
Abstract
Digitalis purpurea L. is one of the main economically viable sources of cardenolides (cardiac glycosides) for the pharmaceutical industry. Nevertheless, production of cardenolides in plants grown by traditional agriculture is not always an efficient process and can be affected by biotic and abiotic factors. This chapter provides two biotechnology strategies for biomass and cardenolide production in D. purpurea. Firstly, we report biomass production using a temporary immersion system (TIS), combined with cardenolide extraction and quantification. Secondly, an efficient protocol for genetic transformation via Agrobacterium tumefaciens is provided. These strategies can be used independently or combined in order to increase the content of cardiac glycosides in D. purpurea and to unravel biosynthetic pathways associated to cardiac glycoside production.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
Digitalis purpurea L. (foxglove ) is an important medicinal plant belonging to the family Plantaginaceae. Leaves of D. purpurea have been used as medicine for many centuries because of the presence of cardenolides , which can increase the force of systolic contractions and regulate heart rhythms in humans. Nowadays plants are still the sole source for commercial acquisition of cardenolide and only D. purpurea and D. lanata are of economic interest [1].
The contents of cardiotonic glycosides in Digitalis spp. obtained through traditional agriculture are generally low and strongly affected by climate, soil conditions, and genotype (reviewed in ref. [1]). For these reasons, many biotechnological strategies have been developed to enhance the production of biomass and valuable cardenolides from Digitalis. Earlier studies demonstrated the use of cell and tissue cultures of members of the Digitalis genus for cardenolide production, such as suspension culture s [2], embryogenic cell culture s [3], as well as root [4] or shoot culture s [5]. However, cardenolide content in such cultures is generally low.
Large-scale tissue culture has been considered as an alternative to the traditional methods of culture for the production of biochemicals. Bioreactors are designed for intensive culture and afford maximal opportunity for monitoring and controlling microenvironmental conditions [6]. However, traditional bioreactors are usually quite expensive and complex as regards cleaning, sterilization, inoculation, and harvesting [7, 8]. In order to reduce the costs, increase simplicity of handling, and improve growth and physiological state of the plant material, bioreactors have been designed based on the principle of temporary immersion in liquid medium [9, 10]. Because such temporary immersion system s (TIS) are suitable for large-scale culture of plant organs, they also represent an attractive alternative for production of plant secondary metabolites [11–13].
Genetic engineering is another alternative that could be used to improve cardenolide production both in vitro and in vivo . This tool can be used to unravel the metabolic pathway involved in cardenolide production. Moreover, through genetic engineering breeders can overexpress genes implicated in cardenolide biosynthesis or silence genes belonging to competitive pathways, resulting in the increase of cardiotonic glycosides.
This chapter describes two biotechnological approaches in Digitalis purpurea, firstly on the production of biomass and cardenolides employing TIS. The second part describes a detailed reproducible protocol for efficient Agrobacterium -mediated transformation, providing a fast and reliable tool for metabolic engineering of cardenolide production and genetic improvement of this species.
2 Material
2.1 Biomass and Cardenolide Production in Temporary Immersion Systems
2.1.1 Plant Material and Surface Sterilization
-
1.
Seeds of D. purpurea cv. Berggold as source of explants.
-
2.
70 % (v/v) ethanol.
-
3.
Sodium hypochlorite (NaOCl) with 5 % of active Cl.
-
4.
Graduated cylinders (1000 mL).
-
5.
50 mL Sterile tube or similar container for disinfection procedure.
-
6.
Flasks with sterile distilled or deionized water for rinses.
-
7.
Tissue culture facilities and tools (laminar flow cabinet, scalpel, forceps, tool sterilizer such as vertical autoclave and glass-bead sterilizer, culture room) and personal protective equipment (i.e., laboratory coat).
2.1.2 Culture Medium
-
1.
Medium based on Murashige and Skoog (MS) salts [14]; see medium formulations in Table 1.
Table 1 Murashige and Skoog (MS) basal medium with vitamins and growth regulators -
2.
Sucrose .
-
3.
Growth regulators: Indole-3-acetic acid (IAA) and 6-benzylaminopurine (6-BAP ).
-
4.
Gelrite .
-
5.
Potassium hydroxide (KOH) (0.5 N).
-
6.
Hydrochloric acid (HCl) (0.5 N).
-
7.
Vessels for plant tissue culture: 500 mL Polycarbonate containers.
-
8.
Tissue culture facilities and tools (technical and analytical balances, magnetic stirrer, pH meter, microwave oven or hot plate, refrigerator and −20 °C freezer, autoclave) and personal protective equipment (i.e., laboratory coat).
2.1.3 Temporary Immersion Systems
-
1.
TIS units of 1 L, comprising glass or plastic vessels with all components, are employed (see diagram in Fig. 1).
Fig. 1 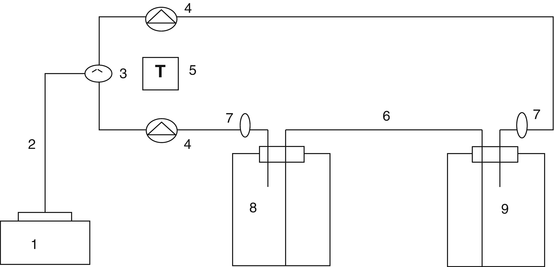
Diagram of the temporary immersion system showing the different components: (1) Air compressor; (2) reinforced PVC tubing (ID 10 mm); (3) pressure regulation station; (4) three-way solenoid electrovalve; (5) programmable timer; (6) autoclavable silicone tubing (ID 6 mm); (7) sterilizable filter (0.22 μm, Midisart 2000, Sartorius AG); (8) culture flask; (9) medium reservoir vessel
-
2.
Individual shoots (1.5–2.0 cm long).
-
3.
Tissue culture facilities and tools (laminar flow cabinet, scalpel, forceps, tool sterilizer such as vertical autoclave and glass-bead sterilizer, culture room) and personal protective equipment (i.e., laboratory coat, latex gloves).

2.1.4 Determination of Cardenolide Content in Shoots Cultured in TIS
2.1.4.1 Extraction Protocol
-
1.
Lyophilizer.
-
2.
Mortars and pestles.
-
3.
Balance.
-
4.
Ultrasonic bath.
-
5.
Micropipettes.
-
6.
Centrifuge.
-
7.
Rotary evaporator.
-
8.
Latex gloves.
-
9.
500 mL Separatory funnels.
-
10.
Funnels.
-
11.
Flasks with caps.
-
12.
50 and 500 mL evaporating flasks.
-
13.
250 mL Erlenmeyer flasks.
-
14.
100 and 500 mL graduated cylinders.
-
15.
Whatman No. 1 filter paper.
-
16.
50 mL Falcon tubes.
-
17.
Wash bottle with bi-distilled water.
-
18.
70 % (v/v) ethanol.
-
19.
Pb(CH2COOH)2·2H2O (15 % w/v).
-
20.
Na2HPO4·2H2O (10 % w/v).
-
21.
Chloroform/isopropanol (3:2 v/v).
-
22.
Absolute ethanol for HPLC analysis.
2.1.4.2 HPLC Analysis
-
1.
HPLC , Agilent 1100 equipped with vacuum degasser, quaternary pump, auto sampler, diode array detector, and an Inertsil ODS-3 column (150 × 4.6 mm; 5 μm).
-
2.
Acetonitrile/water (25:75 v/v).
-
3.
Authentic standards, HPLC -grade digoxin and digitoxin for calibration.
2.2 Agrobacterium tumefaciens-Mediated Genetic Transformation
2.2.1 Equipment, Instruments, and Solutions
-
1.
Micropipette and sterile 1.0 mL and 200 μL pipet tips.
-
2.
Sterile 9 cm Petri dishes.
-
3.
Sterile 9 cm diameter filter paper disks to remove excess Agrobacterium .
-
4.
Parafilm® to seal Petri dishes.
-
5.
Sterile 250 mL Erlenmeyer flasks.
-
6.
Sterile conical centrifuge tubes.
-
7.
50 mL Glass test tubes.
-
8.
Wire loop.
-
9.
Tissue culture chamber.
-
10.
Flasks with bi-distilled or deionized water.
-
11.
70 % (v/v) ethanol.
-
12.
Orbital shaker.
-
13.
Centrifuge.
-
14.
Spectrophotometer.
-
15.
Tissue culture facilities and tools (pH meter, vertical laminar flow cabinet, scalpel, forceps, tool sterilizer such as vertical autoclave and glass-bead sterilizer, culture room) and personal protective equipment (i.e., laboratory coat, latex gloves).
2.2.2 Plant Material
-
1.
Leaf segments (1.0 cm2 adaxial surface to the medium ) from in vitro plants (second to seventh subculture ).
2.2.3 Culture Medium for Bacterial Culture, Cocultivation, and Regeneration of Transformants
-
1.
Antibiotics: Spectinomycin, streptomycin, rifampicin, cefotaxime, timentin, geneticin (G-418).
-
2.
Acetosyringone (4-acetyl-2,6-dimethoxyphenol).
-
3.
Luria Broth (LB) medium and YEP liquid medium for bacterial culture.
-
4.
MS basal medium (Table 1) for inoculation medium, semisolid callus induction medium (CIM), and semisolid regeneration medium.
2.2.4 Agrobacterium tumefaciens Strain and Binary Vector
-
1.
Agrobacterium strain C58C1RifR containing the helper plasmid pMP90 [15].
-
2.
Binary vector pTJK136 [16] that contains an aminoglycoside adenyltransferase marker gene (aadA), which confers bacterial resistance to spectinomycin/streptomycin. Moreover a T-DNA with P35S-uidAint-nos (35S RNA promoter of the cauliflower mosaic virus, β-glucuronidase gene with potato st-ls1 intron, nopaline synthase gene terminator) and Pnos-nptII-ocs (nopaline synthase gene promoter, neomycin phosphotransferase gene, octopine synthase gene terminator) as chimeric plant screenable and selectable marker genes, respectively (Fig. 3a).
2.3 Histochemical β-Glucuronidase Assays
-
1.
X-Gluc solution for histochemical analysis [17]: 1 mM 5-bromo-4-chloro-3-indolyl-β-D-glucuronide (X-Gluc) in 0.1 mM phosphate buffer (pH 7.0) containing 10 mM ethylene diamine tetraacetic acid (EDTA), 0.5 mM potassium ferricyanide, 0.5 mM potassium ferrocyanide, and 0.1 % Triton X-100. Store at −20 °C.
-
2.
70 % (v/v) ethanol.
-
3.
Micropipette and sterile 1.0 mL pipet tips.
-
4.
Sterile conical centrifuge tubes.
-
5.
Sterile scalpels and forceps.
-
6.
Incubator.
2.4 Molecular Analysis
2.4.1 DNA Isolation from Transformed Tissue
-
1.
Sterile mortars and pestles.
-
2.
Liquid nitrogen.
-
3.
Eppendorf tubes.
-
4.
Micropipettes and tips.
-
5.
Gloves.
-
6.
Extraction buffer: 10 mM Tris–HCl pH 8.0, 1.4 M NaCl pH 8.0, 20 mM ethylene diamine tetraacetic acid (EDTA), 4 % (w/v) cetyltrimethylammonium bromide (CTAB), 2 % (w/v) polyvinylpyrrolidone (PVP), 10 mM β-mercaptoethanol. Store at room temperature.
-
7.
TAE buffer (50×): Tris (242 g), glacial acetic acid (57.1 mL), EDTA (0.5 M, pH 8.0, 100 mL), adjust pH to 8.0 and make up volume.
-
8.
Agarose molecular grade.
-
9.
TE buffer (10×): 100 mM Tris, 10 mM EDTA, pH 8.0 (with HCl).
-
10.
24:1 Chloroform-isoamyl alcohol.
-
11.
70 % (v/v) ethanol.
-
12.
RNase solution.
-
13.
Sodium acetate 3 M.
-
14.
Water bath (set at 55 °C).
-
15.
Vortexer.
-
16.
Fume hood.
-
17.
Spectrophotometer.
2.4.2 Polymerase Chain Reaction
-
1.
For the polymerase chain reaction (PCR ) mix: Sterile bi-distilled water, PCR buffer mix, dNTPs, forward primer, reverse primer, Taq DNA polymerase, MgCl2 solution, DNA template.
-
2.
Agarose molecular grade.
-
3.
200 μL PCR tubes.
-
4.
Crushed ice, ice bucket.
-
5.
Micropipettes and tips.
-
6.
Gloves.
-
7.
Thermal cycler.
2.4.3 Southern Hybridization
-
1.
SacII, store enzyme at −20 °C.
-
2.
Agarose molecular grade.
-
3.
Nylon membrane: Hybond N+ (GE Healthcare).
-
4.
Labeled probe: PCR amplification of the 488 bp nptII gene fragment is labeled with DIG-dUTP using a DIG-High Prime DNA Labeling and Detection Kit.
-
5.
Micropipettes and tips.
-
6.
X-ray film, cassette to hold X-ray film, X-ray film developer, clingfilm.
-
7.
Gloves.
-
8.
Others: Materials for Southern hybridization analysis according to standard procedures [18].
3 Methods
3.1 Biomass and Cardenolide Production in Temporary Immersion Systems
3.1.1 Plant Material, Surface Sterilization, and Culture Conditions
-
1.
Add distilled or deionized water up to ½ the final medium volume in a beaker and mix each component of MS basal medium and supplements (Table 1). MS basal medium is supplemented with 30 g/L sucrose for germination phase. For multiplication phase, this medium is also supplemented with vitamins and growth regulators (Table 1) and named MMS. Growth regulators are added to the culture medium prior to sterilization (see Note 1 ). Mix the solution until all components are dissolved. Bring to the final volume with distilled or deionized water. Adjust pH to 5.8 with 0.5 N KOH or 0.5 N HCl.
-
2.
Add 3.0 g/L Gelrite , heat until gelling agent is fully dissolved before autoclaving at 1.2 kg/cm2 and 121 °C for 20 min or add Vitrofural® as chemical sterilization (see Note 2 ), and dispense into autoclavable containers. Use 70 mL in each 500 mL culture vessel for germination and multiplication phases. Store the medium in a clean and dark area at 4 °C. Use within 2 weeks.
-
3.
Sterilize all instruments and the laminar flow cabinet before use.
-
4.
In the laminar flow cabinet, place the seeds (Fig. 2a) in tubes with an aqueous solution of sodium hypochlorite (NaOCl) with 5 % of active Cl and shake during 5 min.
Fig. 2 
In vitro propagation of Digitalis purpurea, (a) seeds as explant source, disinfected with 5 % NaOCl during 5 min, (b) seedlings after 15 days growing on MMS culture medium supplemented with 1.0 mg/L 6-BAP and 0.1 mg/L IAA, (c) in vitro plants ready for subculturing after 28 days on MMS culture medium , (d) 1.5–2.0 cm long individual shoots, (e) temporary immersion system used for shoot multiplication , (f) hyperhydric shoots, (g) plantlets growing on TIS under 2 min of immersion every 4 h, (h) harvested biomass from TIS after 28 days
-
5.
Rinse seeds three times with sterile distilled or deionized water (see Note 3 ).
-
6.
Culture sterile seeds on an MS basal medium and incubate at 27 ± 2 °C under 16-h photoperiod and 125–150 μmol/m2/s photosynthetic photon flux supplied by cool white fluorescent lamps (see Note 4 ).
-
7.
Grow seedlings (Fig. 2b) on MMS culture medium until they have attained 2–4 cm length under the same culture conditions as of step 6. Leaves and roots are eliminated. The plantlets (Fig. 2c) are subcultured every 28 days. Divide shoots individually, cut shoots approximately in 1.5–2.0 cm length (Fig. 2d), and use for multiplication on semisolid medium and TIS. Discard any contaminated shoots.

3.1.2 Temporary Immersion Systems
-
1.
Prepare TIS units (Fig. 2e) and connect all components as shown in Fig. 1.
-
2.
Prior to sterilization, add to each TIS 250 mL MMS supplemented as described (Subheading 3.1.1, step 1) without Gelrite (see Note 5 ).
-
3.
Check functioning of TIS before inoculation. Program the timer for some parameters like time and immersion frequency (2 min every 4 h) (see Note 6 ).
-
4.
Inoculate 12 individual shoots (1.5–2.0 cm long) per TIS in a laminar flow cabinet.
-
5.
Reconnect TIS to the installation in a culture room at 27 ± 2 °C under 16-h photoperiod and 125–150 μmol/m2/s photosynthetic photon flux supplied by cool white fluorescent lamps.
-
6.
Collect biomass after 28 days (Fig. 2g, h), rinse with distilled water, and thereafter blot dry on filter paper.
-
7.
Suggested parameters to evaluate: number of hyperhydric shoots, water content (see Note 7 ), length and the number of shoots per TIS, and fresh and dry weights (g) produced per TIS, which represent biomass production (see Notes 8 and 9 ).
3.1.3 Cardenolide Content
-
1.
Freeze-dry biomass in a lyophilizer (see Note 10 ).
-
2.
Finely grind dried in vitro shoots in a mortar.
-
3.
Extract powdered plant material sample (1.5 g) with 15 mL ethanol (70 %, v/v) in an ultrasonic bath at 70 °C for 15 min.
-
4.
After cooling to room temperature add 25 mL bi-distilled water.
-
5.
Add 10 mL lead acetate (15 %) and divide extract in two 50 mL tubes. Rinse flask twice with bi-distilled water.
-
6.
Centrifuge at 3000 × g for 5 min.
-
7.
Filter samples using Whatman No. 1 filter paper and rinse filter twice with bi-distilled water in Erlenmeyer flask.
-
8.
Add 12 mL Na2HPO4 (10 %), and dispense the extract in four tubes.
-
9.
Centrifuge at 3000 × g for 5 min.
-
10.
Filter samples using Whatman No. 1 filter paper and pass extract to a separatory funnel.
-
11.
Add 30 mL chloroform/isopropanol (3/2) and mix.
-
12.
Extract the organic phase to 500 mL evaporating flask (first extraction).
-
13.
Add 20 mL chloroform/isopropanol (3/2) to extract in separatory funnel and mix. Extract the organic phase and repeat again, and mix fractions (see Note 11 ).
-
14.
Evaporate under reduced pressure using a rotary evaporator equipped with a water bath held at 40 °C to approximately 1.0 mL, then rinse evaporating flask several times with warm ethanol, transfer the extract to a 50 mL evaporating flask, and evaporate again.
-
15.
Dissolve solid residue with 1.0 mL warm ethanol (ultrasonic bath) and transfer to a vial (1.5 mL) for HPLC analysis.
-
16.
Store prepared samples at 4 °C.
-
17.
Prepare a calibration curve by injecting standards of several known concentrations, digoxin (0–256 μg/mL) and digitoxin (0–625 μg/mL). Plot peak area against concentration.
-
18.
Inject 10 μL of sample in Agilent 1100 HPLC system.
-
19.
Use acetonitrile/water (25/75; v/v) mixture as eluent at 1.5 mL/min flow rate. Carry out measurements at 40 °C (see Note 12 ).
-
20.
Detect glycosides at 220 nm wavelength. Compare UV spectra with authentic commercially available standards and identify digitoxin and digoxin on the basis of their retention time.
3.2 Agrobacterium tumefaciens-Mediated Genetic Transformation
3.2.1 Solutions and Culture Media
-
1.
Spectinomycin: Prepare stock solution in bi-distilled water by adding 100 mg/mL. Filter sterilize and store in 1.0 mL aliquots at −20 °C.
-
2.
Streptomycin: Prepare stock solution in bi-distilled water by adding 300 mg/mL. Filter sterilize and store in 1.0 mL aliquots at −20 °C.
-
3.
Rifampicin: Prepare stock solution in bi-distilled water by adding 50 mg/mL. Filter sterilize and store in 1.0 mL aliquots at −20 °C.
-
4.
Timentin (a 15:1 mixture of ticarcillin and clavulanic acid): Prepare stock solution in bi-distilled water by adding 200 mg/mL. Filter sterilize and store in 1.0 mL aliquots at −20 °C.
-
5.
Geneticin (G-418): Prepare stock solution in bi-distilled water by adding 50 mg/mL. Filter sterilize and store in 1.0 mL aliquots at −20 °C. Light sensitive, consequently geneticin-containing medium should be stored in the dark.
-
6.
Cefotaxime: Prepare stock solution in bi-distilled water by adding 500 mg/mL. Filter sterilize and store in 1.0 mL aliquots at −20 °C.
-
7.
Acetosyringone (AS): Prepare stock solution in dimethylsulfoxide at 500 mM (e.g., 0.098 g in 1.0 mL); there is no need to sterilize but solution should always be prepared freshly in a laminar flow.
-
8.
Luria Broth (LB) medium : Dissolve 10 g tryptone, 10 g of yeast extract, and 10 g NaCl in 900 mL of bi-distilled water. Make the volume up to 1.0 L with bi-distilled water. Adjust the pH to 7.5 and add 15 g/L of agar . Supplement with the appropriate antibiotics (100 mg/L spectinomycin and 300 mg/L streptomycin) after cooling the autoclaved medium to 50–55 °C. Dispense 25 mL in sterile 9 cm diameter Petri dishes.
-
9.
YEP liquid medium : Dissolve 5 g/L NaCl, 10 g/L peptone , and 10 g/L yeast extract in bi-distilled water. Adjust pH to 7.5. Supplement with the appropriate antibiotics (100 mg/L spectinomycin and 300 mg/L streptomycin) after cooling the autoclaved medium to 50–55 °C. Dispense 3.0 mL into 15 mL culture tubes and 50 mL into sterile 250 mL Erlenmeyer flasks, according to the scale of the experiment.
-
10.
Inoculation medium : Dissolve 100 % MS salts (Table 1), 20 g/L sucrose , 1.98 g/L D(+)-glucose, and 3.9 g/L 2-[N-morpholino]ethane sulfonic acid (MES) in bi-distilled water and adjust pH to 5.5 before autoclaving. Supplement with 200 μM AS just before inoculation.
-
11.
Semisolid callus induction medium (CIM): Dissolve 100 % MS salts (Table 1), 4.0 mg/L thiamine HCl, 100 mg/L myoinositol, 30 g/L sucrose , and 3.0 g/L Gelrite in bi-distilled water. Adjust pH to 5.8 before autoclaving.
-
12.
Semisolid regeneration medium : Dissolve 100 % MS salts (Table 1), 0.57 μM IAA, 4.4 μM BAP , 30 g/L sucrose , and 2.5 g/L Gelrite . Adjust pH to 5.7 before autoclaving.
3.2.2 Growth of Agrobacterium Cultures and Cocultivation
3.2.2.1 Isolation of Single Bacterial Colonies from a Glycerol Stock
-
1.
Using a 200 μL pipet tip, scratch a small amount from the surface of the frozen glycerol bacterial stock (see Note 13 ) and drop onto the surface of the selective semisolid LB plate. Place back the frozen bacterial stock immediately into the freezer to avoid thawing.
-
2.
Using a flamed and cooled wire loop, streak cells across the selective semisolid LB plate to spread bacteria.
-
3.
Incubate the inoculated selective LB plate upside-down at 28 °C until single colonies are visible.
3.2.2.2 Inoculation with Agrobacterium and Cocultivation
-
1.
Select 2–3 isolated single colonies from the plate using sterile pipet tips.
-
2.
Inoculate one colony per 15 mL tube containing YEP medium supplemented with 100 mg/L rifampicin, 100 mg/L spectinomycin, and 300 mg/L streptomycin.
-
3.
Grow Agrobacterium cultures overnight on an incubating shaker set at 200 rpm and 28 °C. Approximate OD600nm at harvest: 1.5–2.0.
-
4.
Collect 50 μL of grown culture and inoculate 250 mL Erlenmeyer flask containing YEP medium supplemented with 100 mg/L spectinomycin and 300 mg/L streptomycin.
-
5.
Grow Agrobacterium culture overnight on an incubating shaker set at 200 rpm and 28 °C. Approximate OD600nm at harvest: 1.2–1.5.
-
6.
Pellet bacterial cells by centrifugation for 10 min at 3200 × g.
-
7.
Discard supernatant and gently wash the pellet once with inoculation medium described above (Subheading 3.2.1, step 10).
-
8.
Discard washing medium and gently resuspend the pellet in inoculation medium. Measure the OD600 nm and adjust with the above medium to 0.7 units (see Note 14 ).
-
9.
Place 30–40 leaf discs (1 cm2) in capped 50 mL disposable polypropylene conical centrifuge tube containing 30–40 mL of Agrobacterium suspension (see Note 15 , Fig. 3b).
Fig. 3 
Agrobacterium tumefaciens-mediated genetic transformation of Digitalis purpurea L. (a) T-DNA region of a pTJK136 [16], LB and RB, left border and right border, respectively; P35S, cauliflower mosaic virus 35S RNA promoter; uidA-intron, intron-interrupted β-glucuronidase gene; Pnos and Tnos, nopaline synthase gene promoter and polyadenylation signal; nptII, neomycin phosphotransferase gene; Tocs, octopine synthase gene polyadenylation signal, (b) inoculated leaf segments in tubes containing Agrobacterium suspension, (c) transient GUS expression in a leaf inoculated with C58C1RifR (pMP90) (pTJK136), (d) untransformed leaf, (e) callus formation from untransformed leaf on CIM without antibiotics, (f) callus formation from transformed leaf segment on CIM with 70 mg/L G-418, (g) stable GUS expression in transformed callus, (h) callus from untransformed segments without GUS-positive spots, (i) regenerated plants from transformed segments. Stable GUS expression in transformed (j) plantlets and (k) leaf
-
10.
Incubate tubes containing Agrobacterium suspension and leaf segments at room temperature for 15 min with gentle manual agitation every 2 min.
-
11.
Take away leaf segments from Agrobacterium suspension and remove excess bacteria from explants by blotting them on sterile filter paper using sterile forceps (see Note 16 ).
-
12.
Transfer leaf segments (adaxial surface to the medium , five segments per plate) to cocultivation on CIM (Subheading 3.2.1, step 11) supplemented with 200 μM AS. Incubate plates in the dark at 21 °C for 5 days.

3.2.3 Histochemical β-Glucuronidase Assays
Transient GUS expression is determined in leaf segments inoculated with Agrobacterium strain.
-
1.
After cocultivation, dip untransformed control and transgenic leaves into X-Gluc solution and incubate at 37 °C in the dark for 6 h or overnight.
-
2.
After incubation, remove X-Gluc solution, add the same volume of 70 % (v/v) ethanol, and incubate overnight to remove chlorophyll and other pigments prior to visual analysis and photographing. Note blue coloration indicating transient expression of the uidA gene (Fig. 3c, d).
3.2.4 Selection and Regeneration of Transformants
-
1.
Transfer inoculated leaf segments into 50 mL centrifugation tubes; rinse twice with 30–40 mL sterile liquid CIM containing 200 mg/L timentin and 500 mg/L cefotaxime.
-
2.
Blot leaf segments dry on sterile filter paper.
-
3.
Place leaf segments onto CIM containing 70 mg/L G-418 as selection agent and 200 mg/L timentin to inhibit Agrobacterium growth (see Note 17 ).
-
4.
Incubate selection plates for 8–12 weeks at 27 ± 2 °C in the dark with subculture to fresh selective plates every 2 weeks until calli form (Fig. 3e, f).
-
5.
Perform histochemical β-glucuronidase assay of calli as described above (Subheading 3.2.3, Fig. 3g). Non-transgenic calli are simultaneously stained in the same way (Fig. 3h).
-
6.
Transfer the individual putatively transformed callus induced on selection medium to 9 cm Petri dishes containing regeneration medium described above (Subheading 3.2.1, step 12) and keep in a growth chamber at 27 ± 2 °C under 16-h photoperiod and 70 μmol/m2/s photosynthetic photon flux supplied by cool white fluorescent lamps.
-
7.
Transfer differentiated shoots individually (Fig. 3i) to sterile 50 mL glass test tubes containing MMS medium .
-
8.
Stable GUS expression in regenerated plants is assessed as described above (Subheading 3.2.3, Fig. 3j, k). Non-transgenic regenerated plants are simultaneously stained in the same way.
3.3 Molecular Analysis
3.3.1 DNA Isolation from Transformed Tissue
Total DNA is extracted following the protocol described by Khayat et al. [19] with minor modifications.
-
1.
Take 1 g leaf tissue from a transformed Digitalis plant. Grind tissue to fine powder in a mortar with liquid nitrogen.
-
2.
Add 10 mL of extraction buffer (Subheading 2.4.1, step 6) per gram of tissue and shake vigorously using vortexer (see Note 18 ).
-
3.
Incubate the mixture at 55 °C for 30 min with occasional inversion.
-
4.
Cool the mixture on ice for 5 min.
-
5.
Centrifuge at 3200 × g, for 5 min at 4 °C. Transfer the supernatant to a new tube.
-
6.
Remove RNA by adding RNase to a final concentration of 200 μg/mL and incubate at 37 °C for 15 min.
-
7.
Add an equal volume of chloroform-isoamyl alcohol (24:1) to the above mixture and separate the two phases by centrifuging at 12,857 × g for 10 min at 4 °C.
-
8.
Transfer the upper aqueous phase to a fresh tube.
-
9.
Add an equal volume of isopropanol and mix gently by inversion. Incubate for 60 min at −80 °C or overnight at −20 °C.
-
10.
Pellet genomic DNA by centrifugation at 18,500 × g for 15 min at 4 °C.
-
11.
Wash the pellet once with 1.0 mL 70 % (v/v) ethanol.
-
12.
Centrifuge at 18,500 × g for 15 min at 4 °C and discard ethanol.
-
13.
Vacuum dry pellet for 5 min and dissolve in 30 μL of TE buffer (1×).
-
14.
Store at −20 °C.
-
15.
Check DNA by agarose gel electrophoresis and quantify 1:10 dilutions of DNA in spectrophotometer at 260/230 and 260/280 nm. DNA can be used for either PCR or Southern hybridization analysis.
3.3.2 PCR
PCR is performed using genomic DNA of each plant as a target. pTJK136 plasmid isolated from Escherichia coli DH5α-pTJK136 strain is used as positive control. Bacterial culture is grown overnight on an incubating shaker set at 200 rpm and 37 °C. Plasmid DNA is isolated using Purification kit Wizard plus SV Minipreps.
The primer sequences for analyzing tissues transformed with nptII and uidA genes are given in Table 2.
-
1.
Fill out a PCR worksheet with the sample identifiers of all samples to be used.
-
2.
Label PCR tubes with sample identifier numbers from the PCR worksheet.
-
3.
Place PCR tubes in the freezer rack to keep cold.
-
4.
Calculate the amount of each component needed for the total number of samples taking into account that PCR amplification reactions are carried out in 25 μL of total volume (see Note 19 ).
-
5.
Close caps of all tubes firmly and place the tubes into the thermal cycler.
-
6.
Start the program (denaturation at 94 °C for 3 min, followed by 35 cycles of 94 °C for 30 s, 65 °C for 30 s, and 72 °C for 30 or 60 s, with a final extension at 72 °C for 10 min).
-
7.
After the PCR is complete, analyze amplified fragments by electrophoresis at 100 V for 1 h on 1.0 % Tris-acetate-EDTA agarose gel followed by staining in ethidium bromide (5 μg/mL) and detection and photography under UV illumination. An nptII- and uidA-specific band of 488 bp and 1031 bp, respectively, is observed in the transformed plants while being absent in the untransformed control plants and water (Fig. 4a, see Note 20 ).
Fig. 4 
Molecular analysis of putative transformed Digitalis purpurea L. plantlets. (a) PCR analysis for the nptII and uidA genes in transgenic tissues, lane 1–13 genomic DNA from putative transgenic lines obtained with C58C1RifR (pMP90) (pTJK136), (−) untransformed control, (+) pTJK136 plasmid control, MW molecular weight marker Gene Ruler™ DNA Ladder Mix (Fermentas). (b) Southern hybridization of putative transformed D. purpurea L. PCR-positive plantlets, lanes 1–6 genomic DNA from putative transgenic lines obtained with C58C1RifR (pMP90) (pTJK136), (−) untransformed plant, (+) pTJK136 plasmid control, MW digoxigenin-labeled DNA molecular weight marker VII (Roche)

3.3.3 Southern Hybridization
The integration of the T-DNAs of randomly selected PCR -positive lines is analyzed by Southern blot hybridization (see Fig. 4b).
-
1.
Digest 20 μg of genomic DNA with SacII.
-
2.
Separate digested genomic DNA samples on a 0.8 % Tris-acetate-EDTA agarose gel (w/v) run at 25 V for 12 h.
-
3.
Blot DNA to a Hybond-N+ nylon membrane (RPN203B; GE Healthcare) by upward capillarity forces using 20× SSC buffer (3 M NaCl, 0.3 M sodium citrate, pH 7.0) overnight at room temperature.
-
4.
Label the 488-bp nptII gene fragment, amplified from plasmid pTJK136 with the same primers mentioned above (Table 2), with DIG-dUTP using a DIG-High Prime DNA Labeling and Detection Kit.
-
5.
Perform hybridization, washing, and detection according to the manufacturer’s instructions.
-
6.
Expose membrane to X-ray film in the presence of an intensifying screen for 30 min at room temperature.
4 Notes
-
1.
BAP : Stock solution at 1 mM. Dissolve 22.52 mg BAP in a few drops of 1 M sodium hydroxide, dilute to 100 mL with distilled or deionized water, and store at 4 °C. IAA: Stock solution at 1 mM. Dissolve 8.76 mg IAA in a few drops of 1 M sodium hydroxide, dilute to 50 mL with distilled or deionized water, and store at 4 °C.
-
2.
It is possible to use chemical sterilization with Vitrofural® (2-bromo-5-(2-bromo-2-nitrovinyl)-furan) 114 mg/L. Firstly, boil full medium to dissolve Gelrite (3.0 g/L), then dissolve this compound into 1/10 part of hot medium (~90 °C), and immediately add to the rest boiled medium. This compound is used by adding in culture vessel s for seed germination and shoot multiplication on semisolid medium. All the culture vessels used in chemical sterilization with Vitrofural® are previously rinsed in sodium hypochlorite solution at 0.05 % (v/v). Store the medium in a clean area under dark conditions at least 3 days before use. Vitrofural® has therapeutic effect [20] and has allowed the substitution of the autoclave in the sterilization of culture medium used for the micropropagation of banana, plantain, potato, and sugarcane [21, 22]. However, this antimicrobial compound has never been used in Digitalis culture. For shoot multiplication in TIS, the use of Vitrofural® is not convenient taking into account that TIS has several components and that it is very difficult to completely sterilize all of them. On the other hand, previous studies have shown that Digitalis leaves are very sensitive to direct contact with Vitrofural® when liquid medium is used (unpublished data).
-
3.
Be careful, Digitalis seeds are very small and it is necessary to use a filter when they are rinsed. It is possible to obtain less than 1 % of contamination with this simple disinfection protocol.
-
4.
After 15 days, seed germination rate is more than 80 %. In a tropical location it is possible to incubate seeds and seedlings under natural light conditions, photoperiod 13/11 h and 20–45 μmol/m2/s on average.
-
5.
Employ physical sterilization for TIS, and autoclave at 1.2 kg/cm2 and 121 °C for 30 min. Prepare TIS carefully before autoclaving; all components are rinsed in sodium hypochlorite solution at 0.05 % (v/v).
-
6.
Duration and frequency of immersions affect nutrient supply, composition of the internal atmosphere in the culture vessel , and occurrence of hyperhydricity [9]. The latter phenomenon concerns morphological, anatomical, and physiological disorders. Shoots are categorized as hyperhydric shoots according to their external appearance [23–25] (Fig. 2f shows the morphology of hyperhydric shoots). Hyperhydric shoots appear turgid, watery at their surface, and hypolignified, in some cases less green and easily breakable. In D. purpurea, the best biomass production (values for fresh (106.2 g) and dry weight (5.82 g) per 1 L flask) and lowest percentage of hyperhydricity were obtained using 2 min immersion every 4 h [26].
-
7.
Dry weight is recorded after the biomass is dried to constant weight at 60 °C. The water content is an important variable to evaluate because it shows the shoot quality and allows determining favorable conditions to avoid hyperhydricity . Water content (WC) is calculated as [27]
$$ \mathrm{W}\mathrm{C}\%=\frac{\left(\mathrm{F}\mathrm{W}-\mathrm{D}\mathrm{W}\right)}{\mathrm{FW}}\times 100 $$ -
8.
Since cardenolide biosynthesis is basically dependent on morphological differentiation [28], there are attempts to use organ culture, which is easily produced in TIS. The protocol described for D. purpurea biomass production in TIS is also applicable for biomass and cardenolide production in D. lanata shoots. Yields are different because of genotype influence. The most important reason for the efficacy of TIS is that they combine ventilation of the plant tissues and intermittent contact between the entire surface of the tissue and the liquid medium [9], which results in increased growth rates and biomass production.
-
9.
Elicitation is one of the most effective methods to enhance the production of several secondary metabolites from medicinal plant s [29]. Our results suggest that elicitation of Digitalis shoots cultivated in TIS or semisolid medium could influence the competition between biomass and secondary metabolite production [30, 31]. Elicitors as Chitoplant® and Silioplant® are effective to increase cardenolide content in shoots of D. purpurea and D. lanata. In D. lanata, the highest accumulation of lanatoside C was achieved with Chitoplant® (0.1 g/L), which resulted in 316 μg/g-DW, and with Silioplant® (0.01 g/L) giving 310 μg/g-DW; this accounted for a 2.2-fold increase in lanatoside C content compared to non-elicited shoot culture s in TIS [30]. The optimization of elicitor treatment may improve TIS performance. Also, SilioPlant® (0.01 g/L) did not affect biomass production and at the same time increased 3.6-fold and 6.9-fold the digoxin and digitoxin content, respectively, in D. purpurea shoots cultured in semisolid medium [31]. Elicitors are dissolved in distilled or deionized water and added to the culture medium before sterilization. A set of control cultures without elicitors could be included.
-
10.
Alternatively, dry plant material in an incubator at 60 °C during 36 h until constant weight. This drying method is less convenient due to the loss of some organic compounds during the process, although not in significant amounts.
-
11.
Due to the minor contents of pharmaceutical agents we extracted only twice with the 20 mL mixture. If there are high contents, the extraction could be repeated. In order to eliminate water, Na2SO4 could be added, mix it by hand, centrifuge at 11,500 × g for 1 min, and pass the organic phase to new flask. The solution should be clear.
-
12.
The gradient elution profile is 4 → 34 min, A (acetonitrile) = 25 %; 34 → 45 min, A = 37 %; 45 → 60 min, A = 50 %; 60 → 65 min, A = 25 %.
-
13.
Bacterial stocks are maintained in 20 % (v/v) glycerol-containing growing medium with appropriate antibiotics and stored at −80 °C.
-
14.
More dense bacterial cultures may cause overgrowth during cocultivation.
-
15.
Include extra control samples for the transformation and selection process and for assessing transient reporter gene expression. In order to control the selection process place untransformed leaf segments on antibiotic -free CIM as well as on a medium supplemented with both geneticin and timentin. Finally, include several control samples, which will not be infected with Agrobacterium and are cultured on nonselective medium to assess the callus formation and regeneration capacity and to provide untransformed control plants for later analyses.
-
16.
Sterilize paper towels by wrapping in aluminum foil and autoclaving for 20 min at 121 °C.
-
17.
The selection agent used will depend on the selectable marker gene (SMG) present on the T-DNA of the binary vector. Only those plant cells containing the T-DNA harboring the SMG will survive in the presence of the selection agent to which the marker provides resistance. The nptII gene is one of the most commonly used selectable markers; it confers resistance to kanamycin, geneticin (G-418), and paromomycin. For callus induction in D. purpurea geneticin can be used as selective agent at 70 mg/L. Geneticin is light sensitive, so store and incubate selective CIM in the dark. The timentin is incorporated into the medium to kill the Agrobacterium . At a concentration of 200 mg/L, timentin is effective in eradicating several Agrobacterium strains (e.g., C58C1RifR (pMP90), LBA4404, EHA101, EHA105) without evident damage to plant cells.
-
18.
Add β-mercaptoethanol in fume hood just before use.
-
19.
PCR amplification reactions contain 200 ng genomic DNA, 0.5 μM of each primer, 1.5 mM MgCl2, 200 mM dNTP, 1X Taq polymerase reaction buffer, and 0.25 U Taq polymerase. Prepare a master mix without template DNA in a single tube, which can then be aliquoted into individual tubes (number of samples +1, components are provided in excess of the required amount to minimize the possibility of pipetting errors and to save time). Be sure to mix the PCR master mix well before aliquoting it into the sample tubes. Template DNA and primers are added to individual samples. Include a negative control (PCR tube with water instead of genomic DNA) to check contamination. Prepare each sample twice (one for each pair of primers used for both genes: nptII and uidA).
-
20.
A few non-transgenic cells are protected from the selection agent by the high number of calli obtained per leaf segment and escapes occurred. With this protocol, only four GUS-negative lines obtained with strain C58C1RifR (pMP90) showed neither uidA nor nptII genes (4.6 % escapes). Perhaps the increase of selection pressure (e.g., by applying an antibiotic gradient) could prevent this problem. As observed in Fig. 4a, one GUS-negative line (lane 2) did not contain the uidA gene but the nptII gene was present. These results indicate the integration of a truncated T-DNA. All transgenic plants analyzed that were positive for the presence of the uidA gene in the PCR assay also showed constitutive GUS expression in leaves, indicating the presence of a full functional transgene.
References
Sales E, Müller-Uri F, Nebauer SG, Segura J, Kreis W, Arillaga I (2011) Digitalis. In: Kole C (ed) Wild crop relatives: genomic and breeding resources plantation and ornamental crops. Springer, Berlin Heidelberg, pp 73–112
Kreis W, May U, Reinhard E (1986) UDP-Glucose: digitoxin 16’-O-glucosyltransferase from suspension cultured Digitalis lanata cells. Plant Cell Rep 5:442–445
Lindemann P, Luckner M (1997) Biosynthesis of pregnane derivatives in somatic embryos of Digitalis lanata. Phytochemistry 46:507–513
Shimomura K, Yoshimatsu K, Sauerwein M, Christen P, Toda Y, Aoki T (1992) Production of biologically active compounds by transformed cultures. In: Oono R, Hirabayashi T, Kiruchi S, Handa H, Kahwara S (eds) Plant tissue culture and gene manipulation for breeding and formation of phytochemicals. National Institute of Agrobiological Resources, Tsukuba, Japan, pp 293–296
Hagimori M, Matsumoto T, Obi Y (1983) Effects of mineral salts, initial pH and precursors on digitoxin formation by shoot-forming cultures of Digitalis purpurea L. grown in liquid medium. Agri Biol Chem 47:565–571
Paek KY, Chakrabarty D, Hahn EJ (2005) Application of bioreactor systems for large scale production of horticultural and medicinal plants. Plant Cell Tiss Org Cult 81:287–300
Takayama S, Akita M (2005) Practical aspects of bioreactor application in mass propagation of plants. In: Hvoslef-Eide AK, Preil W (eds) Liquid culture systems for in vitro plant propagation. Springer, Dordrecht, pp 61–78
Jiménez E (2005) Mass propagation of tropical crops in temporary immersion system. In: Hvoslef-Eide AK, Preil W (eds) Liquid culture systems for in vitro plant propagation. Springer, Dordrecht, pp 197–211
Berthouly M, Etienne H (2005) Temporary immersion system: a new concept for use liquid medium in mass propagation. In: Hvoslef-Eide AK, Preil W (eds) Liquid culture systems for in vitro plant propagation. Springer, Dordrecht, pp 165–195
Georgiev V, Schumann A, Pavlov A, Bley T (2014) Temporary immersion systems in plant biotechnology. Eng Life Sci 14:607–621
Quiala E, Barbón R, Jiménez E, de Feria M, Chávez M, Capote A, Pérez-Alonso N (2006) Biomass production of Cymbopogon citratus (DC) Stapf., a medicinal plant, in temporary immersion systems. In Vitro Cell Dev Biol-Plant 42:298–300
Sankar-Thomas YD, Lieberei R (2011) Camptothecin accumulation in various organ cultures of Camptotheca acuminata Decne grown in different culture systems. Plant Cell Tiss Org Cult 106:445–454
Schumann A, Berkov S, Claus D, Gerth A, Bastida J, Codina C (2012) Production of galanthamine by Leucojum aestivum shoots grown in different bioreactor systems. Appl Biochem Biotechnol 167:1907–1920
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Koncz C, Schell J (1986) The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimeric genes carried by a novel type of Agrobacterium binary vector. Mol Gen Genet 204:383–396
Kapila J, De Rycke R, Van Montagu M, Angenon G (1997) An Agrobacterium-mediated transient gene expression system for intact leaves. Plant Sci 122:101–108
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98:503–517
Khayat E, Duvdevani A, Lehav E, Ballesteros BA (2004) Somaclonal variation in banana (Musa acuminata cv. Grande Naine). Genetic mechanism, frequency, and application as a tool for clonal selection. In: Jain SM, Swennen R (eds) Banana improvement: cellular, molecular biology, and induced mutation. Science Publishers Inc, Plymouth, UK, pp 99–109
Blondeau JM, Castañedo N, González O, Medina R, Silveira E (1999) In vitro evaluation of G-1: a novel antimicrobial compound. Int J Antimicrob Agents 11:163–166
Quiala E, Jimenez E, Feria M, Alvarado Y, Chávez M, Agramonte D, Ramírez D, Acosta M, Pérez N, Capote A (2002) Empleo del Vitrofural en la esterilización química del endospermo artificial de los embriones encapsulados de Saccharum spp. Híbrido var Cuba 87-51. Biotechnol veg 2(4):221–226
Reyes M, Gómez R, Moreno L, Dion D (2014) Secondary multiplication of somatic embryos in banana (Musa spp. AAA) in semisolid medium: effect of the type of culture vessel and sterilization method. J Adv Biotechnol 4:352–357
Ziv M (1990) Vitrification: morphological and physiological disorders of in vitro plants. In: Debergh PC, Zimmerman RH (eds) Micropropagation: technology and application. Kluwe Academic Publishers, Dordrecht, The Netherlands, pp 45–69
Debergh P, Aitken-Christie J, Cohen D, Grout B, Von Arnold S, Zimmerman R, Ziv M (1992) Reconsideration of the term ‘vitrification’ as used in micropropagation. Plant Cell Tiss Org Cult 30:135–140
Kevers C, Franck T, Strasser RJ, Dommes J, Gaspar T (2004) Hyperhydricity of micropropagated shoots: a typically stress-induced change of physiological state. Plant Cell Tiss Org Cult 77:181–191
Pérez-Alonso N, Wilken D, Gerth A, Jahn A, Nitzsche HM, Kerns G, Capote A, Jiménez E (2009) Cardiotonic glycosides from biomass of Digitalis purpurea L. cultured in temporary immersion systems. Plant Cell Tiss Org Cult 99:151–156
Bandyopadhyay T, Gangopadhyay G, Poddar R, Mukherjee K (2004) Trichomes their diversity, distribution and density in acclimatization of Teak (Tectona grandis L.) plants grown in vitro. Plant Cell Tiss Org Cult 78:113–121
Eisenbeiβ M, Kreis W, Reinhard E (1999) Cardenolide biosynthesis in light- and dark-grown Digitalis lanata shoot cultures. Plant Physiol Biochem 37:13–23
Namdeo AG (2007) Plant cell elicitation for production of secondary metabolites: a review. Pharmacog Rev 1:69–79
Pérez-Alonso N, Capote A, Gerth A, Jiménez E (2012) Increased cardenolides production by elicitation of Digitalis lanata shoots cultured in temporary immersion systems. Plant Cell Tiss Org Cult 110:153–162
Pérez-Alonso N, Arana F, Capote A, Pérez A, Sosa R, Mollineda A, Jiménez E (2014) Stimulation of cardenolides production in Digitalis purpurea L. shoot cultures by elicitors addition. Rev Colomb Biotechnol 16:51–56
Acknowledgments
The authors wish to thank the support of the EU through the ALFA Network CARIBIOTEC (project AML/B7-311/97/0666/II-0201), the German Ministry for Education and Research (BMBF), the Cuban Ministry of Science, Technology and Environment (CITMA), and the Institutional University Collaboration programme with Universidad Central “Marta Abreu” de Las Villas funded by the Flemish Interuniversity Council (VLIR-IUC UCLV).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Pérez-Alonso, N. et al. (2016). Biotechnological Approaches for Biomass and Cardenolide Production in Digitalis purpurea L.. In: Jain, S. (eds) Protocols for In Vitro Cultures and Secondary Metabolite Analysis of Aromatic and Medicinal Plants, Second Edition. Methods in Molecular Biology, vol 1391. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3332-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3332-7_6
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3330-3
Online ISBN: 978-1-4939-3332-7
eBook Packages: Springer Protocols