
Abstract
There is keen interest to define gene therapies aimed at restoration of auditory and vestibular function in the diseased or damaged mammalian inner ear. A persistent limitation of regenerative medical strategies that seek to correct or modify gene expression in the sensory epithelia of the inner ear involves efficacious delivery of a therapeutic genetic construct. Our approach is to define methodologies that enable fetal gene transfer to the developing mammalian inner ear in an effort to correct defective gene expression during formation of the sensory epithelia or during early postnatal life. Conceptually, the goal is to atraumatically introduce the genetic construct into the otocyst-staged mouse inner ear and transfect otic progenitors that give rise to sensory hair cells and supporting cells. Our long-term goal is to define therapeutic interventions for congenital deafness and balance disorders with the expectation that the approach may also be exploited for therapeutic intervention postnatally.
In the inaugural volume of this series, we introduced electroporation-mediated gene transfer to the developing mouse inner ear that encompassed our mouse survival surgery and transuterine microinjection protocols (Brigande et al., Methods Mol Biol 493:125–139, 2009). In this chapter, we first briefly update our use of sodium pentobarbital anesthesia, our preferred anesthetic for mouse ventral laparotomy, in light of its rapidly escalating cost. Next, we define a rapid, cost-effective method to produce recombinant adeno-associated virus (rAAV) for efficient gene transfer to the developing mouse inner ear. Our immediate goal is to provide a genetic toolkit that will permit the definition and validation of gene therapies in mouse models of human deafness and balance disorders.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Recombinant adeno-associated virus (rAAV) vectors
- Virus-mediated gene transfer
- Transuterine microinjection
- Mouse experimental embryology
- Fetal gene transfer
1 Introduction
We previously demonstrated electroporation-mediated gene transfer to the developing mouse inner ear [1] and now establish an adeno-associated virus (AAV)-mediated gene transfer paradigm. AAV is a non-enveloped, 25 nm diameter, single-stranded DNA virus belonging to the Parvoviridae family [2]. Recombinant AAV (rAAV) lacks the ability to self-propagate, and serves as an attractive vector for fetal gene therapy because of its low immunogenicity, its ability to direct long-term gene expression in a variety of tissues, and a lack of disease association in humans [3, 4]. AAV tropism is determined by the viral capsid proteins through interaction with cell surface receptors [5]. Coating the virus with different AAV capsid serotypes alters rAAV host range and can be exploited to optimize the infection efficiency for specific applications such as transducing otic epithelial progenitors in the otocyst-stage mouse inner ear [6].
The goal of this chapter is to provide a modified AAV production protocol to rapidly and cost-effectively produce rAAV vector of sufficient purity and titer to enable efficient gene delivery into the developing mouse inner ear. The key advantages of this protocol are: (1) a streamlined production process that obviates the need for further virus purification by the time-consuming iodixanol gradient ultracentrifugation methodology [7], (2) optimized polyethylenimine transfection conditions that produce high virus yield [8], and (3) a small-scale, “one flask per vector” transfection scheme that routinely yields enough concentrated viral inoculum for at least 100 mouse otocyst injections.
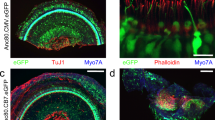
We demonstrate early postnatal transgene expression after transuterine microinjection of rAAV into the otocyst-stage mouse inner ear without toxicity and at comparable efficiencies to rAAV purified through a standard iodixanol gradient ultracentrifugation methodology (Fig. 1). Since rAAV preparations generated by our rapid method are still significantly contaminated with cellular proteins, this approach is primarily useful for studies in which the purity of the viral vector is not a confounding factor. For instance, we have introduced viral inocula produced by this rapid rAAV protocol to evaluate promoter sequences that might guide cell-specific expression and to test the efficacy of epitope-tagging a fluorescent reporter (Fig. 2). The overarching strength of the approach is that it will enable feasibility-level pilot, proof-of-concept, and screening studies that require sustained gene expression in the inner ear from late embryogenesis through early postnatal stages. We predict that this method will be useful for other target tissues and organ systems accessible by fetal gene transfer provided AAV serotype, vector functionality, and potential toxicity are rigorously evaluated.

rAAV produced by the rapid, single flask method demonstrates similar infection dynamics in the mouse inner ear as rAAV purified by iodixanol gradient ultracentrifugation. rAAV1-chick beta actin (CBA)-GFP was produced by the (A) iodixanol and the (B) single flask methods in which the culture supernatant and cellular lysate are pooled and purified by gradient ultracentrifugation or only partially purified by Amicon filtration, respectively. Titers of 7.3 × 1012 and 7.9 × 1012 vg/mL were achieved with the iodixanol and single flask methods, respectively. Viral inocula were introduced into the embryonic day 12.5 (E12.5) mouse otic vesicle by transuterine microinjection until it swelled [9]. The embryos were carried to term, born naturally, and the inner ears of postnatal day 6 (P6) pups were harvested for immunofluorescence localization of myosin 7A (MYO 7A), a hair cell marker [17]. The iodixanol (a) and the single flask (b) purified rAAVs result in GFP expression in inner and outer hair cells as demonstrated in these confocal microscopic projections. In over 200 E12.5 mouse otocyst injections, no embryonic lethality was observed and cochlear maturation to early adult stages is indistinguishable from that of wild-type embryos injected with rAAV purified by iodixanol gradient ultracentrifugation

The single flask method enables rapid evaluation of differentially epitope-tagged GFP constructs for efficacy of expression in the early postnatal mouse inner ear. Four pAAV1-CBA-GFP-[epitope tag] constructs were produced to independently evaluate Myc, 6x Histidine (6x-His), hemagglutinin (HA), and Flag tag detection of GFP. Two copies of the Myc, HA, and Flag tag coding sequences were used against only one 6x-His tag sequence. Four rAAV1-CBA-GFP-[epitope tag] vectors were produced by the single flask method and titers in vg/mL were: Myc—9.2 × 1011; 6x-His—2.5 × 1012; HA—8.3 × 1011; and Flag—2.4 × 1012. The viral inocula were independently injected into E12.5 mouse otocysts and inner ears were harvested at P6 for whole mount immunofluorescence with anti-Myc antibody (Abcam #1162); anti-6x-His tag antibody (Abcam #9108); anti-HA tag antibody (Abcam #9110); and anti-DDDDK tag antibody (Abcam #1162) at 1:100 dilution. Representative confocal projections of the apical cochlea for each vector are presented in the images. Column (a), GFP expression in inner (ihc) and outer hair cells (ohc) in P6 apical cochlea. Column (b), Immunofluorescence detection of the four epitope tags in the organs of Corti in Column (a). Column (c), Merged images of (a) and (b). The C-terminal epitope tags were detected by their respective antisera in GFP+-[epitope-tagged] hair cells with extremely high efficiency. Finally, the 6x-Hisx vector was consistently less robustly expressed. These data indicate that the Myc-, HA-, and Flag-tagged GFP are robustly expressed in sensory hair cells by P6 via rAAV-mediated gene transfer to the developing mouse inner ear. Moreover, the single flask method enables a rapid testing scheme to empirically determine which epitope tag to deploy at the N- or C-terminal of the ion channel or transcription factor of interest, for example. Once bioactivity of the tagged protein is confirmed, a fully purified, high-titer iodixanol gradient preparation of only the validated rAAV vector is performed, ultimately saving time and resources
2 Materials
2.1 General Anesthesia for Mouse Ventral Laparotomy: Update
-
1.
50 mg/mL Nembutal (pentobarbital sodium solution, United States Pharmacopeia [USP]) stock solution. Pentobarbital sodium anesthesia remains the anesthetic of choice to provide the depth and duration of anesthesia required for ventral laparotomy without adversely affecting the developing embryos subjected to transuterine microinjection [9]. The recent escalation in Nembutal cost has necessitated an alternative approach to its purchase from commercial sources to preserve its use for mouse survival surgery (see Note 1 ).
2.2 Cell Culture, Transfection, and rAAV Vector Production
-
1.
Human Embryonic Kidney (HEK) 293A cells (Life Technologies), or HEK293 cells (American Type Culture Collection; ATCC) (see Note 2 ).
-
2.
Dulbecco’s Phosphate-Buffered Saline (DPBS) without Ca2+/Mg2+.
-
3.
1× Trypsin EDTA Solution: 0.05 % trypsin, 0.53 mM EDTA, without, NaHCO3/Ca2+/Mg2+, in 1× Hanks’ Balanced Salt Solution (HBSS, Corning Life Sciences).
-
4.
Complete media: Prepare Dulbecco’s Modification of Eagle’s Medium (DMEM) containing 4.5 g/L glucose, l-glutamine, and sodium pyruvate (Corning Cellgro; Media Tech) with 10 % Fetal Bovine Serum (FBS). Add 1× Penicillin/Streptomycin/l-Glutamine (100 international units [I.U.]/mL penicillin, 100 μg/mL streptomycin, and 292 μg/mL l-glutamine).
-
5.
Post-transfection media: DMEM containing 2 % FBS and 1× Penicillin-Streptomycin-l-Glutamine.
-
6.
Transfection media (serum-free, antibiotic-free): DMEM.
-
7.
AAV rep-cap serotype plasmid (~1 μg/μL) expressing the appropriate viral capsid serotype, e.g., pAAV2/1 (see Notes 3 – 6 ).
-
8.
AAV vector transgene plasmid(~1 μg/μL). This is the plasmid containing the target gene flanked by viral inverted terminal repeats (ITRs), e.g., pAAV-CAG-GFP (Addgene #37825) (see Notes 4 – 7 ).
-
9.
Adenoviral helper plasmid, e.g., pAd DeltaF6 (Penn Vector Core, Gene Therapy Program, University of Pennsylvania School of Medicine) (~1 μg/μL) (see Notes 4 – 6 ).
-
10.
High Potency Linear Polyethylenimine (PEI) “Max” (Mw 40,000) stock solution (Polysciences, Inc.): 1 mg/mL in water, pH 7.1. Sterile filter using 0.2 μm filter (see Notes 8 and 9 ).
-
11.
T-150 culture flasks (Corning), 2 mL aspirating pipette, 10 mL pipette, 15 mL conical tubes.
-
12.
Hemocytometer.
-
13.
Class II Biological Safety Cabinet (BSC) (see Note 10 ).
-
14.
Designated cell culture incubator set at 37 °C and 5 % CO2.
-
15.
Inverted microscope: 4× and 10× objectives.
2.3 Cell Harvest, Lysis, and Pre-clearing
-
1.
0.5 M EDTA. Sterile filter using an 0.2 μm filter.
-
2.
Benzonase nuclease (≥250 units/μL (see Note 11 ).
-
3.
95–100 % ethanol, molecular biology grade.
-
4.
Dry ice (pelleted).
-
5.
37 °C water bath.
-
6.
Beckman J2-21M/E centrifuge or a centrifuge capable of 3836 × g (e.g., 5000 rpm in a Beckman Coulter JA-14 rotor).
2.4 AAV Concentration and Buffer Exchange
-
1.
AAV storage buffer: DPBS, 35 mM NaCl, 5 % glycerol (v/v). Sterile filter using an 0.2 μm filter.
-
2.
Amicon Ultra-15 (MWCO 100,000) centrifugal filter (Millipore, Bedford, MA).
-
3.
Pipettors: P-200 μL and P-1000 μL with sterile pipette tips.
-
4.
Allegra 6R centrifuge or equivalent that supports a swinging bucket rotor capable of 2056 × g (e.g., 3000 rpm in a Beckman Coulter GH-3.8 rotor).
2.5 AAV DNA Preparation and Titration by Quantitative Polymerase Chain Reaction (qPCR)
-
1.
Heating Blocks.
-
2.
DNase I and 10× DNase buffer (Life Technologies).
-
3.
10 mM Tris, pH 9.0.
-
4.
50 mM EDTA.
-
5.
Proteinase K (20 mg/mL; Amresco).
-
6.
Chicken beta actin (CBA) promoter, forward primer (100 μM): 5′-CCA CGT TCT GCT TCA CTC TCC-3′ (see Note 12 ).
-
7.
Chicken beta actin (CBA) promoter, reverse primer (100 μM): 5′-CCC CAT CGC TGC ACA AAA TA-3′ (see Note 12 ).
-
8.
Chicken beta actin (CBA) Probe (10 μM) (Biosearch Technologies): 5′d FAM-CCC CCT CCC CAC CCC CAA TTT TGT ATT TAT-BHQ-1 3′ (see Note 12 ).
-
9.
TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA).
-
10.
Standard plasmid: pAAV-CAG-GFP (Addgene, A gift from Edward Boyden) linearized with EcoRI (5442 base pairs). The CAG promoter is identical to the CBA promoter. Standard dilution series: 107, 106, 105, 104, 103 copies/reaction.
-
11.
7500 Real Time PCR System (Applied Biosystems) or equivalent.
3 Methods
3.1 Seeding HEK293A Cells
-
1.
Plate HEK293A cells in 20 mL of complete media at 7.5 × 106 cells/T-150 flask 24 h prior to transfection in the BSC (see Notes 2 , 10 and 13 ).
-
2.
Transfer cells to an incubator at 37 °C and 5 % CO2. This results in approximately 1.5 × 107 cells/flask on the day of transfection.
3.2 Triple Transfection of HEK293A Cells
On the day of transfection:
-
1.
Check cells under a microscope to ensure they are growing evenly in a monolayer and are 80–90 % confluent (see Note 14 ).
-
2.
Pre-warm 15 mL of post-transfection media in a 37 °C water bath and equilibrate the transfection media to room temperature.
-
3.
Set up two 15 mL tubes, labeled “PEI” and “Plasmids”, in the BSC.
-
4.
To each tube, add 1.5 mL of transfection media.
-
5.
Add 30 μL of 1 mg/mL PEI “Max” Stock solution to the “PEI” tube (see Notes 8 and 9 ).
-
6.
Add 3.75 μg of AAV serotype plasmid, 3.75 μg of the vector transgene ITR plasmid, and 7.5 μg of adenoviral helper plasmid, to the “Plasmids” tube. Mix well by vortexing.
-
7.
Add the PEI-transfection media solution to the Plasmid-transfection media solution in the “Plasmids” tube. Mix well by vortexing.
-
8.
To form the transfection complex, incubate the PEI:Plasmid solution at room temperature for 15 min. In the meantime, remove the flask containing the HEK293A cells and aspirate the media. Add 15 mL of pre-warmed post-transfection media to the flask (see Note 15 ).
-
9.
After the transfection complex has formed, vortex the PEI:Plasmid solution again and then add 3 mL of the PEI:Plasmid complex dropwise to the flask containing 15 mL of post-transfection media without disturbing the cells. Gently tilt the flask back and forth so that the transfection complex is evenly distributed over the cells.
-
10.
Return the flask to the incubator at 37 °C and 5 % CO2 (see Note 16 ).
3.3 Harvesting Cells and Supernatant with Lytic Release of rAAV
-
1.
Harvest cells ~72 h post-transfection. To aid detachment of the cells from the flask surface, add 300 μL of 0.5 M EDTA per T-150 flask for a final concentration of 8–10 mM EDTA. Return the flask to the 37 °C incubator and incubate for at least 15 min. Gently tilt the flask back and forth about 10 times to dislodge the cells. Carefully take up the solution containing cells with a sterile 10 mL serological pipette and release without creating any air bubbles in the media (e.g., maintain a 1 mL residual volume in the pipette until final ejection). Transfer both supernatant containing secreted AAV and the cells containing cell-associated AAV (SN + cells) into a sterile 50 mL conical centrifuge tube (see Note 17 ).
-
2.
Freeze–thaw the SN + cells solution 3 times by cycling between a dry-ice ethanol bath and 37 °C water bath (see Note 11 ). Vortex the cell suspension vigorously after each thaw to encourage the cells to lyse and release any cell-associated AAV into the solution (SN + lysate).
-
3.
Add 6–7 μL Benzonase nuclease (≥250 units/μL) to achieve a 100 unit/mL final concentration in the SN + lysate solution. Mix gently and incubate for 1 h in a 37 °C water bath (see Note 11 ). Re-mix the solution every 15 min during the incubation.
-
4.
Centrifuge the SN + lysate at 3836 × g (e.g., 5000 rpm in a Beckman Coulter JA-14 rotor) for 15 min at 4 °C. Separate the supernatant containing the rAAV particles from the pellet with a sterile 25 mL serological pipette and transfer to a sterile 50 mL conical centrifuge tube. Discard the pellet containing the cellular debris into a biohazardous waste receptacle.
3.4 Concentration and Buffer Exchange
-
1.
Prepare the Amicon Ultra-15 (MWCO 100,000) centrifugal filter for concentration and buffer exchange by first adding 10 mL of sterile water to the filter membrane and centrifuging at 2056 × g (e.g., 3000 rpm in a Beckman Coulter GH-3.8 rotor) for 10 min at room temperature. Discard the flow through. The water wash removes impurities left on the membrane from the manufacturing process.
-
2.
Add 15 mL of AAV storage buffer to the membrane. Centrifuge at 2056 × g for 10 min at room temperature. Discard the flow through. The buffer wash equilibrates the membrane prior to concentration and buffer exchange.
-
3.
Add 15 mL of the rAAV solution to the equilibrated Amicon Ultra-15 (MWCO 100,000) centrifugal filter (see Note 18 ). Centrifuge at 2056 × g for 10 min at room temperature. Discard the flow through.
-
4.
Visually estimate the volume of the retentate, which contains the rAAV particles. If more than 200 μL of retentate is present, gently pipette the solution up and down three times using a P-1000 pipettor to disturb the concentration gradient of rAAV particles that forms near the membrane. Avoid touching the membrane with the pipette tip, which could mechanically compromise the membrane resulting in loss of rAAV in the flow through. Continue centrifugation with resuspension of retentate in ~10 min intervals until the ~200 μL target volume is achieved.
-
5.
Mix 15 mL of AAV storage buffer with ~200 μL of retentate by gently pipetting 3 times with a sterile 5 mL serological pipette.
-
6.
Centrifuge at 2056 × g for 10 min at room temperature, assess retentate volume, and continue Amicon filtration until the 200 μL target volume is achieved.
-
7.
Repeat the concentration and buffer exchange procedure at least three times using the ~200 μL volume as the endpoint for each cycle. The final centrifugation step strives to achieve a final volume of 100–120 μL without evidence of viral precipitation (see Note 18 ).
-
8.
Transfer the concentrated rAAV vector with a P-200 μL pipettor into a 1.5 mL microfuge tube.
-
9.
Briefly centrifuge to remove any precipitate. Transfer the rAAV supernatant to a new 1.5 mL microfuge tube.
-
10.
Aliquot the rAAV vector in 10 μL volumes and freeze at −80 °C. Leave at least one 10 μL aliquot of rAAV vector at 4 °C for quantification of viral genomes present.
3.5 Preparation of rAAV DNA and Titer Determination by Quantitative Polymerase Chain Reaction (qPCR)
rAAV DNA is prepared from the concentrated rAAV vector for quantification by qPCR according to the method described by Rohr and colleagues [10]. Only critical steps are summarized below.
-
1.
DNase treatment: Digest a 1 μL aliquot of concentrated rAAV with 1 unit DNase I (Life Technologies) in the supplied buffer at 1× concentration in a final volume of 50 μL for 30 min at 37 °C. Inactivate DNase I by addition of 5 μL 50 mM EDTA and heating for 10 min at 65 °C.
-
2.
Proteinase K digestion: Add 45 μL of DPBS containing 0.2 μg/μL of Proteinase K to the 55 μL DNase-treated sample to achieve a final concentration of 9–10 μg Proteinase K in the 100 μL reaction volume. Incubate samples at 56 °C for 1 h. Inactivate proteinase K by heating at 95 °C for 20 min.
-
3.
Dilute samples 100-fold in 10 mM Tris, pH 9.0 for qPCR (e.g., add 5 μL to 495 μL of 10 mM Tris, pH 9.0).
-
4.
Titer Determination: For titration by qPCR, 10 μL aliquots of sample dilutions and standards are assayed in duplicate using the CBA promoter primer-probe in a 7500 Real-Time PCR system (Applied Biosystems) (see Note 12 ). Use 10 μL of PCR-grade water as negative control.
-
5.
The viral genomes (vg)/mL are calculated from the dilution factor and the mean copies of rAAV per reaction determined from the standard curve consisting of 107 -103 copies of the standard (see Note 19 ).
4 Notes
-
1.
The official transcript (version 4/17/2013) of the Office of Laboratory Animal Welfare (OLAW) Webinar on March 1, 2012 entitled “Use of Non-Pharmaceutical-Grade Chemicals and Other Substances in Research with Animals” included an appendix that addressed a comment submitted in writing after the Webinar, regarding the limited availability and extreme cost of Nembutal [11]. OLAW, the United States Department of Agriculture Animal Care, and the Association for Assessment and Accreditation of Laboratory Animal Care jointly issued the following response: “The Regulatory guidance on this matter specifically allows for use of non-pharmaceutical-grade compounds due to non-availability and with Institutional Animal Care and Use (IACUC) approval. The exorbitant cost of this product has placed it logistically into the unavailable category. Pentobarbital from a reagent or analytical-grade powder, properly prepared by your pharmacist or other knowledgeable individual (e.g., chemist, veterinarian, researcher, with assurance of appropriate storage and handling, and approval by the IACUC is acceptable. IACUC approval can be institution-wide for the drug prepared in this fashion for all approved users.” The Brigande Laboratory has secured Oregon Health & Science University (OHSU) IACUC approval to compound a 50 mg/mL solution of pentobarbital sodium solution for mouse survival surgery. Reagent and analytical-grade pentobarbital sodium powder is available from a compounding pharmacist with appropriate DEA licensure and potentially from other commercial sources. Stability testing of the compounded anesthetic should be conducted to determine safe storage conditions that preserve potency and efficacy. Our formulation is 50 mg/mL pentobarbital sodium in 40 % propylene glycol, (USP), 10 % ethanol, (USP), and the balance water for injection, (USP), and is stable in the dark at room temperature for 12 months. Consult your IACUC for their preferred method of compounding pentobarbital sodium, which typically differs between institutions.
-
2.
HEK293 cells contain integrated adenovirus sequences that provide essential helper functions for AAV production. Transfection efficiency can diminish after prolonged passage in culture or if cells are allowed to become over-confluent. For routine passage, we recommend splitting the cells every 3–4 days before they reach confluence. To maintain cell quality and consistency between virus productions, we recommend creating a low-passage working cell bank from the initial cell stock. A passage limit for efficient transfection should be determined empirically (usually no more than 20–30 passages). After the appropriate cell passage limit is reached, a new cell vial should be thawed from the working cell bank.
-
3.
All stock solutions and buffers are made with endotoxin-free water with a resistivity of 18.2 MΩ cm.
-
4.
The biosafety classification and attendant biosafety procedures required to work with rAAV vectors is dependent on a number of factors that have to be carefully evaluated by the investigator prior to beginning the work. The Institutional Biosafety Committee (IBC) and IACUC at OHSU work in tandem to ensure that appropriate biosafety protocols are in place to protect both investigators and their animal model systems. According to the November 6, 2013 issue of the Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules, wild-type and recombinant AAVs of all serotypes are generally classified as Risk Group 1 (RG-1) agents (i.e., are not associated with disease in healthy human adults). Therefore, work with RG-1 rAAV requires at a minimum Biosafety Level (BSL)-1 and Animal Biosafety Level (ABSL)-1 containment. Additional modifications may raise the risk profile. For example, BSL-2 and ABSL-2 containment is usually required if the rAAV is produced using a live helper virus and/or encodes a potentially toxic or tumorigenic transgene. In addition, the human-derived HEK293A cell line and the significant cellular material present in unpurified rAAV preparations generally requires BSL-2 as per the blood-borne pathogens standard set by the Occupational Safety & Health Administration (OSHA). All in vivo work with rAAV in the Brigande Laboratory is conducted under BSL-2 protocols, though our dams with inoculated embryos are housed at ABSL-1 with permission of the OHSU IACUC. However, it is essential to consult your institutional biosafety officials and your IACUC for guidance on formulating an appropriate institutional biosafety protocol appropriate for the production and in vivo use of rAAV.
-
5.
The basic AAV component plasmids can be obtained from a variety of sources, such as the Penn Vector Core at the University of Pennsylvania, or from one of several commercial vendors. Also, the non-profit Addgene plasmid repository (www.addgene.org) stocks a growing collection of specific AAV vector transgene plasmids deposited by researchers. The AAV triple transfection method outlined here requires three plasmids: a transgene plasmid with flanking inverted-terminal repeats (ITRs); a serotype-specific plasmid expressing AAV rep-cap proteins; and an adenoviral plasmid providing essential virus helper functions [12–14]. The separation of virus functions onto different plasmids makes the system very flexible as plasmids of interest can be swapped into the transfection protocol in order to change virus characteristics without having to alter the virus production methodology. For example, the serotype-specific plasmid determines the tissue tropism of the rAAV vector and influences virus production yields and cellular transduction efficiency. The triple transfection scheme allows screening of several AAV capsid serotypes enabling identification of AAV1 as highly effective for gene delivery to the developing inner ear. Similarly, the transgene plasmid sequence can be modified or exchanged to alter the genetic payload.
-
6.
The serotype-specific plasmid and the adenoviral helper plasmid are grown in common E. coli strains (such as TOP10) as high-copy plasmids in Luria Broth containing 50 μg/mL carbenicillin at 37 °C. The inverted-terminal repeat (ITR)-containing plasmids (i.e., the cis or vector transgene plasmids) contain an appropriate transgene sequence flanked by the AAV ITR on each side. The ITRs are critical for proper vector transgene packaging into virus particles, but are prone to deletions and rearrangements. Therefore, ITR-containing plasmids should be grown in recombination-deficient (rec-) E. coli strains (such as Sure (Agilent Technologies), Stbl2, or Stbl3 (Life Technologies)) as low-copy plasmids at 30 °C. Using these conditions the plasmid yields are significantly lower, hence we recommend using a rich media like 2XYT and doubling the growth volumes. For example, for a Qiagen Maxi Prep, set up two 100 mL overnight cultures. Use twice the volumes of P1, P2, and P3 buffers for initial resuspension and cell lysis. Then purify over a single Maxi Prep column following the standard Qiagen endotoxin-free Maxi Kit protocol. In addition, to avoid failed rAAV vector productions, the integrity of the ITRs in each new plasmid preparation should be carefully verified in advance by restriction digests using enzymes that cut within the ITRs (such as XmaI, MscI, AhdI, PauI, and PvuII).
-
7.
The wild-type AAV genome size is about 4.7 kb. All viral sequences (except for the two 145 bp inverted terminal repeats (ITRs)) are deleted in rAAV vectors. That provides a transgene capacity of up to ~4.5 kb. Even though slightly larger transgene cassettes can sometimes still be packaged efficiently, there is a risk of non-packaging or incomplete packaging with oversized genomes (i.e., absent or low vector titers, respectively). Therefore, it is recommended not to exceed the AAV packaging capacity.
-
8.
The use of PEI “Max” as a transfection agent eliminates the need to change media post-transfection since it is non-toxic to cells. In addition, it is chemically stable and functions over a wide pH range.
-
9.
PEI “Max” is built on the same linear backbone of PEI, but contains more than 11 % additional free nitrogens. The amino groups make it a highly potent cationic polymer allowing it to form ionic interactions with the phosphate backbones of DNA. PEI:DNA complexes are transported into the cell via endocytosis and escape endosomes via a proton sponge effect that promotes osmotic swelling and disruption of the endosomal membrane. In PEI:DNA complexes, the ratio between molar units of PEI nitrogen atoms to units of DNA phosphate atoms, known as the N/P ratio, was found to correlate with transfection efficiency [8, 15]. We optimized our AAV transfection and production protocol using PEI “Max” by assaying various N/P ratios and DNA amounts. An N/P ratio of 15.3 for PEI “Max” coupled with 1.0 μg DNA per mL of media (15 μg DNA/T-150 flask) produces the highest AAV yields. The volume (μL) of PEI was calculated based on the following equation [15]: 3 × D × R/S, where D = total amount of plasmid DNA used (15 μg), R = N/P ratio (ratio of nitrogen content in PEI to phosphorous content in DNA= 15.3), S = concentration of the PEI stock (23.2 mM, monomer unit).
-
10.
The BSC is a vertical type of laminar flow hood whose class 100 environment protects the user from exposure to biohazards. The primary route of exposure to rAAV particles is through aerosolization during pipetting. Confining all procedures involved in the production of rAAV to the BSC minimizes the potential for exposure, protecting the investigator and laboratory staff. The Environmental Health and Radiation Safety group at OHSU requires annual recertification of all biological safety cabinets used for production of viral vectors. Check institutional policy regarding all biosafety requirements to safely work with rAAV vectors.
-
11.
The freeze–thaw cycles initiate cell lysis and the release of cellular DNA and RNA along with unencapsidated viral DNA. Benzonase is an endonuclease that digests all forms of DNA and RNA to 5′-monophosphate terminated oligonucleotides 3–5 bases in length. Cellular debris is cleared from the preparation by a subsequent centrifugation step. Benzonase-treated solutions exhibit reduced viscosity, which facilitates concentration and buffer exchange by Amicon filtration.
-
12.
The PCR primers and probe are specific to the vector transgene sequence (e.g., the CBA promoter), in order to accurately quantify the number of vector genomes in a large background of cellular DNA. If other transgene sequences are used, the primer and probe sequences need to be appropriately designed and the qPCR assay validated.
-
13.
It is also possible to plate HEK293A cells up to 5 days prior to transfection. In this case, plate at 0.9 × 106 cells/T-150 flask in 20 mL of complete growth media. This results in ~1.5 × 107 cells/flask on the day of transfection. Twenty-four hours prior to transfection, aspirate off the old growth media, feed the cells with 15 mL of fresh complete growth media, and return the flask to the 37 °C incubator.
-
14.
Eighty to ninety percent confluence is equivalent to approximately 12–15 × 106 cells per T-150 flask. The number of cells present at the time of transfection is critical given that the expected productivity per cell is about 1 × 104 viral genomes. Vector titers may be reduced if cells are over- or under-confluent.
-
15.
Complete DMEM containing 10 % FBS is used for seeding HEK293A cells, but the amount of FBS in the media is reduced to 2 % post-transfection. The reduced FBS concentration adequately supports cell growth, though at a slower rate; helps boost virus production compared to serum-free growth media; and is comparable to virus production with 10 % FBS. In addition, 2 % FBS compared to 10 % FBS is cost-effective, decreases the viscosity of the media, and prevents clogging of the Amicon ultrafilters due to precipitation of FBS proteins during the concentration and buffer exchange step.
-
16.
rAAV does not produce a clear cytopathic effect (CPE) and this may make it difficult to monitor virus production. Nevertheless, there are general signs that can be observed, e.g., rounded up cells, increased cellular debris, and the presence of floating cells, which suggest that the transfection was successful. When performing this protocol for the first time, we recommend using a marker gene (e.g., GFP) that is expressed from the viral cassette to assess transfection efficiency by visualizing the number of fluorescent cells present. Transfection is considered efficient when 80–90 % of the cells express the fluorescent marker protein.
-
17.
The prototypical AAV2 virus accumulates to about 90 % inside cells, and so AAV has traditionally been harvested from cells rather than the media. In contrast, many of the more recently discovered AAV serotypes (including AAV1) tend to release a significant and variable amount of virus particles into the media within the first 72 h post-transfection [16]. Furthermore, AAV particles tend to leak into the media during prolonged cell culture; hence harvesting both the cells and supernatant (i.e., the production media) can significantly boost yields and enhance viral titer.
-
18.
The Amicon Ultra-15 (MWCO 100,000) centrifugal filter has three advantages. First, contaminants such as media components and proteins <100,000 kDa pass through the membrane as part of the flow through. For example, Benzonase at 30 kDa is removed from the virus preparation in this step. Second, Amicon filtration concentrates the virus preparation 100-fold by reducing the volume from ~20 mL to ~200 μL or less. Third, the ultrafilter exchanges the rAAV media with a buffer that is compatible for delivery into the embryonic mouse inner ear and suitable for long-term rAAV storage at −80 °C with no significant drop in viral titer or infectivity.
-
19.
Typical titers obtained from a single flask AAV production from supernatant plus cells ranged from 7.3 × 1011 to 7.7 × 1012 vg/mL. The average titer ± the standard error of the mean (SEM) obtained from 16 single flask preparations was 3.2 × 1012 ± 6.0 × 1011 vg/mL. The average volume of rAAV concentrate ± SEM from these 16 single flask preparations was 107 ± 2.4 μL. In practice, 100 μL of viral inoculum can be managed to enable approximately 100 E12.5 otocyst injections.
References
Brigande JV, Gubbels SP, Woessner DW, Jungwirth JJ, Bresee CS (2009) Electroporation-mediated gene transfer to the developing mouse inner ear. Methods Mol Biol 493:125–139
Berns KI (1990) Parvovirus replication. Microbiol Rev 54(3):316–329
Grimm D, Kleinschmidt JA (1999) Progress in adeno-associated virus type 2 vector production: promises and prospects for clinical use. Hum Gene Ther 10(15):2445–2450
Jooss K, Yang Y, Fisher KJ, Wilson JM (1998) Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J Virol 72(5):4212–4223
Wu Z, Asokan A, Samulski RJ (2006) Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther 14(3):316–327
Bedrosian JC, Gratton MA, Brigande JV, Tang W, Landau J, Bennett J (2006) In vivo delivery of recombinant viruses to the fetal murine cochlea: transduction characteristics and long-term effects on auditory function. Mol Ther 14(3):328–335
Lock M, Alvira M, Vandenberghe LH, Samanta A, Toelen J, Debyser Z, Wilson JM (2010) Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum Gene Ther 21(10):1259–1271
Huang X, Hartley AV, Yin Y, Herskowitz JH, Lah JJ, Ressler KJ (2013) AAV2 production with optimized N/P ratio and PEI-mediated transfection results in low toxicity and high titer for in vitro and in vivo applications. J Virol Meth 193(2):270–277
Wang L, Jiang H, Brigande JV (2012) Gene transfer to the developing mouse inner ear by in vivo electroporation. J Vis Exp (64). doi: 10.3791/3653
Rohr UP, Wulf MA, Stahn S, Steidl U, Haas R, Kronenwett R (2002) Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time PCR. J Virol Methods 106(1):81–88
Transcript of OLAW On-line Seminar broadcast on March 1, 2012—Use of non-pharmaceutical grade chemicals and other substances in research with animals (v4/17/2013)
Grimm D, Kern A, Rittner K, Kleinschmidt JA (1998) Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther 9(18):2745–2760
Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, Iwaki Y, Colosi P (1998) Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 5(7):938–945
Xiao X, Li J, Samulski RJ (1998) Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 72(3):2224–2232
Reed SE, Staley EM, Mayginnes JP, Pintel DJ, Tullis GE (2006) Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. J Virol Methods 138(1–2):85–98
Vandenberghe LH, Xiao R, Lock M, Lin J, Korn M, Wilson JM (2010) Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum Gene Ther 21(10):1251–1257
Gubbels SP, Woessner DW, Mitchell JC, Ricci AJ, Brigande JV (2008) Functional auditory hair cells produced in the mammalian cochlea by in utero gene transfer. Nature 455(7212):537–541
Acknowledgements
We thank the members of the Molecular Virology Support Core (MVSC) laboratory for their support, in particular Dr. Don Siess for his technical insights. Research funding for the development of AAV preparation methods was provided by NIH P51 center grant OD011092 to the Oregon National Primate Research Center. The Brigande lab gratefully acknowledges funding from the National Institute on Deafness and other Communication Disorders: R01DC008595, R01DC014160, and R21DC012916 (JVB); P30DC005983 (Oregon Hearing Research Center); and the Hearing Health Foundation through the Hearing Restoration Project.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Gomes, M.M., Wang, L., Jiang, H., Kahl, C.A., Brigande, J.V. (2016). A Rapid, Cost-Effective Method to Prepare Recombinant Adeno-Associated Virus for Efficient Gene Transfer to the Developing Mouse Inner Ear. In: Sokolowski, B. (eds) Auditory and Vestibular Research. Methods in Molecular Biology, vol 1427. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3615-1_3
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3615-1_3
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3613-7
Online ISBN: 978-1-4939-3615-1
eBook Packages: Springer Protocols