2max) increases VEGF mRNA expression by approximately 4.5-fold [30], whereas a 60 min bout of cycling at approximately 60% of VO2max increased the concentration of interstitial VEGF by >6-fold compared to resting levels [77]. These data are also supported by Rullman et al. [78] who demonstrated a 1.5-fold increase in VEGF protein content in subjects who exercised for 65 min at ~65 VO2max. The type and intensity of acute endurance exercise may also influence the VEGF response to exercise. In fact, high intensity sprint interval exercise has recently been investigated and appears to be at least as effective as prolonged moderate intensity exercise training for inducing a VEGF response [79]. Specifically, a greater increase in VEGF protein was observed when subjects participated in four 30 s “all out” exercise bouts which were each followed by 5 min of passive rest when compared to subjects who completed 1 h of cycling at 50% of their peak power output. With the high intensity protocol, there was a 90 pg/mL increase in VEGF 4 h after exercise, whereas there was only a 35 pg/mL increase after moderate intensity exercise. Notably, this physiological difference in VEGF content occurred even though the absolute amount of time spent exercising was 97% lower in the high intensity sprint interval group compared to the low intensity group (i.e., 2 min vs. 60 min) [79].
There is also some evidence to suggest that acute resistance exercise induces a VEGF response. In this regard, three sets of ten repetitions at 60–80% of one-repetition maximum (1RM) induce a threefold increase in skeletal muscle VEGF mRNA and 1.5-fold increase in VEGF protein 4 h post-exercise compared to rest [80]. However, this response may be dependent on the type of resistance training (i.e., isotonic, isometric, or isokinetic). Specifically, three repetitions of either 40% of 1RM or maximal isokinetic knee extension did not increase serum VEGF [81]. High volume resistance training of 3 × 30 repetitions in combination with a 30% muscle blood flow restriction also induces a VEGF response [82]. Notably, the VEGF response is attenuated 30 min after exercise when muscle blood flow is normal following exercise training (i.e., a 1.5-fold increase), as compared to the same exercise condition when muscle blood flow is restricted (i.e., a 2.5-fold increase). This observation suggests that intermittent hypoxia or ischemia may be a key factor in regulating exercise-induced angiogenesis.
There appears to be an attenuated VEGF response to an acute bout of exercise following chronic exercise training. For example, the arterial and venous plasma levels of VEGF in response to an acute bout of exercise decreased by 12% and 15%, respectively, after seven exercise training sessions over a 10-day period consisting of 45-min of knee extension exercise at 60–70% maximum work rate, as compared to baseline [29]. However, basal VEGF mRNA and protein expression increased twofold after the 7-day training period. The VEGF response after acute exercise appears to be further attenuated after a longer training period. Particularly, 8 weeks of single-leg knee extension exercise three times per week for 60 min at 50% maximum work rate showed an attenuated VEGF response compared to the untrained muscle. This could be explained by the 18% increase in capillary to fiber ratio of the quadriceps and therefore a further angiogenic response may not be required for the given training stimulus [75].
Receptors VEGFR1, VEGFR2, and NRP-1 are known to increase after a single bout of exercise [30, 33, 34, 83], but respond differently after chronic exercise training, which is characterized by a selective increase in NRP-1 and VEGFR2 [34, 83]. Intermittent hypoxia or ischemia may also influence the regulation of VEGF receptors. For example, resting VEGF protein and VEGFR2 protein content increased threefold under restricted blood flow conditions and remained unchanged under the non-restricted blood flow conditions in response to 5 weeks of knee extension exercise training for 45 min per day, four times per week under 20% restricted blood flow on one leg and non-restricted blood flow to the other leg [34]. This is an interesting finding because VEGFR2 is essential for a majority of the angiogenic actions of VEGF [76, 84]. Moreover, subjects who were “high responders” (i.e., an improvement of +0.71 ± 0.1 L/min of O2 improvement) after 6 weeks of cycling exercise at 75% of their peak VO2 for 45 min four times per week had an eightfold increase in NRP-1and a threefold increase in VEGFR-2, while “non-responders” (i.e., +0.17 ± 0.1 L/min of O2 improvement) had no change [83].
3.2 Matrix Metalloproteinases in Acute Exercise and Chronic Exercise Training
It is now well known that MMPs play an important role in the formation, remodeling, and degradation of the extracellular matrix [85]. They are members of the protease family and 25 enzymes of the MMP family have been identified and are expressed in most tissues [86–88], including skeletal muscle. Specifically, MMP-2 and MMP-9 have been shown to be upregulated in response to exercise [77, 78, 89–93]. They are both collagenases which degrade collagen type IV, which is the most prevalent protein in skeletal muscle basal lamina [89]. It has been suggested that MMP-2 plays a role in the cleavage of the basal lamina and extracellular matrix during angiogenesis, making a path for the migration of endothelial sprouts and the formation of new capillaries [88, 94]. The promoter region of MMP-9 contains a variety of response elements sensitive to a variety of growth factors and cytokines [95] and may also be a regulator of growth factor bioavailability via proteolytic release. There is also evidence indicating that MMP-1, -3, and -14 also increase in response to chronic exercise training [78, 89, 93].
Evidence indicates that the MMP system is activated during an acute bout of exercise as well as following exercise in skeletal muscle [77, 78]. The same exercise protocol used by Hoier et al. [77] discussed in the VEGF section increased MMP-9 mRNA by three and fourfold at 1 and 3 h post-exercise, respectively. However, there were no changes in MMP-2 mRNA. Furthermore, 65 min of cycle ergometer exercise, consisting of 20 min at 50% of VO2max, 40 min at 65% of VO2max, and 5 min at the subject’s maximal tolerable work rate elevated vastus lateralis MMP-9 protein content by ~2.5-fold during exercise and MMP-9 mRNA was elevated ~2.5-fold 2 h after the exercise bout [78]. These data indicate that MMP activity occurs before any measurable changes in MMP-9 transcription, suggesting a post-transcriptional mechanism. As observed with VEGF, the intensity of exercise may influence the MMP response to an acute bout of exercise. Moreover, it has been hypothesized that shear stress is increased during body vibration (as reviewed by Mester et al. [96]) and has been hypothesized that the utilization of vibration training in conjunction with hypoxia and cycling exercise will induce an angiogenic response [92]. Specifically, subjects performed 90 min of cycling; 10 min warm up at 50% VO2max followed by ten intervals of 3 min at a workload of 85% VO2max and 5 min recovery at 60% VO2max. This protocol was carried under four different conditions: normal conditions without vibration; normal conditions with vibration; hypoxic conditions without vibration; and hypoxic conditions with vibration. With the exception of normal conditions with vibration, circulating MMP-2 was increased by approximately 10% immediately after an acute bout of exercise. On the other hand, circulating MMP-9 was shown to increase by approximately twofold in all training conditions 4 h post-exercise, therefore indicating a different activating mechanism for these proteins.
MMP-2 and MMP-9 mRNA and protein levels are known to be regulated by chronic exercise training. For example, 10 days of single-leg exercise training (which consisted of 45 min at the subjects’ maximum tolerable workload four times per week) increased MMP-2 mRNA and protein content by 3.5- and 5.5-fold, respectively [89]. However, partial leg ischemia (i.e., 20%) did not exert an additional effect above that of exercise training alone [89]. Even so, basal MMP-2 levels remained elevated after 5 weeks of training in both exercise training protocols. Rullman et al. [89] also found a ~3–7-fold increase in MMP-9 in the normal and restricted blood flow conditions, respectively, after an acute bout of exercise, but the group changes were not different. Moreover, basal MMP-9 mRNA after 10 days of training increased ~5- and ~13-fold after 5 weeks of training in both groups, with no change in protein. Interestingly, basal MMP-14, which activates MMP-2 [94], mRNA, was increased ~5-fold after 10 days of training in both groups and was maintained after 5 weeks.
Chronic exercise training consisting of aerobic and sprint training in conjunction with either calisthenic or resistance training has been shown to increase circulating MMPs [93]. Calisthenic exercises consist of body weight exercises such as push-ups and pull-ups and squats and lunges. Both the calisthenic and resistance exercise groups exercised 5 days per week for 8 weeks. Exercises were performed until exhaustion in the calisthenic group; whereas the resistance exercise group performed three sets of eight repetitions at 80% of the subject’s 1RM followed by 30 s of rest between sets. Immediately after an acute resistance exercise test, MMP-1 concentration increased ~1.2-fold and MMP-2 remained unchanged in both groups compared to baseline. Interestingly, MMP-3 concentration decreased ~1.5-fold and MMP-9 concentration increased ~4-fold in response to an acute resistance exercise test after exercise training in the calisthenic exercise group, but no changes were observed in the resistance training group. However, the resistance training group increased basal MMP-1 ~1.2-fold and MMP-2 increased twofold after chronic exercise training, whereas the calisthenic exercise-trained group demonstrated a ~1.4-fold decrease in basal MMP-2, a 1.4-fold decrease in basal MMP-3, and a 1.3-fold increase in MMP-9. These data suggest that the mode of exercise elicits a specific training adaptation of MMPs following chronic exercise training.
3.3 Angiopoietins in Acute Exercise and Chronic Exercise Training
The angiopoietin family (Ang-1 and Ang-2) has been suggested to play a role in the angiogenic process. Angiopoietins are vascular endothelial-specific factors that are activated via RTK pathways and modulate vessel development and remodeling [97]. Ang-1 is expressed in many tissues including the myocardium and perivascular cells and plays a role in the maturation of new blood vessels and vascular stabilization [97]. Ang-2 promotes vascular remodeling by either facilitating growth factor actions or vascular regression when no growth signal is present [98] and is thought to lead to vessel degradation in the absence of VEGF. Ang-1 and Ang-2 compete for the tyrosine kinase receptor Tie2, where Ang-1 acts as an agonist for Tie2 [99] and Ang-2 acts as an antagonist [98]. An increase in the Ang-2/Ang-1 ratio is suggested to have a permissive effect on angiogenesis [100].
Evidence indicates that an acute aerobic exercise bout induces a higher Ang-2/Ang-1 ratio in skeletal muscle [77]. The protocol utilized by Hoier et al. [77] found that Ang-2 mRNA was increased ~2-fold 3 h post-exercise while Ang-1 remained unchanged. As a result, the Ang-2/Ang-1 ratio was increased by ~3.5-fold. Moreover, Tie-2 mRNA was increased ~2-fold 3 h post-exercise. However, these data contradict previous findings by Gustafsson et al. [34], who reported no changes in Ang-1 or Ang-2 mRNA following exercise. These different observations could be explained by differences in exercise mode and intensity utilized in each of the two studies (i.e., cycling at 60% VO2max utilized by Hoier et al. [77] vs. one-legged knee extension at the highest tolerable workload utilized by Gustafsson et al. [34]).
Endurance-trained athletes may have a different angiopoietin response to an acute bout of exercise. Moreover, the response to a lower-intensity, longer duration (e.g., marathon running) vs. a high-intensity, short bout (e.g., 1,500 m sprint) may induce different Ang-1 and Ang-2 responses. Specifically, circulating Ang-1 increased ~2.2-fold immediately after a marathon and remained unchanged after a 1,500 m field test [101]. However, immediately after the marathon and 1,500 m field test, Ang-2 increased 2- and 1.3-fold, respectively. Even so, the Ang-2/Ang-1 ratio remained unchanged after both events. Receptor Tie2 expression may also be altered by resistance exercise. The protocol utilized by Gavin et al. [80] found that vastus lateralis Tie2 mRNA increased twofold 4 h after the acute resistance exercise test. However, Ang-1, Ang-2, and the Ang-2/Ang-1 ratio remained unchanged.
Chronic exercise training appears to alter the response of Ang-1 and Ang-2 to an acute exercise bout and elicits a change in resting levels. Forty-five minutes of cycling four times per week for 6 weeks at 75% VO2max increased vastus lateralis Ang-1 mRNA approximately 2.5-fold in subjects identified as “high responders” by Timmons et al. [83], while “low responders” had no change. In contrast, Ang-2 remained unchanged in both groups and therefore indicating a reversal in the Ang-2/Ang-1 ratio after an acute exercise bout prior to exercise training. However, Hoier et al. [77] demonstrated that Ang-2 mRNA after 4 weeks of aerobic training increased ~1.8-fold 3 h after an acute bout of exercise compared to rest. These data suggest that angiopoietins Ang-1 and Ang-2 tend to change in a time-dependent manner in response to an acute bout of exercise after chronic exercise training. It should also be pointed out that single-leg knee extension under non-restricted and restricted blood flow conditions for 45 min four times per week for 5 weeks did not alter vastus lateralis Ang-1, Ang-2 mRNA, or the Ang-2/Ang-1 ratio, while basal vastus lateralis Ang-1 mRNA was reduced 10 days after training and returned to baseline after 5 weeks [34]. Moreover, basal Ang-2 mRNA and protein increased ~1.5-fold after 10 days of training. Although Ang-2 mRNA remained elevated, Ang-2 protein returned to baseline levels after 5 weeks of training. Even so, basal Ang-2/Ang-1 ratio was elevated ~1.5-fold after 10 days of training and remained elevated after 5 weeks of training.
3.4 Other Factors That Stimulate Angiogenesis
Endothelial cells and mural cells produce and activate transforming growth factor-β (TGF-β) which plays a critical role in the maturation of blood vessels by covering the vessel with mural cells (see review by Jain [40]). Mice lacking the TGF-β gene die from defects of the vasculature in utero, suggesting an important role in angiogenesis prior to birth [102]. One hour of one-leg kicking exercise at 67% maximum workload increased TGF-β in the connective tissue of the vastus lateralis approximately twofold [103]. A maximal graded exercise test has also shown to increase serum TGF-β 2.7-fold immediately after exercise and 1.9-fold 2 h after exercise compared to baseline [104]. To our knowledge, TGF-β in response to chronic exercise training has not been investigated. Although basic fibroblast growth factor (bFGF or FGF-2), a 18–23 kDa cationic mitogen, has been shown to upregulate VEGF [105] as well as NO production [106], some studies have failed to demonstrate its upregulation in response to acute or chronic exercise training in humans [32, 79, 107].
4 Factors That Inhibit Angiogenesis in Response to Exercise
In addition to several pro-angiogenic molecules, angiogenesis is also regulated by several key anti-angiogenic molecules; however, their response to acute exercise and chronic exercise has been investigated in less depth than the pro-angiogenic factors. Some of these anti-angiogenic factors are described below.
4.1 Endostatin in Acute and Chronic Exercise Training
Endostatin, a 20-kDa fragment released from collagen XVIII C-terminal fragment, blocks angiogenesis by preventing the release of VEGF and VEGF receptor signaling [108, 109] as well as MMP-2 activity [110]. It can thus be seen to restrict endothelial cell proliferation and migration. Endostatin also plays a role in relaxation of the vessel walls through endothelial nitric oxide synthase (eNOS) phosphorylation and prostacyclin release [111, 112]. A 65 min bout of cycling at 65% VO2max utilized by Rullman et al. [78] increased plasma arterial endostatin ~1.7-fold 17 min into the exercise protocol, which was higher than plasma venous endostatin, which increased ~1.6-fold but then returned to resting values at 57 min into exercise and remained at basal levels 120 min post-exercise. Interestingly, plasma venous endostatin continued to increase at 57 min by ~2-fold and was significantly higher compared to plasma arterial endostatin, suggesting an uptake from the circulation into the muscle. Cycling under hypoxic conditions in conjunction with vibration training, utilized by Suhr et al. [92], increased circulating endostatin by 1.1–1.25-fold immediately after exercise, with the greatest increase found under normal conditions and the lowest increase under hypoxic conditions with vibration. Circulating endostatin remained elevated 30 and 60 min post-exercise only in the normal conditions protocol. A maximal exercise test may induce a greater endostatin response. Specifically, plasma endostatin concentrations increased at 30, 120, and 360 min after exercise by 43%, 73%, and 33%, respectively, compared to rest [107]. These data indicate that endostatin increases to a greater extent following heavy intensity exercise, as compared to moderate intensity exercise. On the other hand, chronic exercise training may reduce resting plasma endostatin, which would thereby promote angiogenesis. Participants who either ran for 60 min or cycled for 90 min three times per week at moderate intensity for 6 months reduced basal circulating endostatin levels by 14% in both groups [113]. Furthermore, participants in both exercise groups significantly reduced basal endostatin levels when compared to a sedentary control group. Nonetheless, further research is needed to more clearly demonstrate the influence of chronic exercise training on the regulation of endostatin mRNA and protein content in humans.
4.2 Thrombospondin-1 in Acute and Chronic Exercise Training
Another endogenous inhibitor of angiogenesis is thrombospondin-1 (TSP-1), which is a large 450 kDa matrix glycoprotein produced by a number of cells, including platelet α-granules, smooth muscle cells, endothelial cells, fibroblasts, neutrophils, and macrophages and located in the extracellular compartment of several organs and tissues, including skeletal muscle, bone, skin, lung, and connective tissue [114, 115]. Its primary anti-angiogenic role is to inhibit cell migration and to induce apoptosis in endothelial cells [115–117]. Few data exist on the response of TSP-1 to acute exercise in humans. A 60 min bout of treadmill running at 20 m/min at 10o incline increased rat gastrocnemius TSP-1 mRNA 3.5-fold immediately after exercise and was further increased 6.2-fold 1 h after exercise, compared to resting levels [118]. More recently, 60 min of cycling at 60% VO2max was shown to increase human vastus lateralis TSP-1 mRNA ~2.75-fold at 1 h after exercise and remained elevated for up to 3 h after exercise [77]. Exercise-trained rats, under both normoxic and hypoxic conditions for 60 min a day, 5 days per week for 8 weeks at 18 m/min and 10o incline, were shown to upregulate gastrocnemius TSP-1 mRNA 3.4 and 3.3-fold, respectively [118]. However, this response was not altered by hypoxia. Humans who completed 4 weeks of cycling for 60 min at 60% VO2max also showed a ~2.5-fold increase in TSP-1 mRNA in the vastus lateralis when sampled 1 h after an acute bout of exercise; however, this acute effect was not different from the pre-training response, suggesting that TSP-1 may not be affected by chronic exercise training [77].
4.3 Tissue Inhibitors of Matrix Metalloproteinases in Acute Exercise and Chronic Exercise Training
TIMPs are polypeptides containing 184–194 amino acids and are shown to regulate MMP activity by suppressing extracellular matrix turnover and activation of proteins on the cell surface [119]. TIMPs also have other anti-angiogenic properties without inhibiting MMPs through TIMP-2 integrin-mediated binding to endothelial cells (see review by Stetler-Stevenson et al. [120]). Human studies indicate that TIMPs are not upregulated in the circulation [121, 122] or skeletal muscle [77, 78, 89] in response to acute exercise. This is in contrast to the evidence from an animal study indicating that rat soleus TIMP-1 and -2 mRNA increased twofold 6 h after a bout of eccentric exercise consisting of 17 m/min for 130 min at 13.5o decline, which was sustained for 2 days after running and soleus TIMP-1 protein increased 1.5-fold [123]. The lack of research examining the influence of exercise on the regulation of TIMPs suggests that this area may be investigated more fully in the future. Chronic exercise training appears to increase TIMP concentration in human skeletal muscle. Rullman et al. [89] found that 10 days of single-leg knee extension training increased vastus lateralis TIMP-1 mRNA ~4- and 6-fold 24 h after an acute bout of exercise in the non-restricted and restricted blood flow conditions, respectively, compared to pre-training levels. Moreover, Hoier et al. [77] found that 4 weeks of cycle ergometer training increased vastus lateralis TIMP-1 mRNA ~2-fold 1 and 3 h after an acute bout of exercise. These data indicate that chronic exercise training increases the expression of TIMP-1 mRNA after an acute bout of exercise compared to pre-training states, suggesting that the inhibition of extracellular remodeling is greater after training when there is sufficient capillary growth, which was demonstrated by Hoier et al. [77].
4.4 Other Factors That Inhibit Angiogenesis
Angiostatin was the first discovered anti-angiogenic factor found to block tumor neovascularization and growth of metastases [124]. Angiostatin is a 38-kDa proteolytic fragment derived from degradation of MMP. It is plausible that angiostatin is upregulated by exercise because MMP is known to increase in response to acute exercise and chronic exercise training, but this possibility remains to be investigated. Another factor, namely vasohibin-1 (VASH-1), is secreted by endothelial cells and contributes to vessel stabilization and maturation [125] and is more highly expressed in less vascularized muscle compared to more vascularized muscle [126]. Rats that completed a maximal running treadmill test that lasted 60–90 min increased VASH-1 by ~1.5-fold immediately after exercise in the hindlimb but returned to baseline 4 h after exercise [126]. In contrast, myocardial VASH-1 did not increase immediately after exercise but increased 4 h after the acute exercise bout. The influence that chronic exercise training has on VASH-1 remains to be fully elucidated because this study indicated that 5 days of training for 60 min at 25–30 m/min at 12o incline did not increase VASH-1 levels in rat hindlimb or heart, as compared to baseline.
5 Endothelial Progenitor Cells in Acute Exercise and Chronic Exercise Training
Bone marrow contains EPCs which have the ability to multiply and differentiate into endothelial cells (see review by Ribatti [127]) and were first discovered in adult peripheral blood [128]. EPCs have also been shown to reside in interstitial tissue surrounding distressed tissues and organs in need of vascular regeneration where the EPCs produce a variety of pro-angiogenic cytokines and growth factors, including VEGF, hepatic growth factor (HGF), Ang-1, stroma-derived growth factor (SDF)-1α, insulin-like growth factor (IGF)-1, and eNOS [129]. They have also been suggested to play an important role in the regulation of angiogenesis through the differentiation and maturation to endothelial cells and by coordination of paracrine signaling (see review by Carmeliet and Jain [37]). Moreover, endothelial progenitors may play a role in exercise-induced angiogenesis; however, the mechanism of their migration from bone marrow is not fully understood.
Several markers of EPCs have been identified to increase after an acute bout of exercise including CD34+VEGFR-2+ cells, CD34+CD133+ cells, CD133+VE-cadherin+ cells, and monocyte/macrophage-derived angiogenic cells (CACs) in healthy subjects [101, 130–132] and appear to be associated with intensity of exercise. Specifically, immediately after a marathon run circulating CD34+VEGFR-2+, CD133+VE-cadherin+, and CAC cell count was found to increase ~1.8-, 3.5-, and 4-fold, respectively, compared to rest, and returned to baseline 20 h after the race [101]. After completion of a 1,500 m field test, CD34+VEGFR-2+, CD133+VE-cadherin+, and CAC increased ~2.25-, 5-, and 4-fold, respectively, compared to rest. Moreover, group differences were observed, where CD34+VEGFR-2+ was ~1.8-fold higher in the 1,500 m group compared to the marathon group and CAC cell count was ~2.3-fold higher in the marathon group compared to the 1,500 m group [101]. After a maximal graded exercise test on a cycle ergometer, CD34+VEGFR-2+ increased by ~1.7-fold, compared to rest [131]. Thirty minutes of running at anaerobic threshold and 80% of anaerobic threshold increased circulating endothelial progenitors by 235% and 263%, respectively [133]. However, no changes in circulating EPCs were observed after 10 min of running at 80% of anaerobic threshold. Therefore, these data suggest that EPCs may be mobilized differently from the bone marrow in response to different intensities of exercise. Moreover, duration of exercise may play an important role in the release of endothelial progenitors from the bone marrow. To our knowledge, there is no data that exists to determine if chronic exercise training increases circulating EPCs in healthy subjects. However, several studies have demonstrated that endothelial progenitors increase after chronic exercise training in patients with cardiovascular disease [132, 134–136].
6 miRNAs in Acute Exercise and Chronic Exercise Training
miRNAs are highly conserved 20–30 nucleotide RNAs that regulate the translation of proteins and enhance messenger RNA (mRNA) degradation (see review by Bartel [137]). miRNAs are formed by two major enzymes; Dicer and Drosha. Drosha regulates nuclear processing of the primary miRNAs into precursors, or pre miRNAs [138], and Dicer cleaves pre-miRNA into the mature miRNA [139]. A number of miRNAs have been identified within cardiac and skeletal muscle which may be involved in cardiogenesis during embryonic development [140] and skeletal muscle myogenesis [141] and remodeling [142]. Several miRNAs including miR-20a [143], miR-210 [144], miR-221/miR-222 [145], and miR-328 [146] have also been identified which have anti-angiogenic effects. Specifically, miR-20a expression is increased by VEGF but has an inhibitory effect on endothelial cell migration and overall capillary formation [147]. Increased miR-328 expression is found to reduce cell adhesion, aggregation, and migration and also regulates the formation of the capillary structure [146]. miR-210 upregulation under normoxic conditions increases endothelial cell tubulogenesis and migration, but under hypoxic conditions, miR-210 decreases capillary formation [144]. It has also been shown that supplementation of miR-210 to the myocardium to the pre-infarct zone induces angiogenesis in a mouse model [148]. miR-221/miR-222 mediate the angiogenic activity of stem-cell factor (SCF) [145]. These miRNAs have only more recently been investigated in response to an acute bout of exercise and chronic exercise training. The response of miRNAS to an acute bout of exercise has been investigated in competitive rowers [149]. Specifically, it has been shown that circulating miR-221 and miR-222 are increased ~3.6- and 2.5-fold immediately after a maximal, incremental cycling exercise test, respectively. Further investigations may be warranted to determine if acute exercise has similar effects on miRNA regulation in non-athlete populations.
Baggish et al. [149] investigated the effects of chronic exercise training in rowing athletes. After 90 days of training for an average 13.1 h per week, miR-221 and miR-222 increased ~5.8- and 2.4-fold compared to baseline resting levels, respectively. After the chronic exercise training period, miR-20a was 3.1-fold higher after an acute bout of exercise compared to baseline resting levels. An additional increase in circulating miR-222 was found (fourfold increase) after an acute bout of exercise after training but not in miR-221. These data indicate a different pattern in regulation between several miRNAs before and after chronic exercise training and further investigations are needed among different populations.
7 Exercise-Induced Angiogenesis in Chronic Disease
From the foregoing discussion, it is evident that chronic exercise training in healthy individuals may promote angiogenesis by affecting the expression of several angiogenic factors (Table 11.1). The role of chronic exercise training in the primary and secondary prevention of different diseases has also been investigated extensively. Interestingly, regular exercise may reduce the risk of developing over 25 chronic conditions [150] including coronary heart disease, stroke, hypertension, breast and colon cancer, type 2 diabetes, and osteoporosis. It is likely that research examining the biological mechanisms that regulate exercise-induced angiogenesis may guide the identification of novel therapeutic targets for the treatment of chronic diseases. For example, patients with cardiovascular disease who exercise train to a level sufficient to stimulate angiogenesis and the formation of collateral vessels in ischemic areas of the heart and skeletal muscle are known to decrease the severity of ischemic events [151–153]. The potential of chronic exercise training to upregulate and normalize circulating pro-angiogenic growth factors and EPCs in patients with cardiovascular disease has been documented previously [132, 135, 154–162]; it also has the potential to improve collateral flow in the heart [163] and increase capillary density in skeletal muscle [160]. Various studies examining the role of chronic exercise training and its ability to upregulate several pro- and anti-angiogenic growth factors as well as EPCs are listed in Table 11.2. Similar health benefits may also be attained by peripheral arterial disease patients, which is a population characterized with ischemia of the limbs that often leads to tissue necrosis requiring amputation [164]. Notably, chronic exercise training consisting of 50 min of walking exercise at an intensity below that which induces claudication symptoms twice a week for 6 months significantly elevated EPCs and VEGF in patients diagnosed with peripheral artery disease [135]. Moreover, exercise-induced angiogenesis may have an important therapeutic role in heart failure. It may be noted that patients with heart failure often experience dyspnea and exercise intolerance, which is not only due to insufficient cardiac output and impaired lung function but also due to peripheral pathophysiological maladaptations such as endothelial dysfunction [165], abnormal skeletal morphology and metabolism [166] and ventilatory control [167]. Chronic exercise training consisting of 55 min of upright cycling three times a week for 8 weeks at an intensity that elevated blood lactate to 2–2.5 mmol/L markedly increased circulating EPCs and pro-angiogenic factors, including VEGF amongst a cohort of heart failure patients. This beneficial effect of exercise training washed out 8 weeks after the heart failure patients stopped exercise training [136].
One goal of cancer therapy is to prevent the blood supply of a tumor using anti-angiogenic drugs [168]. Interestingly, even though chronic exercise training induces a positive angiogenic response in the heart and skeletal muscle, it may have an anti-angiogenic effect on tumors. To our knowledge there are currently no human models for investigating the effects of chronic exercise training and tumor blood vessel supply. Furthermore, the potential mechanisms to explain how exercise training exerts an anti-angiogenic effect on tumors still remain to be elucidated. Nonetheless, the effects of chronic exercise training have been examined using a Dalton’s lymphoma mouse model [169]. Data from that study [169] indicate that blood supply to the peritoneal cavity was significantly reduced in mice that exercised 30 min per day three times per week at approximately 80% VO2max [169]. Moreover, mice that exercised 60 min a day had a further decrease in blood supply to the peritoneal cavity compared to the 30 min group. One of the potential mechanisms for the reduced blood supply was that VEGF was significantly reduced in the exercise-trained mice vs. the sedentary controls. However, this may be specific to the type of cancer, as it was found that 44 days of voluntary wheel running did not reduce blood supply or tumor growth in a mouse model of human breast cancer [170]. Further studies should continue to examine the influence that chronic exercise training exerts on tumor growth and angiogenesis.
8 Conclusions
Angiogenesis is a process regulated by pro- and anti-angiogenic factors and plays an important role for normal growth and physiological function. Several disease states are also characterized by deficient or dysregulated angiogenesis. In general, the literature indicates acute and chronic exercise training stimulates angiogenesis by upregulating pro- and anti-angiogenic factors that ultimately increase capillary density in both heart and skeletal muscle. Chronic exercise training may also have an important therapeutic role for regulating angiogenesis healthy as well as clinical populations.
References
Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9:653–660
Krogh A (1919) The number and distribution of capillaries in muscles with calculations of the oxygen pressure head necessary for supplying the tissue. J Physiol 52:409–415
Krogh A (1919) The rate of diffusion of gases through animal tissues, with some remarks on the coefficient of invasion. J Physiol 52:391–408
Krogh A (1919) The supply of oxygen to the tissues and the regulation of the capillary circulation. J Physiol 52:457–474
Krogh A (1922) The anatomy and physiology of capillaries. Yale University Press, New Haven, CT
Hertig A (1935) Angiogenesis in the early human chorion and in the primary placenta of the macaque monkey. Contrib Embryol 25:37–81
Sandison JC (1932) Contraction of blood vessels and observations on the circulation in the transparent chamber in the rabbit’s ear. Anat Rec 24:105–127
Findlay JK (1986) Angiogenesis in reproductive tissues. J Endocrinol 111:357–366
Hudlicka O, Wright AJ, Ziada AM (1986) Angiogenesis in the heart and skeletal muscle. Can J Cardiol 2:120–123
Kawamata T, Speliotes EK, Finklestein SP (1997) The role of polypeptide growth factors in recovery from stroke. Adv Neurol 73:377–382
Kumar S, West D, Shahabuddin S et al (1983) Angiogenesis factor from human myocardial infarcts. Lancet 13:364–368
Burgos H, Herd A, Bennett JP (1989) Placental angiogenic and growth factors in the treatment of chronic varicose ulcers: preliminary communication. J R Soc Med 82:598–599
Baird A (1994) Fibroblast growth factors: activities and significance of non-neurotrophin neurotrophic growth factors. Curr Opin Neurobiol 4:78–86
Folkman J, Merler E, Abernathy C et al (1971) Isolation of a tumor factor responsible for angiogenesis. J Exp Med 1:275–288
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1:27–31
Christou H, Yoshida A, Arthur V et al (1998) Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 18:768–776
Patz A (1978) Current concepts in ophthalmology. Retinal vascular diseases. N Engl J Med 298:1451–1454
Laughlin MH, Bowles DK, Duncker DJ (2012) The coronary circulation in exercise training. Am J Physiol Heart Circ Physiol 302:H10–H23
Savard R, Smith LJ, Palmer JE et al (1988) Site specific effects of acute exercise on muscle and adipose tissue metabolism in sedentary female rats. Physiol Behav 43:65–71
Dohm GL, Huston RL, Askew EW et al (1973) Effects of exercise, training, and diet on muscle citric acid cycle enzyme activity. Can J Biochem 51:849–854
Egginton S (2009) Invited review: activity-induced angiogenesis. Pflugers Arch 457:963–977
Hudlicka O, Brown M, Egginton S (1992) Angiogenesis in skeletal and cardiac muscle. Physiol Rev 72:369–417
Sundberg CJ, Kaijser L (1992) Effects of graded restriction of perfusion on circulation and metabolism in the working leg; quantification of a human ischaemia-model. Acta Physiol Scand 146:1–9
Milkiewicz M, Ispanovic E, Doyle JL et al (2006) Regulators of angiogenesis and strategies for their therapeutic manipulation. Int J Biochem Cell Biol 38:333–357
Dvorak HF, Orenstein NS, Carvalho AC et al (1979) Induction of a fibrin-gel investment: an early event in line 10 hepatocarcinoma growth mediated by tumor-secreted products. J Immunol 122:166–174
Dvorak HF, Brown LF, Detmar M et al (1995) Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 146:1029–1039
Gustafsson T, Puntschart A, Kaijser L et al (1999) Exercise-induced expression of angiogenesis-related transcription and growth factors in human skeletal muscle. Am J Physiol 276:H679–H685
Richardson RS, Wagner H, Mudaliar SR et al (1999) Human VEGF gene expression in skeletal muscle: effect of acute normoxic and hypoxic exercise. Am J Physiol 277:H2247–H2252
Gustafsson T, Knutsson A, Puntschart A et al (2002) Increased expression of vascular endothelial growth factor in human skeletal muscle in response to short-term one-legged exercise training. Pflugers Arch 444:752–759
Gavin TP, Robinson CB, Yeager RC et al (2004) Angiogenic growth factor response to acute systemic exercise in human skeletal muscle. J Appl Physiol 96:19–24
Jensen L, Pilegaard H, Neufer PD et al (2004) Effect of acute exercise and exercise training on VEGF splice variants in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 287:R397–R402
Jensen L, Bangsbo J, Hellsten Y (2004) Effect of high intensity training on capillarization and presence of angiogenic factors in human skeletal muscle. J Physiol 557:571–582
Gustafsson T, Ameln H, Fischer H et al (2005) VEGF-A splice variants and related receptor expression in human skeletal muscle following submaximal exercise. J Appl Physiol 98:2137–2146
Gustafsson T, Rundqvist H, Norrbom J et al (2007) The influence of physical training on the angiopoietin and VEGF-A systems in human skeletal muscle. J Appl Physiol 103:1012–1020
Egginton S, Zhou AL, Brown MD et al (2001) Unorthodox angiogenesis in skeletal muscle. Cardiovasc Res 49:634–646
Gustafsson T (2011) Vascular remodelling in human skeletal muscle. Biochem Soc Trans 39:1628–1632
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307
Folkman J, Klagsbrun M (1987) Angiogenic factors. Science 235:442–447
Folkman J (1985) Toward an understanding of angiogenesis: search and discovery. Perspect Biol Med 29:10–36
Jain RK (2003) Molecular regulation of vessel maturation. Nat Med 9:685–693
Prior BM, Yang HT, Terjung RL (2004) What makes vessels grow with exercise training? J Appl Physiol 97:1119–1128
Qutub AA, Mac Gabhann F, Karagiannis ED et al (2009) Multiscale models of angiogenesis. IEEE Eng Med Biol Mag 28:14–31
Yancopoulos GD, Davis S, Gale NW et al (2000) Vascular-specific growth factors and blood vessel formation. Nature 14:242–248
Egginton S (2011) Physiological factors influencing capillary growth. Acta Physiol (Oxf) 202:225–239
Djonov V, Baum O, Burri PH (2003) Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res 314:107–117
Gianni-Barrera R, Trani M, Reginato S et al (2011) To sprout or to split? VEGF, Notch and vascular morphogenesis. Biochem Soc Trans 39:1644–1648
Styp-Rekowska B, Hlushchuk R, Pries AR et al (2011) Intussusceptive angiogenesis: pillars against the blood flow. Acta Physiol (Oxf) 202:213–223
Makanya AN, Hlushchuk R, Djonov VG (2009) Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 12:113–123
Burri PH, Djonov V (2002) Intussusceptive angiogenesis—the alternative to capillary sprouting. Mol Aspects Med 23:S1–S27
Hobson B, Denekamp J (1984) Endothelial proliferation in tumours and normal tissues: continuous labelling studies. Br J Cancer 49:405–413
Haas TL, Milkiewicz M, Davis SJ et al (2000) Matrix metalloproteinase activity is required for activity-induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol 279:H1540–H1547
Hansen-Smith FM, Hudlicka O, Egginton S (1996) In vivo angiogenesis in adult rat skeletal muscle: early changes in capillary network architecture and ultrastructure. Cell Tissue Res 286:123–136
Paku S, Paweletz N (1991) First steps of tumor-related angiogenesis. Lab Invest 65:334–346
Ausprunk DH, Folkman J (1977) Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res 14:53–65
Gerhardt H, Golding M, Fruttiger M et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161:1163–1177
Benjamin LE, Golijanin D, Itin A et al (1999) Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 103:159–165
Dor Y, Djonov V, Abramovitch R et al (2002) Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J 21:1939–1947
Caduff JH, Fischer LC, Burri PH (1986) Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat Rec 216:154–164
Patan S, Haenni B, Burri PH (1996) Implementation of intussusceptive microvascular growth in the chicken chorioallantoic membrane (CAM): 1. pillar formation by folding of the capillary wall. Microvasc Res 51:80–98
Kauffman SL, Burri PH, Weibel ER (1974) The postnatal growth of the rat lung. II. Autoradiography. Anat Rec 180:63–76
Lloyd PG, Prior BM, Li H et al (2005) VEGF receptor antagonism blocks arteriogenesis, but only partially inhibits angiogenesis, in skeletal muscle of exercise-trained rats. Am J Physiol Heart Circ Physiol 288:H759–H768
Williams JL, Cartland D, Rudge JS et al (2006) VEGF trap abolishes shear stress- and overload-dependent angiogenesis in skeletal muscle. Microcirculation 13:499–509
Olfert IM, Howlett RA, Wagner PD et al (2010) Myocyte vascular endothelial growth factor is required for exercise-induced skeletal muscle angiogenesis. Am J Physiol Regul Integr Comp Physiol 299:R1059–R1067
Forsythe JA, Jiang BH, Iyer NV et al (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16:4604–4613
Shweiki D, Itin A, Soffer D et al (1992) Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 29:843–845
Richardson RS, Duteil S, Wary C et al (2006) Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol 571:415–424
Richardson RS, Noyszewski EA, Kendrick KF et al (1995) Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J Clin Invest 96:1916–1926
Mason SD, Howlett RA, Kim MJ et al (2004) Loss of skeletal muscle HIF-1alpha results in altered exercise endurance. PLoS Biol 2:e288
Ameln H, Gustafsson T, Sundberg CJ et al (2005) Physiological activation of hypoxia inducible factor-1 in human skeletal muscle. FASEB J 19:1009–1011
Fraisl P, Mazzone M, Schmidt T et al (2009) Regulation of angiogenesis by oxygen and metabolism. Dev Cell 16:167–179
Hardie DG (2003) Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144:5179–5183
Ouchi N, Shibata R, Walsh K (2005) AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res 29:838–846
Jensen L, Schjerling P, Hellsten Y (2004) Regulation of VEGF and bFGF mRNA expression and other proliferative compounds in skeletal muscle cells. Angiogenesis 7:255–267
Arany Z, Foo SY, Ma Y et al (2008) HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 451:1008–1012
Richardson RS, Wagner H, Mudaliar SR, Saucedo E, Henry R, Wagner PD (2000) Exercise adaptation attenuates VEGF gene expression in human skeletal muscle. Am J Physiol Heart Circ Physiol 279:H772–H778
Neufeld G, Cohen T, Gengrinovitch S et al (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13:9–22
Hoier B, Nordsborg N, Andersen S et al (2012) Pro- and anti-angiogenic factors in human skeletal muscle in response to acute exercise and training. J Physiol 590:595–606
Rullman E, Rundqvist H, Wagsater D et al (2007) A single bout of exercise activates matrix metalloproteinase in human skeletal muscle. J Appl Physiol 102:2346–2351
Wahl P, Zinner C, Achtzehn S et al (2011) Effects of acid–base balance and high or low intensity exercise on VEGF and bFGF. Eur J Appl Physiol 111:1405–1413
Gavin TP, Drew JL, Kubik CJ et al (2007) Acute resistance exercise increases skeletal muscle angiogenic growth factor expression. Acta Physiol (Oxf) 191:139–146
Rojas Vega S, Knicker A, Hollmann W et al (2010) Effect of resistance exercise on serum levels of growth factors in humans. Horm Metab Res 42:982–986
Takano H, Morita T, Iida H et al (2005) Hemodynamic and hormonal responses to a short-term low-intensity resistance exercise with the reduction of muscle blood flow. Eur J Appl Physiol 95:65–73
Timmons JA, Jansson E, Fischer H et al (2005) Modulation of extracellular matrix genes reflects the magnitude of physiological adaptation to aerobic exercise training in humans. BMC Biol 3:19
Olsson AK, Dimberg A, Kreuger J et al (2006) VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol 7:359–371
Chen X, Li Y (2009) Role of matrix metalloproteinases in skeletal muscle: migration, differentiation, regeneration and fibrosis. Cell Adh Migr 3:337–341
Baum O, Ganster M, Baumgartner I et al (2007) Basement membrane remodeling in skeletal muscles of patients with limb ischemia involves regulation of matrix metalloproteinases and tissue inhibitor of matrix metalloproteinases. J Vasc Res 44:202–213
Carmeli E, Moas M, Lennon S et al (2005) High intensity exercise increases expression of matrix metalloproteinases in fast skeletal muscle fibres. Exp Physiol 90:613–619
Deryugina EI, Quigley JP (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25:9–34
Rullman E, Norrbom J, Stromberg A et al (2009) Endurance exercise activates matrix metalloproteinases in human skeletal muscle. J Appl Physiol 106:804–812
Deus AP, Bassi D, Simoes RP et al (2012) MMP(−2) expression in skeletal muscle after strength training. Int J Sports Med 33:137–141
Heinemeier KM, Olesen JL, Haddad F et al (2007) Expression of collagen and related growth factors in rat tendon and skeletal muscle in response to specific contraction types. J Physiol 582:1303–1316
Suhr F, Brixius K, de Marees M et al (2007) Effects of short-term vibration and hypoxia during high-intensity cycling exercise on circulating levels of angiogenic regulators in humans. J Appl Physiol 103:474–483
Urso ML, Pierce JR, Alemany JA et al (2009) Effects of exercise training on the matrix metalloprotease response to acute exercise. Eur J Appl Physiol 106:655–663
Haas TL, Davis SJ, Madri JA (1998) Three-dimensional type I collagen lattices induce coordinate expression of matrix metalloproteinases MT1-MMP and MMP-2 in microvascular endothelial cells. J Biol Chem 273:3604–3610
Yan C, Boyd DD (2007) Regulation of matrix metalloproteinase gene expression. J Cell Physiol 211:19–26
Mester J, Kleinoder H, Yue Z (2006) Vibration training: benefits and risks. J Biomech 39:1056–1065
Davis S, Aldrich TH, Jones PF et al (1996) Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 87:1161–1169
Maisonpierre PC, Suri C, Jones PF et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55–60
Holash J, Maisonpierre PC, Compton D et al (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284:1994–1998
Asahara T, Chen D, Takahashi T et al (1998) Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res 83:233–240
Bonsignore MR, Morici G, Riccioni R et al (2010) Hemopoietic and angiogenetic progenitors in healthy athletes: different responses to endurance and maximal exercise. J Appl Physiol 109:60–67
Dickson MC, Martin JS, Cousins FM et al (1995) Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 121:1845–1854
Heinemeier KM, Bjerrum SS, Schjerling P et al (2011) Expression of extracellular matrix components and related growth factors in human tendon and muscle after acute exercise. Scand J Med Sci Sports 23:1–12
Czarkowska-Paczek B, Bartlomiejczyk I, Przybylski J (2006) The serum levels of growth factors: PDGF, TGF-beta and VEGF are increased after strenuous physical exercise. J Physiol Pharmacol 57:189–197
Stavri GT, Zachary IC, Baskerville PA et al (1995) Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells. Synergistic interaction with hypoxia. Circulation 92:11–14
Babaei S, Teichert-Kuliszewska K, Monge JC et al (1998) Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circ Res 82:1007–1015
Gu JW, Gadonski G, Wang J et al (2004) Exercise increases endostatin in circulation of healthy volunteers. BMC Physiol 4:2
Hajitou A, Grignet C, Devy L et al (2002) The antitumoral effect of endostatin and angiostatin is associated with a down-regulation of vascular endothelial growth factor expression in tumor cells. FASEB J 16:1802–1804
Kim YM, Hwang S, Kim YM et al (2002) Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem 277:27872–27879
Lee SJ, Jang JW, Kim YM et al (2002) Endostatin binds to the catalytic domain of matrix metalloproteinase-2. FEBS Lett 519:147–152
Li C, Harris MB, Venema VJ et al (2005) Endostatin induces acute endothelial nitric oxide and prostacyclin release. Biochem Biophys Res Commun 329:873–878
Wenzel D, Schmidt A, Reimann K et al (2006) Endostatin, the proteolytic fragment of collagen XVIII, induces vasorelaxation. Circ Res 98:1203–1211
Brixius K, Schoenberger S, Ladage D et al (2008) Long-term endurance exercise decreases antiangiogenic endostatin signalling in overweight men aged 50–60 years. Br J Sports Med 42:126–129
Bornstein P (1992) Thrombospondins: structure and regulation of expression. FASEB J 6:3290–3299
Iruela-Arispe ML, Luque A, Lee N (2004) Thrombospondin modules and angiogenesis. Int J Biochem Cell Biol 36:1070–1078
Bonnefoy A, Moura R, Hoylaerts MF (2008) The evolving role of thrombospondin-1 in hemostasis and vascular biology. Cell Mol Life Sci 65(5):713–727
Kazerounian S, Yee KO, Lawler J (2008) Thrombospondins in cancer. Cell Mol Life Sci 65:700–712
Olfert IM, Breen EC, Gavin TP, Wagner PD (2006) Temporal thrombospondin-1 mRNA response in skeletal muscle exposed to acute and chronic exercise. Growth Factors 24:253–259
Murphy G, Houbrechts A, Cockett MI et al (1991) The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry 30:8097–8102
Stetler-Stevenson WG, Seo DW (2005) TIMP-2: an endogenous inhibitor of angiogenesis. Trends Mol Med 11:97–103
Frederiksen CB, Lomholt AF, Lottenburger T et al (2008) Assessment of the biological variation of plasma tissue inhibitor of metalloproteinases-1. Int J Biol Markers 23:42–47
Koskinen SO, Heinemeier KM, Olesen JL et al (2004) Physical exercise can influence local levels of matrix metalloproteinases and their inhibitors in tendon-related connective tissue. J Appl Physiol 96:861–864
Koskinen SO, Wang W, Ahtikoski AM et al (2001) Acute exercise induced changes in rat skeletal muscle mRNAs and proteins regulating type IV collagen content. Am J Physiol Regul Integr Comp Physiol 280:R1292–R1300
O’Reilly MS, Holmgren L, Shing Y et al (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79:315–328
Sonoda H, Ohta H, Watanabe K et al (2006) Multiple processing forms and their biological activities of a novel angiogenesis inhibitor vasohibin. Biochem Biophys Res Commun 342:640–646
Kishlyansky M, Vojnovic J, Roudier E et al (2010) Striated muscle angio-adaptation requires changes in Vasohibin-1 expression pattern. Biochem Biophys Res Commun 399:359–364
Ribatti D (2007) The discovery of endothelial progenitor cells. An historical review. Leuk Res 31:439–444
Asahara T, Murohara T, Sullivan A et al (1997) Isolation of putative progenitor endothelial cells for angiogenesis. Science 275:964–967
Ii M, Nishimura H, Iwakura A et al (2005) Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via “imported” nitric oxide synthase activity. Circulation 111:1114–1120
Rehman J, Li J, Parvathaneni L, Karlsson G et al (2004) Exercise acutely increases circulating endothelial progenitor cells and monocyte-/macrophage-derived angiogenic cells. J Am Coll Cardiol 43:2314–2318
Van Craenenbroeck EM, Vrints CJ, Haine SE et al (2008) A maximal exercise bout increases the number of circulating CD34+/KDR+endothelial progenitor cells in healthy subjects. Relation with lipid profile. J Appl Physiol 104:1006–1013
Laufs U, Werner N, Link A et al (2004) Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation 109:220–226
Laufs U, Urhausen A, Werner N et al (2005) Running exercise of different duration and intensity: effect on endothelial progenitor cells in healthy subjects. Eur J Cardiovasc Prev Rehabil 12:407–414
Steiner S, Niessner A, Ziegler S et al (2005) Endurance training increases the number of endothelial progenitor cells in patients with cardiovascular risk and coronary artery disease. Atherosclerosis 181:305–310
Schlager O, Giurgea A, Schuhfried O et al (2011) Exercise training increases endothelial progenitor cells and decreases asymmetric dimethylarginine in peripheral arterial disease: a randomized controlled trial. Atherosclerosis 217:240–248
Sarto P, Balducci E, Balconi G et al (2007) Effects of exercise training on endothelial progenitor cells in patients with chronic heart failure. J Card Fail 13:701–708
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Lee Y, Ahn C, Han J et al (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425:415–419
Lund E, Guttinger S, Calado A et al (2004) Nuclear export of microRNA precursors. Science 303:95–98
Zhao Y, Samal E, Srivastava D (2005) Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436:214–220
Chen JF, Mandel EM, Thomson JM et al (2006) The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38:228–233
Keller P, Vollaard NB, Gustafsson T et al (2011) A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J Appl Physiol 110:46–59
Dews M, Homayouni A, Yu D et al (2006) Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38:1060–1065
Fasanaro P, D’Alessandra Y, Di Stefano V et al (2008) MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 283:15878–15883
Poliseno L, Tuccoli A, Mariani L et al (2006) MicroRNAs modulate the angiogenic properties of HUVECs. Blood 108:3068–3071
Wang CH, Lee DY, Deng Z et al (2008) MicroRNA miR-328 regulates zonation morphogenesis by targeting CD44 expression. PLoS One 3:e2420
Pin AL, Houle F, Guillonneau M et al (2012) miR-20a represses endothelial cell migration by targeting MKK3 and inhibiting p38 MAP kinase activation in response to VEGF. Angiogenesis 14:1–14
Hu S, Huang M, Li Z et al (2010) MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 122:S124–S131
Baggish AL, Hale A, Weiner RB et al (2011) Dynamic regulation of circulating microRNA during acute exhaustive exercise and sustained aerobic exercise training. J Physiol 589:3983–3994
Warburton DE, Katzmarzyk PT, Rhodes RE et al (2007) Evidence-informed physical activity guidelines for Canadian adults. Can J Public Health 98:S16–S68
Elsman P, van’t Hof AW, de Boer MJ et al (2004) Role of collateral circulation in the acute phase of ST-segment-elevation myocardial infarction treated with primary coronary intervention. Eur Heart J 25:854–858
Habib GB, Heibig J, Forman SA et al (1991) Influence of coronary collateral vessels on myocardial infarct size in humans. Results of phase I thrombolysis in myocardial infarction (TIMI) trial. The TIMI Investigators. Circulation 83:739–746
Sabia PJ, Powers ER, Ragosta M et al (1992) An association between collateral blood flow and myocardial viability in patients with recent myocardial infarction. N Engl J Med 327:1825–1831
Beck EB, Erbs S, Mobius-Winkler S et al (2012) Exercise training restores the endothelial response to vascular growth factors in patients with stable coronary artery disease. Eur J Prev Cardiol 19:412–418
Onkelinx S, Cornelissen V, Defoor J et al (2011) The CAREGENE study: genetic variants of the endothelium and aerobic power in patients with coronary artery disease. Acta Cardiol 66:407–414
Gustafsson T, Bodin K, Sylven C et al (2001) Increased expression of VEGF following exercise training in patients with heart failure. Eur J Clin Invest 31:362–366
Testa M, Ennezat PV, Vikstrom KL et al (2000) Modulation of vascular endothelial gene expression by physical training in patients with chronic heart failure. Ital Heart J 1:426–430
Gatta L, Armani A, Iellamo F et al (2012) Effects of a short-term exercise training on serum factors involved in ventricular remodelling in chronic heart failure patients. Int J Cardiol 155:409–413
Hansen AH, Nielsen JJ, Saltin B et al (2010) Exercise training normalizes skeletal muscle vascular endothelial growth factor levels in patients with essential hypertension. J Hypertens 28:1176–1185
Duscha BD, Robbins JL, Jones WS et al (2011) Angiogenesis in skeletal muscle precede improvements in peak oxygen uptake in peripheral artery disease patients. Arterioscler Thromb Vasc Biol 31:2742–2748
Lee BC, Hsu HC, Tseng WY et al (2009) Effect of cardiac rehabilitation on angiogenic cytokines in postinfarction patients. Heart 95:1012–1018
Sandri M, Adams V, Gielen S et al (2005) Effects of exercise and ischemia on mobilization and functional activation of blood-derived progenitor cells in patients with ischemic syndromes: results of 3 randomized studies. Circulation 111:3391–3399
Zbinden R, Zbinden S, Meier P et al (2007) Coronary collateral flow in response to endurance exercise training. Eur J Cardiovasc Prev Rehabil 14:250–257
Troidl K, Schaper W (2012) Arteriogenesis versus angiogenesis in peripheral artery disease. Diabetes Metab Res Rev 28:27–29
Ferrari R, Bachetti T, Agnoletti L et al (1998) Endothelial function and dysfunction in heart failure. Eur Heart J 19:G41–G47
Drexler H, Riede U, Munzel T et al (1992) Alterations of skeletal muscle in chronic heart failure. Circulation 85:1751–1759
Piepoli M, Ponikowski P, Clark AL et al (1999) A neural link to explain the “muscle hypothesis” of exercise intolerance in chronic heart failure. Am Heart J 137:1050–1056
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285:1182–1186
Verma VK, Singh V, Singh MP et al (2009) Effect of physical exercise on tumor growth regulating factors of tumor microenvironment: implications in exercise-dependent tumor growth retardation. Immunopharmacol Immunotoxicol 31:274–282
Jones LW, Viglianti BL, Tashjian JA et al (2010) Effect of aerobic exercise on tumor physiology in an animal model of human breast cancer. J Appl Physiol 108:343–348
Acknowledgments
TAD is supported by research funding from Manitoba Health and Research Council (MHRC) and the Canadian Institutes for Health Research (CIHR). DSK is supported by a MHRC Graduate Studentship. The infrastructural support during this study was provided by the St. Boniface Hospital Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Kehler, D.S., Dhalla, N.S., Duhamel, T.A. (2013). Biochemical Mechanisms of Exercise-Induced Angiogenesis. In: Mehta, J., Dhalla, N. (eds) Biochemical Basis and Therapeutic Implications of Angiogenesis. Advances in Biochemistry in Health and Disease, vol 6. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5857-9_11
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5857-9_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5856-2
Online ISBN: 978-1-4614-5857-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)