
Abstract
The α/β-hydrolase domain-containing 6 (ABHD6) enzyme is a newly found serine hydrolase whose substrate profile resembles that of monoacylglycerol lipase (MAGL), the major 2-arachidonoyl glycerol (2-AG) hydrolase in the brain. Here, we describe a sensitive fluorescent assay of ABHD6 activity in a 96-well-plate format that allows parallel testing of inhibitor activities of up to 40 compounds in a single assay. The method utilizes lysates of HEK293 cells transiently overexpressing human ABHD6 as the enzymatic source, and kinetically monitors glycerol liberated in the hydrolysis of 1(3)-AG, the preferred arachidonoyl glycerol isomer. Glycerol output is coupled to an enzymatic cascade generating the fluorescent end-product resorufin. The approach has major benefits compared to laborious traditional mass spectrometric methods and liquid scintillation-based assays, or approaches using unnatural substrates.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- 2-AG hydrolase
- Endocannabinoid
- Fluorescence
- Glycerol
- Inhibitor
- Lipase
- Monoacylglycerol
- Natural substrate
- Screening
1 Introduction
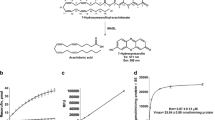
The endocannabinoid 2-arachidonoyl glycerol (2-AG) is a unique bioactive lipid that, based on recent evidence, has three major hydrolyzing enzymes in the body: monoacylglycerol lipase (MAGL), α/β-hydrolase domain-containing 6 (ABHD6), and 12 (ABHD12) [1]. The newly characterized ABHD6 has been associated with 2-AG metabolism both in the central nervous system [2] and in peripheral tissues [3]. It is estimated that at the bulk brain level, ~4 % of 2-AG hydrolysis is due to ABHD6 [4]. The MAG substrate profile of human ABHD6 (hABHD6) closely resembles that of hMAGL with the exception that hABHD6 prefers the MAG 1(3)-isomers over 2-isomers, as shown in our previous in vitro study [5]. Retrospectively, development of sensitive and rapid methods to monitor endocannabinoid hydrolysis has appeared challenging, probably mostly because of instability and lipophilic nature of 2-AG and other endocannabinoids. Mass spectrometric approaches in this field appear rather laborious. On the other hand, relying on radioactive-based liquid scintillation analysis has many disadvantages. Here, we describe in detail a sensitive fluorescence-based method for assessment of hABHD6 activity and its inhibition in 96-well-plate format, based on our recent publication [5]. The method relies on detection of glycerol liberated from the natural MAG substrate 1(3)-AG as a result of ABHD6 hydrolytic activity. Glycerol is then converted by a coupled-enzyme system to generate the fluorescent end-product resorufin. The principle of the coupled-enzyme system to detect glycerol formed by ABHD6 activity is detailed in Fig. 1.

Principle of the coupled-enzyme system to detect ABHD6 activity. ABHD6-catalyzed hydrolysis of 1(3)-AG generates equimolar amounts of arachidonic acid (AA) and glycerol. In the coupled-enzyme system, glycerol is converted to glycerol-1-phosphate (G-1-P) in the presence of ATP, in a reaction catalyzed by glycerol kinase (GK). Glycerol 3-phosphate oxidase (GPO)-catalyzed oxidation of G-1-P generates H2O2 which in the presence of horseradish peroxidase (HRP) converts Amplifu™ Red into the fluorescent product resorufin whose fluorescence (λ ex 530 nm; λ em 590 nm) is kinetically monitored
The method is highly sensitive, allowing detection of low picomolar quantities of the enzymatic product (water-soluble glycerol), and requires only a small amount of lysate (0.3 μg protein/well) prepared from HEK293 cells transiently overexpressing hABHD6. The kinetic assay format has been validated for the three 2-AG hydrolases (hMAGL, hABHD6, and hABHD12) [5], and has been recently modified further to allow diacylglycerol lipase (DAGL) activity measurements [6].
2 Materials
Prepare all solutions using Millipore-purified, deionized high-quality water (HQW). Store buffer stocks at room temperature (RT) and other reagents at −80 °C (see Note 1 ).
2.1 Basic Incubation Cocktail
-
1.
500 mM Tris–HCl, pH 7.4: Dissolve 60.55 g Tris in 800 ml of HQW with constant stirring for at least 2 h. Adjust pH to 7.40 with strong HCl, adjust volume to 1 l with HQW. Store at RT in a dark bottle.
-
2.
100 mM EDTA: Dissolve 3.7224 g Na2EDTA in 100 ml of HQW. Store at 4 °C in a dark bottle.
-
3.
500 mM MgCl2: Dissolve 20.33 g MgCl2∙6H2O in 200 ml of HQW. Store at 4 °C in a dark bottle.
-
4.
4 M NaCl: Dissolve 23.376 g NaCl in 100 ml of HQW. Store at 4 °C in a dark bottle.
-
5.
Basic incubation cocktail (BIC): Prepare BIC from the fresh stocks by pipetting 50 ml of 500 mM Tris–HCl (pH 7.4) in a 250 ml volumetric flask. Add 5 ml of 100 mM EDTA, 5 ml of 500 mM MgCl2, and 12.5 ml of 4 M NaCl, and adjust the volume to 250 ml with HQW. Final concentrations of the buffer components are 100 mM Tris–HCl (pH 7.4), 2 mM EDTA, 10 mM MgCl2, and 200 mM NaCl. Store at RT in a dark bottle.
2.2 Components of the Coupled-Enzyme System
For the preparations of coupling enzyme stocks (GPO, GK, HRP, see details below) in HQW, calculate the amount of HQW needed for the stock preparation, which depends on the specific activity of each enzyme batch. Mix well and divide in 25 μl single-use aliquots for storage at −80 °C. Thaw one aliquot of each just prior to the experiment and keep on ice until use.
-
1.
200 U/ml glycerol 3-phosphate oxidase (GPO) in HQW: GPO from Pediococcus sp. (Sigma), 100 U, 40–80 U/mg solid, pH 8.1.
-
2.
200 U/ml glycerokinase (GK) in HQW: GK from Cellulomonas sp. (Sigma), 1000 U, 25–75 U/mg protein.
-
3.
240–250 U/ml horseradish peroxidase (HRP) in HQW: HRP (Sigma), 5000 U, 45 mg solid.
2.3 Other Reagents
-
1.
10 mM Amplifu Red™ in DMSO: Dissolve the vial content of Amplifu Red™ (Fluka) in DMSO (e.g., 5 mg in 1944 μl) and divide in 25 μl single-use aliquots to be stored at −80 °C. Thaw one aliquot just prior to the experiment and protect from light (see Note 2 ).
-
2.
10 mM ATP in HQW: This needs to be prepared fresh for each experiment. Weigh ATP and dissolve in HQW just prior to use, e.g., 2.536 mg/0.5 ml of HQW. Keep on ice until use.
-
3.
5 % BSA (w/v) in HQW: Carefully dissolve BSA (essentially fatty acid free) with constant stirring, e.g., 10 g in 200 ml of HQW. Divide in 1 ml aliquots for storage at −20 °C (see Note 3 ).
-
4.
10 mM 1(3)-AG in ethanol: 1(3)-AG is commonly supplied in acetonitrile. Transfer 5 mg 1(3)-AG into a preweighed Eppendorf tube, evaporate the solvent under a stream of nitrogen, weight the residue, and dissolve in ethanol (1321 μl/5 mg). Divide in 100 μl aliquots for storage at −80 °C.
-
5.
DMSO for control samples and for dilution of ABHD6 inhibitors.
-
6.
Phosphate-buffered saline (PBS) for the preparation and dilution of cellular lysates.
-
7.
10 mM Tetrahydrolipstatin (THL) in DMSO: Weigh, e.g., 2.55 mg THL and dissolve it in 514 μl of DMSO. Divide in 10 μl aliquots for storage at −80 °C. THL serves as a reference inhibitor and is included at 10−5 and 10−7 M concentrations in each experiment to monitor assay performance (see Note 4 ). Prepare 1 mM and 10 μM intermediate dilutions in DMSO; these will give 10−5 and 10−7 M final concentrations during the preincubation with the enzyme (1:100 ratio).
2.4 Glycerol Standards and Glycerol Quality Control
-
1.
Glycerol (liquid with a density of 1.25 g/ml): 1 μl corresponds to 13.57 μmol glycerol (13.57 M stock). This needs to be diluted several times, in order to reach the pmol concentration range.
-
2.
Std-A: Dilute glycerol stock 1:100 with HQW as follows: Weigh 125 mg of glycerol into a 10 ml volumetric flask, and dilute to 10 ml ⇒ 1 μl corresponding to 135.7 nmol glycerol. This can be divided into 0.5 ml aliquots for storage at −20 °C.
-
3.
Std-B: Dilute Std-A 1:100 with HQW (10 μl + 990 μl) ⇒ 1 μl corresponding to 1.357 nmol glycerol.
-
4.
Glycerol quality control (GQC): Prepare a larger volume (50 ml) by pipetting 25 ml of BIC + 5 ml of 5 % (w/v) BSA + 50 μl Std-B + HQW. Divide in 900 μl aliquots for storage at −20 °C. In the assay, pipette 99 μl of GQC together with DMSO or inhibitors (1 μl). This equals to 134 pmol glycerol per well (see Note 5 ).
2.5 Reagents for Cell Culture
-
1.
Dulbecco’s modified Eagle’s medium (DMEM).
-
2.
Fetal bovine serum (FBS).
-
3.
Antibiotics (penicillin and streptomycin).
2.6 Equipment
-
1.
Black 96-well plates suitable for fluorescence reading.
-
2.
Automatic (or manual) multichannel pipettes for rapid pipetting of enzyme dilutions and the glycerol assay mix (containing substrate and the coupling enzymes). Normal set of pipettes is needed for preparation of inhibitor dilutions and other assay components.
-
3.
Plastic pipetting reservoirs for multichannel pipetting substrate mix and enzyme dilutions (see Note 1 ).
-
4.
A fluorescence reader capable of kinetic measurements (λ ex 530 nm; λ em 590 nm) (see Note 6 ).
-
5.
Equipment for cell culture.
3 Methods
We have validated the assay by using lysates of HEK293 cells transiently overexpressing hABHD6 [5]. HEK293 cells have negligible endogenous ABHD6 activity and thus serve as excellent host cells for transient overexpression of the enzyme. The fluorescent assay is highly sensitive requiring merely 0.3 μg of lysate protein/well [5]. Under these conditions, 1(3)-AG hydrolysis by the cellular background is low and the signal-to-noise is therefore optimal [5]. We recommend including also mock-transfected HEK293 cells as negative control, in order to facilitate verification of successful ABHD6 expression (see Note 7 ).
3.1 Enzyme Preparation
-
1.
Transfection: Culture HEK293 cells as monolayers in DMEM containing 10 % FBS under antibiotics (penicillin/streptomycin) at 37 °C in a humidified atmosphere of 5 % CO2/95 % air. Use plasmids containing ABHD6 for introducing cDNAs to cells by a standard transient transfection procedure [5]. Culture mock cells (i.e., cells transfected with an empty vector) in parallel to facilitate verification of successful enzyme expression and activity.
-
2.
Preparation of cell lysates: Wash cells twice with ice-cold PBS. Scrape off and pellet cells at 250 × g for 10 min at 4 °C. Freeze-thaw cell pellets three times, resuspend in ice-cold PBS, briefly sonicate, and aliquot for storage at −80 °C. Determine protein concentration by using BSA as a standard. For the assay, dilute an aliquot to a protein concentration of 3 mg/ml using PBS. Total volume required for a full 96-well plate is 10 μl (see Note 8 ).
-
3.
Analysis of ABHD6 expression: We recommend verifying successful expression of ABHD6 before routine use in the glycerol assay, preferentially by using activity-based protein profiling (ABPP), as this method discloses a catalytically active enzyme with proper size [5]. If this methodology is not available, Western blotting can be used to detect immunoreactive protein [5].
3.2 Activity Assay
Carry out all procedures at RT unless otherwise specified. Here, we detail our routine assay platform for 96-well plates allowing determination of hABHD6 hydrolytic activity together with activity screening of potential hABHD6 inhibitors. With this setup, up to 40 distinct inhibitors (or different concentrations of selected inhibitors) can be tested in duplicates in a single full-plate assay. The procedure can be completed within 3–4 h.
Overview of the method. Final volume per well is 200 μl. The solvent (DMSO) or the inhibitors are first pipetted in a volume of 1 μl into appropriate wells of the 96-well plate (Fig. 2). After this, 99 μl of blank buffer (without enzyme), hABHD6-HEK lysate (0.3 μg protein/well), or GQC (134 pmol glycerol/well) will be added to the indicated wells using a multichannel pipette, and the plate is vortexed and incubated for 30 min at RT. Following this, glycerol assay mix (100 μl/well) containing the substrate and the coupled-enzyme system is added to all wells using a multichannel pipette, the plate is vortexed, and fluorescence is kinetically monitored at 10-min intervals for 90 min at RT.

Platform allowing simultaneous monitoring of ABHD6 hydrolytic activity and its inhibition in a 96-well-plate format. Final volume is 200 μl per well. DMSO/inhibitors (1 μl) are added first into the appropriate wells, and then blank buffer, glycerol quality control (GQC), or the diluted enzyme preparation are added in 99 μl of assay buffer. Following 30-min preincubation of enzyme with inhibitors, 100 μl of glycerol assay mix containing the substrate and the coupling enzymes is added by using a multichannel pipette. The plate is vortexed and the fluorescence (λ ex 530 nm; λ em 590 nm) is kinetically monitored at 10-min intervals for 90 min at RT
-
1.
Pipetting DMSO/inhibitors into the wells: Prepare 10 mM inhibitor stocks in DMSO (see Note 9 ). The stocks are further diluted in DMSO, so that the solutions to be pipetted into the wells (1 μl) are 100-fold concentrated over the desired final concentrations during the preincubation. For wells without inhibitor, use 1 μl of DMSO (see Note 10 ). In each experiment, include THL at the two final concentrations (10−7 and 10−5 M) as the reference inhibitor, in order to monitor assay performance (see Note 4 ).
-
2.
Preincubation of enzyme with DMSO/inhibitors: Prepare the blank buffer and the hABHD6-HEK lysate working dilution into test tubes according to Table 1. Transfer the solutions into plastic reservoirs, and by using a multichannel pipette add 99 μl into indicated wells (Fig. 2). Finally, pipette 99 μl of GQC into indicated wells (Fig. 2) (see Note 11 ). At this stage, all wells should have a total volume of 100 μl. Vortex the plate and incubate at RT for 30 min with the plate covered.
Table 1 Preparation of blank and hABHD6-HEK dilutions -
3.
Kinetic assay with the substrate: Just prior to use (i.e., during the final 5–10 min of the preincubation step), prepare 11 ml of glycerol assay mix containing the substrate and the coupled-enzyme system (Table 2). Amplifu™ Red should be added last in order to minimize decomposition (see Note 2 ). Mix the cocktail well by vortexing several times, and avoid bubbles (see Note 3 ), as well as unnecessary delays. Transfer into a plastic reservoir, and by using a multichannel pipette add 100 μl into all wells (see Note 11 ), vortex the plate (see Note 12 ), and in order to obtain initial fluorescence reading at time point 0 min, start the kinetic measurement without delay. Record fluorescence unit (FU) readings at 10-min intervals for 90 min at RT (see Note 13 ).
Table 2 Preparation of the glycerol assay mix
3.3 Guidelines for Calculations
We use well-based changes in fluorescence (FU90min − FU0min) as the basis to calculate all results, as explained below (see Note 14 ).
-
1.
Calculate the net fluorescence (NetFU) (FU90min − FU0min) for individual wells by subtracting the mean value (FU90min − FU0min) of the blank wells from the (FU90min − FU0min) values of all other wells.
-
2.
Calculate ABHD6 activity in the DMSO-ABHD6 wells (representing total activity in the absence of any inhibitor), based on the mean NetFU of these wells obtained above and that of the GQC wells (see Note 5 ):
ABHD6 activity (pmol/well) = (NetFUABHD6 × 134)/NetFUGQC.
-
3.
Calculate substrate consumption, based on ABHD6 activity and the amount of substrate. Final 1(3)-AG concentration in wells is initially 12.5 μM; thus 2500 pmol substrate is initially available:
% substrate consumption = (ABHD6 activity/2500 pmol) × 100.
Ideally, substrate consumption during the 90-min incubation should be <20 %.
-
4.
For wells with inhibitors, calculate percentage of remaining activity (see Note 9 ):
% remaining activity = (NetFUinhibitor/NetFUABHD6-DMSO) × 100.
4 Notes
-
1.
Any detergent residues (containing glycerol) from the washing-up steps of laboratory glassware and plastic reservoirs are potential contaminators of the assay. Therefore, careful washing of containers and plastic pipetting reservoirs with HQW is recommended before use.
-
2.
Amplifu™ Red is light sensitive, and thus protect the reagent from light and cover the plate after substrate mix addition. Check also the color of the substrate mix after preparation: if it is reddish, the reagent may be giving too high values and rise the background.
-
3.
Fatty acid-free BSA is included in this assay to minimize nonspecific binding of hydrophobic compounds to the plastic. Be aware that strong vortexing of BSA solutions may cause bubbles that disturb fluorescence reading. Also, in the black wells used in these experiments small bubbles are difficult to see.
-
4.
Using this methodology, the IC50 value for THL towards hABHD6 is 47.9 nM [5]; the two concentrations are expected to produce ~60 % (10−7 M) and 100 % (10−5 M) inhibition.
-
5.
The inclusion of GQC is important for the following reasons: (1) Fixed amount of glycerol added to the wells (134 pmol/well in the case of GQC) allows to monitor from assay to assay that the coupled-enzyme system properly coverts glycerol to resorufin; that is, the net fluorescence of the GQC wells should be comparable between different assays. If fluorescence dramatically drops, component(s) of the coupled-enzyme system are not working properly and fresh stock(s) may need to be prepared. (2) Using GQC as a standard, it is possible to calculate the amount of glycerol formed (pmol/well) for each individual well. The assay is highly linear in the range of 0–500 pmol glycerol/well [5]. Inclusion of GQC makes it possible to determine the amount of glycerol formed (pmol/well) for each well also at various time points of the kinetic assay [5]. (3) In inhibitor discovery, it is important to rule out that the coupled-enzyme system is not the target of the inhibitor. Wells with GQC ± inhibitor giving the same net fluorescence indicate no interference with the coupled-enzyme system.
-
6.
When optimizing condition to detect fluorescent signal from fluorescence reader, test suitable gain values to prevent overflow values during the kinetic analysis. We use Tecan Infinite M200 plate reader with a gain setting of 60.
-
7.
An identical protocol using lysates of HEK293 cells with transient overexpression of hMAGL or hABHD12 can be used to assess hMAGL or hABHD12 activity, respectively [5]. Furthermore, the assay format can, in principle, be adapted for hABHD6 produced in other host cell lines and different biological samples (tissue and cell membranes, cytosolic and nuclear samples), but elaborate validation is needed in each case to demonstrate that the assay specifically reports ABHD6 activity. This is because the relative expression of ABHD6 may be low in cell lines or tissues with endogenous expression of ABHD6, and multiple enzymes possessing MAG hydrolytic activity could contribute to the pool of glycerol formed.
-
8.
Although ABHD6 is predicted to be an integral membrane protein [4], we found no particular enrichment of ABHD6 activity in HEK293 cell membrane preparations when compared to lysates [5]. As only a fraction of cellular material is needed to produce the same amount of protein from lysates as compared to membranes, this economy aspect also favors the use of lysates in these assays. Lysates may sometimes contain material that is difficult to pipette after freezing. This DNA-containing material can be removed by sucking it into the pipette tip but after this step the protein content needs to be determined again.
-
9.
This point is relevant in the case that inhibitors are to be tested in the assay. If not, total number of wells as well as volumes of enzyme preparation (Table 1) and assay mix (Table 2) can be scaled down to fit the size of the experiment. On the other hand, for initial validation purposes it may be useful to construct dose-response curves and to determine IC50 values for inhibitors (e.g., MAFP, IDFP, HDSF, THL, RHC-80267, WWL70) that have been previously evaluated in this assay [5]. At the very least, we advice to include THL at the two concentrations (see Note 4 ). Noteworthy, it may be important to periodically test that for samples receiving the same treatment, fluorescence readings are uniform throughout the plate regardless of the position. Especially when pipetting cell lysates, the wells receiving the lysate mix last may contain slightly more enzymatic activity due to tendency of the lysates to sediment in the reservoirs. The assay is highly sensitive and therefore robustly reports inaccuracies in pipetting, especially those concerning the enzyme preparation. We recommend to test multiple DMSO-hABHD6-HEK samples distributed randomly throughout the plate, and to confirm uniform fluorescence readings for these samples to rule out the possibility that any gradient could exist.
-
10.
When dispending individual inhibitors, pipette (by touching the bottom of the well) 1 μl of compound in the middle of the well. The drop is readily visible to the naked eye, so this serves as a useful control step to check that all wells have received DMSO/inhibitor. DMSO is not readily evaporated, so this step is not rate limiting. However, use parafilm to protect the wells.
-
11.
We recommend to use inverse pipetting when adding the enzyme dilution and glycerol assay mix.
-
12.
When vortexing the plate after addition of the glycerol assay mix, use medium-speed settings to avoid splashing.
-
13.
Prepare in advance the fluorometer for the assay before starting the pipetting steps. This ensures that initial fluorescence readings (approximating time point 0 min) can be obtained without unnecessary delays immediately after the addition of the glycerol assay mix.
-
14.
We recommend to prepare an Excel sheet to facilitate calculations of the results.
References
Savinainen JR, Saario SM, Laitinen JT (2012) The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf) 204:267–276
Naydenov AV, Horne EA, Cheah CS et al (2014) ABHD6 blockade exerts antiepileptic activity in PTZ-induced seizures and in spontaneous seizures in R6/2 mice. Neuron 83:361–371
Thomas G, Betters JL, Lord CC et al (2013) The serine hydrolase ABHD6 is a critical regulator of the metabolic syndrome. Cell Rep 5:508–520
Blankman JL, Simon GM, Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356
Navia-Paldanius D, Savinainen JR, Laitinen JT (2012) Biochemical and pharmacological characterization of human α/β-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12). J Lipid Res 53:2413–2424
Van der Wel T, Janssen FJ, Baggelaar MP et al (2015) A natural substrate-based fluorescence assay for inhibitor screening on diacylglycerol lipase α. J Lipid Res 56:927–935
Acknowledgement
This work was supported by the Academy of Finland (Grant 139620 to J.T.L.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Savinainen, J.R., Navia-Paldanius, D., Laitinen, J.T. (2016). A Sensitive and Versatile Fluorescent Activity Assay for ABHD6. In: Maccarrone, M. (eds) Endocannabinoid Signaling. Methods in Molecular Biology, vol 1412. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3539-0_18
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3539-0_18
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3537-6
Online ISBN: 978-1-4939-3539-0
eBook Packages: Springer Protocols