
Abstract
Bacopa monnieri L. (common name brahmi) is a traditional and renowned Indian medicinal plant with high commercial value for its memory revitalizer potential. Demand for this herb has further escalated due to popularization of various brahmi-based drugs coupled with reported anticancer property. Insufficient seed availability and problems associated with seed propagation including short seed viability are the major constraints of seed conservation in the gene banks. In vitro clonal propagation, a prerequisite for in vitro conservation by enhanced axillary branching was standardized. We have developed a simple, single step protocol for in vitro establishment, propagation and medium-term conservation of B. monnieri. Single node explants, cultured on Murashige and Skoog’s medium supplemented with BA (0.2 mg/L), exhibited shoot proliferation without callus formation. Rooting was achieved on the same medium. The in vitro raised plants were successfully transferred to soil with ~80 % survival. On the same medium, shoots could also be conserved for 12 months with high survival and genetic stability was maintained as revealed by molecular markers. The protocol optimized in the present study has been applied for culture establishment, shoot multiplication and medium-term conservation of several Bacopa germplasm, procured from different agro-ecological regions of India.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Bacopa monnieri
- Brahmi
- Clonal propagation
- Genetic stability
- ISSR
- In vitro conservation
- In Vitro Genebank
- Medicinal plant
- RAPD
1 Introduction
Bacopa monnieri L. (synonym B. monniera), commonly known as “brahmi ,” “jalanimba,” or “the thinking man’s herb” is an ancient and important “Medhyarasayana” drug in the traditional system of Indian medicine—the Ayurveda. In India, it grows in damp areas up to 1320 m. It forms an important ingredient of a number of Ayurvedic preparations, such as “Brahmighrit,” “ Brahmi -rasayana,” “Sarasvatarisht,” and “Brahmivati.” The whole plant is used as a drug to cure epilepsy, relieve mental stress, improve intelligence and memory power, and anxiety neurosis [1, 2]. Besides having anti-inflammatory, analgesic, and antipyretic properties, the plant is known to also have anticancer and antioxidant properties. The saponins—bacoside A and B have been indicated for nervine tonic properties. Additionally, it has a good market especially for Brahmi oil due to its high medicinal value. The demand for Bacopa has further escalated owing to popularization of brahmi-based drugs like “Mentat,” “Memory Plus,” and “Memory Perfect” in the Indian and global market, and the recent report of anticancer activity of the herb extracts using Sarcosoma cell culture . Lack of concerted efforts regarding its cultivation/improvement, coupled with high demand of this medicinal herb by pharmaceutical companies has led to collection of material from wild population. A large amount of plant material is required for drug extraction primarily due to drug content of the plant being very low (~0.2 %). Further, the plant requires very specific growing conditions. Thus conservation of this precious herb needs immediate attention to ensure its availability and sustainable utilization now, and in future [3].
Despite the medicinal potential and high commercial value and realization of the need for conservation , there are no reports regarding conservation of this precious herb in the natural habitat. Application of seed conservation methods is constrained due to insufficient seed availability, difficult seed propagation and short seed viability. Thus, use of in vitro techniques, increasingly applied for rapid clonal propagation and conservation of valuable and threatened germplasm of medicinal importance with varying degrees of success [4, 5], appeared to be a viable conservation strategy for this plant. Standardization of efficient in vitro propagation protocol is an essential prerequisite for application of in vitro conservation . We have developed a method for in vitro clonal multiplication and medium -term conservation of Bacopa [2, 6].
There are few published reports on regeneration of Bacopa from various explants through axillary bud break, adventitious shoot formation, somatic embryo genesis and/or callus regeneration [2, 7–10]. For conservation , enhanced axillary branching is the preferred mode of regeneration as it ensures genetic stability of the conserved germplasm. However, genetic stability of the conserved cultures needs to be tested, while developing new protocol for conservation, using available molecular tools. This chapter describes various steps of a reliable in vitro clonal propagation and conservation protocol developed in our laboratory [2, 6]. This protocol would be of great commercial uses and for large-scale production of high-quality planting material and/or secondary metabolite production as also for conservation of this valuable medicinal species.
2 Materials
2.1 Plant Material
Healthy young shoots of Bacopa plants maintained in pots in the net house for culture initiation and young leaves for genetic stability studies.
2.2 Media Preparation
-
1.
Equipment: Magnetic stirrer cum hot plate, Weighing balances, Analytical balance to weigh from 0.1 mg up to a few grams, Top pan balance from few to hundreds of grams, Microwave, Gas stove, pH meter, Micropipettes, Autoclave, Refrigerator.
-
2.
Chemicals/reagents: Salts required for Murashige and Skoog’s (MS) [11] medium (see Table 1) and distilled/reverse osmosis (RO) water.
Table 1 Media composition for Murashige and Skoog’s [11] for stock preparation 4.0, 7.0 and 9.2 pH buffers and 1 N NaOH/HCl for setting pH of medium, ethanol.
-
3.
Glassware/plasticware: Reagent bottles, conical flasks (250, 1000 ml capacity), beakers, measuring cylinders (varying capacities), culture tubes (25 × 150 mm), culture tube enclosures (cotton plug/plastic caps/screw caps), glass rods.
2.3 Surface Sterilization and Inoculation
-
1.
Equipment: Laminar air flow, Bead sterilizer.
Tissue culture tool kit comprising 8″ to 12″ rust proof stainless steel forceps, surgical scalpels with supply of removable sterile surgical blades (straight and sharp), tissue paper, cotton wool, etc.
-
2.
Chemicals/reagents: 0.01–0.1 % Mercuric chloride (HgCl2), teepol, 80 % ethanol, sterile distilled water, culture medium .
-
3.
Glassware/plasticware: Conical flasks (100, 250 ml capacity), beakers (250 ml capacity), sterile petri dishes.
2.4 Hardening and Transfer Out
-
1.
Chemicals/reagents: Water, bavistin, ½ strength MS (liquid without sucrose ).
-
2.
Glassware/plasticware: Large petri dishes, forceps 12″, brush to remove agar , plastic and earthen pots, polythene bags, potting mix, soil.
2.5 In Vitro Conservation
-
1.
Equipment: Same as listed in Subheadings 2.2, item 1 and 2.3, item 1, culture incubation chambers at different low temperatures (4, 10, 15, 20, 25 °C), stereo microscope.
-
2.
Chemicals/reagents: Culture medium (see Subheading 2.2, item 2), 80 % ethanol, mannitol, alginic acid (low viscosity), calcium chloride (CaCl2).
-
3.
Glassware/plasticware: Conical flasks (100 ml capacity), beakers (250 ml capacity), sterile petri dishes, cryovials.
2.6 Genetic Stability Assessment
2.6.1 DNA Isolation
-
1.
Equipment: High speed centrifuge, Microfuge, Autopipettes (2–20 μl, 20–200 μl, 200–1000 μl), Water bath, −20 °C Deep freezer, Refrigerator, Freeze-drier (optional), Fume hood.
-
2.
Chemicals/reagents: CTAB (Cetyl Trimethyl Ammonium Bromide) Buffer [1.4 M NaCl, 100 mM Tris, 20 mM EDTA (Ethylene diamine tetra acetic acid), 0.2 % β-Mercaptoethanol, 1.5 % CTAB, 1 % PVP (Polyvinyl pyrrolidone)], Isopropanol, Saturated phenol, NaOAc (Sodium acetate), Chloroform–isoamyl alcohol (24:1) mixture, 10:1 TE (10 mM Tris 1 mM EDTA), RNase A (10 mg/ml), 70 % ethanol.
-
3.
Glassware/plasticware: Conical flasks, beakers, centrifuge tubes (50 ml), Eppendorf tubes, mortar and pestle, liquid nitrogen.
2.6.2 Randomly Amplified Polymorphic DNA (RAPD ) Analysis
-
1.
Equipment: DNA Thermocycler, Microfuge, Autopipettes of range: 2–20 μl, 20–200 μl, 200–1000 μl, DNA electrophoresis unit with power supply, −20 °C Deep Freezer, Refrigerator, Laminar air flow.
-
2.
Chemicals/reagents: 50× TAE, pH 8.0 (2 M Tris–acetate, pH 8.0, 0.05 M EDTA, pH 8.0); 10× Loading Buffer (Bromophenol Blue: 0.25 % Xylene Cyanol FF: 0.25 % Glycerol: 50 % TAE: 1×); Ethidium bromide stock 10 mg/ml ethidium bromide in double distilled water; AmpliTaq DNA Polymerase, MgCl2, Primers, Reaction buffer.
-
3.
Glassware/plasticware: conical flasks, beakers, PCR tubes.
2.7 Intersimple Sequence Repeats (ISSR ) Analysis
3 Methods
3.1 Media Preparation
Media is generally prepared by diluting concentrated stock solutions.
3.1.1 Stock Solution Preparation
Prepare and store separate stock solutions for macronutrients, micronutrients, iron and vitamins (Tables 1 and 2, Note 1 ). Weigh and dissolve each medium component in a small quantity of water and make up the final volume by adding distilled water.
-
1.
For preparation of iron stock, dissolve Na2EDTA∙2H2O in 100 ml hot water. Add FeSO4∙7H2O and shake well till iron gets properly dissolved and forms a clear light yellow solution. Add distilled water to make final volume.
-
2.
Stock of plant growth regulator is prepared by dissolving BAP (a cytokinin) in a minimum volume of 0.5 N NaOH or NAA (an auxin) in 0.5 N NaOH/ethanol and then making up the volume with distilled water.
3.1.2 Preparation and Sterilization of Media
Protocol to make 1 L MS medium (see Note 4 ).
-
1.
Pour 500 ml distilled water in a 1-L flask with a magnetic stirring bar.
-
2.
Add 30 g sucrose and dissolve.
-
3.
Add required stock solutions in the order (see Table 1).
-
4.
Add aliquot of auxin stock/cytokinin stock solution (as per requirement).
-
5.
Adjust the pH to 5.8 using pH meter and 1 N NaOH or HCl, with constant stirring.
-
6.
After adjusting the pH, makeup the volume to 1 L using a graduated cylinder or graduated beaker, pour back in to flask; add 8.0 g agar and keep for melting.
-
7.
Dispense about 15–20 ml medium per culture tube and cap the tube (see Note 5 ).
-
8.
Autoclave at 121 °C with a pressure of 15 psi (1.06 kg/cm2), for 15 min (see Notes 2 – 4 ).
-
9.
Take out from autoclave and allow the medium to cool at room temperature. For preparing slants, keep the tubes tilted (at an angle of 45°) during cooling.
The powdered medium is dissolved in distilled water (10 % less than the final volume), and after adding sugar and other supplements, the final volume is made up with distilled water (see Note 5 ).
3.2 Explant Preparation
-
1.
Collect healthy young shoots with three to four nodes, from plants maintained in the net house (Fig. 1a), in a beaker containing distilled water.
-
2.
Remove leaves and wash in 1 % (v/v) Labolene detergent for 10 min on a magnetic stirrer.
-
3.
Wash these thoroughly under running tap water for 2 h.
3.3 Surface Sterilization and Establishment of In Vitro Cultures
-
1.
Disinfect the surface of the laminar flow with 75 % alcohol after UV light exposure for 15 min.
-
2.
Arrange surface sterilized culture containers and tools on the bench.
-
3.
Dip forceps/scalpels in ethanol followed by flaming after each operation.
-
4.
Sterilize the stem segments with 0.1 % (w/v) mercuric chloride for 10 min under laminar air flow followed by rinsing (5 times) with sterile de-ionized water (see Notes 6 and 7 ).
-
5.
Using sterilized forceps, transfer the explants into a sterile container and rinse 2–3 times with sterile water.
-
6.
Transfer the segment to a sterile petri plate and with the help of a scalpel (with sterile blade), cut the edge (exposed ends) of the explants.
-
7.
Trim single node segments to ca. 0.8 cm and culture on MS medium supplemented with 0.2 mg/L BAP .
-
8.
Inoculate single explant in each culture tube and incubate at 25 ± 2 °C under 16/8 h photoperiod (30 μmol/m2/s) light/dark provided by cool white fluorescent lamps (see Notes 8 and 9 ).
3.4 Shoot Multiplication and Rooting
For shoot multiplication contamination-free cultures are sub-cultured every 4–6 weeks on shoot multiplication medium (Fig. 1b) (see Notes 10 and 11 ).

(a–d) In vitro propagation of Bacopa monnieri. (a) Source of explants; (b) In vitro establishment and shoot multiplication on MS + 0.2 mg/L BAP (L–R); (c) 8-week-old cultures of some accessions showing multiple shoots and roots on MS + 0.2 mg/L BAP; (d) Plantlets established in pots
-
1.
Gently take the cultures out of culture tube and dissect nodal explants to required size, on a petri plate using sterile surgical blade and forceps.
-
2.
Using aseptic forceps, transfer the explants vertically onto shoot multiplication medium .
-
3.
Hold the opening of the tube in the flame for few seconds, and then cap it.
-
4.
Label appropriately using a standardized format and seal with Para film.
-
5.
Maintain cultures at 25 ± 2 °C, 16/8 h photoperiod (30 μmol/m2/s) light/dark provided by cool white fluorescent tubes under controlled relative humidity of 50 to 60 %.
-
6.
Monitor the cultures periodically for contamination, hyperhydricity or growth abnormalities (after each subculture cycle).
On shoot multiplication medium 100 % cultures form roots hence avoid separate rooting medium (Fig. 1b, c).
3.5 Acclimatization and Transfer Out
-
1.
Transfer the rooted shoots (after 8 weeks of culture) to pots for hardening .
-
2.
Gently remove well-rooted plantlets from the culture tubes, keeping the roots intact.
-
3.
Wash off the adhering agar from roots very carefully with a brush, under running tap water.
-
4.
Moisten the sterile vermiculite in plastic pots (7.5 cm diameter), transfer plantlets to pots and water lightly.
-
5.
Cover with perforated polythene bags to maintain high humidity (~90 %).
-
6.
Water the plants every 3 days with half-strength MS salt solution (pH 5.8) for 3 weeks.
-
7.
Remove the polythene bags after 3 weeks and transfer the plants to earthen pots (20 cm diameter) containing garden soil, after 4–6 weeks and allow the plants to adjust to ambient conditions (Fig. 1d) (see Notes 12 and 13 ).
3.6 In Vitro Conservation : Slow Growth
-
1.
Use cultures multiplied for three passages as source of explants
-
2.
Transfer single nodal segment (0.8 cm) from 8-week-old cultures to glass tubes (25 × 150 mm) containing 15 ml of shoot multiplication medium solidified with 0.8 % agar .
-
3.
Incubate all the cultures in culture room for 10 days to detect and eliminate contamination (Fig. 2a).
Fig. 2 
(a–g) In vitro conservation of Bacopa monnieri using slow growth strategies. (a) A view of the In Vitro Genebank at NBPGR, India conserving multi-crops including Bacopa. Effect of various slow growth strategies (b–f). (b) Effect of culture tube enclosure; (c) Effect of mineral oil; (d) Effect of minimal media; (e) Regrowth of conserved cultures; (f) Storage of encapsulated shoot tip s in a cryovial without nutrient medium ; (g) Conserved shoot culture s of various accessions after 12 months on MS + 0.2 mg/L BAP
-
4.
To standardize the most suitable strategy for conservation of germplasm in a gene bank, the following slow growth strategies (see Note 14 ) (Fig. 2a–f) are tested:
-
(a)
To study effect of enclosures, close the tubes with either cotton plugs or polypropylene caps. Maintain cultures in the culture room.
-
(b)
For low temperature storage, transfer healthy cultures to low temperature, 10, 15, and 20 °C to select optimum responding temperature (see Note 15 ).
-
(c)
To test the effect of media modifications, various osmotica (sucrose , mannitol) are included in the media. Maintain cultures under culture room conditions.
-
(d)
To test the effect of mineral oil, culture single node in each culture tube, and using a micropipette, pour ca. 3 ml mineral oil (10 mm depth) (autoclaved separately and cooled to 25 °C) over the explants to cover the explants completely. Maintain cultures in the culture room.
-
(e)
To test the effect of storing encapsulated shoot tip s in vials without any nutrient medium ,
-
Collect apical shoot tips from the shoot culture of the same subculture age (8 weeks after last subculture).
-
Using fine forceps and scalpel dissect out shoot tip s (1–2 mm) under the stereomicroscope, in a laminar air flow cabinet. The explants should contain the apical meristem dome along with one to three non-expanded leaf primordia.
-
Make encapsulated shoot tip s (referred as beads) by using 3 % sodium alginate as gel matrix and 100 mM calcium chloride (CaCl2∙H2O) for complexation. Suspend excised shoot tips for ~20 min in sodium alginate (low viscosity alginic acid in liquid MS medium without calcium, pH 5.7). Explant along with sodium alginate are sucked (aspirated) into a sterile disposable Pasteur pipette and dropped one by one into calcium chloride solution. Allow beads to stand in the solution for 30 min with gentle shaking, for polymerization. Then take them out by decanting off calcium chloride solution, rinsing with sterile water followed by surface drying on sterile filter paper.
-
Transfer five uniform beads with one explant each, in a cryovial without any medium , and maintain them under culture room conditions.
-
-
(a)
-
5.
Periodically assess survival of cultures by visual examination and/or by the ability of explant to resume growth on fresh medium (Figs. 2 and 3) (see Notes 15 – 20 ).
Fig. 3 
Survival of some accessions of Bacopa monnieri after conservation for 12 months on MS + 0.2 mg/L BA, under culture room conditions (see Note 21 )
-
6.
For testing regrowth of conserved beads, transfer the beads onto shoot multiplication medium periodically after 2 weeks and score for emergence of shoots.


3.7 Genetic Stability Assessment
3.7.1 Preparation of Extraction Buffers
-
1.
Add calculated amount of CTAB, NaCl, EDTA, Tris–HCl, and Polyvinyl pyrrolidone PVP in 200 ml of water and make the final volume to 1 L.
-
2.
Adjust pH to 8.0 with HCl and autoclave before use.
-
3.
Warm at 60 °C in a water bath for 30 min.
-
4.
Add 0.2 % β-mercaptoethanol to the extraction buffer under fume hood
3.7.2 Plant DNA Isolation and Purification
Method for Total genomic DNA extraction [12] (see Note 21 ):
-
1.
Weigh 5 g of clean young leaf tissue and grind to fine powder with a pestle and mortar after freezing in liquid nitrogen.
-
2.
Transfer to 50 ml centrifuge tube with 20 ml CTAB buffer maintained at 60 °C in a water bath.
-
3.
Mix vigorously or vortex. Incubate at 60 °C for one hour. Mix intermittently.
-
4.
Add 20 ml chloroform: isoamyl alcohol. Mix gently by inverting for 5 min.
-
5.
Spin at 34,541 × g for 10 min with SS34 rotor in Sorval RC-5C centrifuge at 25 °C Transfer aqueous phase to a fresh centrifuge tube.
-
6.
Add equal amount of isopropanol and let the DNA to settle down for 20 min.
-
7.
Spool out the DNA or spin at 17,210 × g for 15 min and drain liquid phase.
-
8.
Add 0.5 ml 70 % ethanol. Mix gently and incubate for 30 min.
-
9.
Spin at 17,210 × g for 15 min and decant and repeat the 70 % ethanol treatment.
-
10.
Decant off and dry the pellet under vacuum. Dissolve DNA in minimum volume of 10:1 TE buffer.
-
11.
Add RNAse (0.2 ml) and incubate at 37 °C for 1 h. Add proteinase (0.2 ml) and incubate at 37 °C for 1 h (see Note 22 ).
-
12.
Add equal volume of phenol–chloroform (1:1), mix properly for at least 2 min and spin for 5 min.
-
13.
Take out the DNA supernatant and after this perform two chloroform: isoamyl alcohol extractions as before. Spin at 17,210 × g for 15 min after each extraction.
-
14.
Precipitate DNA by adding 1/10 volume of 3 M NaOAc and 2.5 times of the total volume chilled ethanol.
-
15.
Mix and spool out the DNA. Remove extra salts by two washings with 70 % ethanol.
-
16.
Dry under vacuum. Add minimum volume of TE (10:1).
-
17.
Dissolve at room temperature. Store frozen at −20 °C.
3.7.3 Randomly Amplified Polymorphic DNA (RAPD ) Analysis (See Note 23 )
-
1.
Switch on the thermocycler at least 15 min earlier.
-
2.
Pipette out accurately using appropriate autopipettes into sterile 0.5 ml microtubes the reagents in the following order:
-
dd H2O: 13.80 μl
-
10× Reaction buffer: 2.50 μl
-
25 mM MgCl2: 2.50 μl
-
10 μM Primer: 1.00 μl
-
10 mM mix of dNTPs: 2.50 μl
-
Taq DNA polymerase (5 U/μl): 0.2 μl
-
Template DNA (10 ng/μl): 2.50 μl
-
Total reaction volume: 25.00 μl
-
-
3.
Mix by repeated pipetting.
-
4.
Spin down the contents (2–5 s).
-
5.
Place the tubes firmly in the wells.
-
6.
Start thermocycler.
-
The temperature cycling conditions are:
-
Step 1. Denature at 94 °C for 4 min Step
-
Step 2. Denature at 94 °C for 1 min Step
-
Step 3. Annealing at 37 °C for 1 min Step
-
Step 4. Polymerization at 72 °C for 2 min
-
Step 5. Repeat from step 2–4: 39 times
-
Step 6. Extended polymerization at 72 °C for 8 min
-
Step 7. Refrigerate.
-
-
7.
At the end of the run take out the tubes and add 2.5 μl 10× loading dye. Spin for 2–5 s. Store at 4 °C till electrophoresis.
-
8.
Agarose Gel Electrophoresis : The amplified products in RAPD analysis are usually smaller than 4 kb size. Hence, they are separated by electrophoresis in 1.4–1.8 % agarose gels and visualized by staining with ethidium bromide and viewing under UV light.
-
(a)
Prepare the gel tray by taping the open ends. Place the comb and level the tray.
-
(b)
Boil and prepare 1.4 % agarose gel in 1× TAE buffer. Cool to 60 °C. Pour into gel tray avoiding air bubbles. Allow setting for 30–40 min.
-
(c)
Remove the adhesive tapes. Place in electrophoresis tank filled with 1× TAE buffer. Remove the comb carefully. Pour 1× TAE buffer till the gel is fully immersed.
-
(d)
Load the samples carefully. Take care to load suitable DNA size markers (about 200 ng). Connect the leads and start electrophoresis run at constant 60 V.
-
(e)
Stop the run when bromophenol blue dye has traveled less than 2/3 the length of gel.
-
(f)
Stain in 0.5–1 μg/ml ethidium bromide in distilled water for 30–40 min. Wash briefly in distilled water (see Note 24 ).
-
(g)
View under UV light. Photograph the gel (see Note 24 ).
-
(h)
Score the amplified products across the lanes comparing their respective molecular weight (Fig. 4). The data can be analyzed statistically using suitable packages (see Note 25 ).
Fig. 4 
Representative RAPD profiles (primer OPC-15) generated from culture (1–4) and ex vitro transferred plants (5–9) of Bacopa monnieri, L—denotes GeneRuler™ 100 bp DNA ladder
-
(a)

3.7.4 Intersimple Sequence Repeats (ISSR ) Analysis (See Note 26 )
-
1.
Switch on the thermocycler at least 15 min earlier (Table 2).
Table 2 Recommended storage duration of stock solutions (see Note 1) -
2.
Pipette out accurately using appropriate autopipettes into sterile 0.5 ml microtubes the reagents in the following order:
-
H2O: 14.30 μl
-
10× Reaction buffer: 2.50 μl
-
25 mM MgCl2: 1.00 μl
-
5 μM Primer: 3.00 μl
-
2.5 mM mix of dNTPs: 2.00 μl
-
Taq DNA polymerase (5 U/μl): 0.20 μl
-
Template DNA (10 ng/μl): 2.00 μl
-
Total reaction volume: 25.00 μl
-
-
3.
Mix by repeated pipetting.
-
4.
Spin down the contents (2–5 s).
-
5.
Place the tubes firmly in the wells.
-
6.
Start thermocycler.
The following general thermocycling steps are followed
-
Step 1: Initial denaturation at 94 °C for 5.0 min
-
Step 2: Denaturation at 94 °C for 1.0 min
-
Step 3: Primer annealing at 52–59 °C (depending upon primer) for 45 s
-
Step 4: Primer extension at 72 °C for 2.0 min
-
Step 5: Go to step 2, 39 times
-
Step 6: Final extension at 72 °C for 7 min
-
Step 7: 4 °C for ever
-
-
7.
After the run is completed turn off the machine and remove the samples. Add 2.5 μl gel loading dye in each tubes and carry out electrophoresis or store samples at 4 °C until electrophoresis.
-
8.
Agarose gel electrophoresis and analysis (Fig. 5): same as RAPD .
Fig. 5 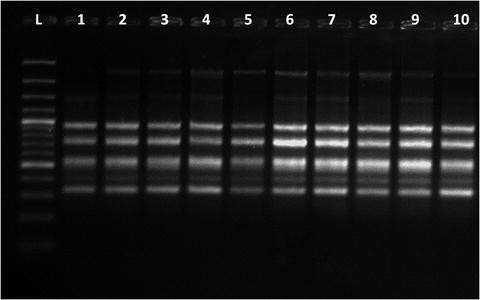
Representative ISSR profiles (primer UBC-808) generated from culture (1–5) and ex vitro transferred plants (6–10) of Bacopa monnieri, L—denotes GeneRuler™ 100 bp DNA ladder

4 Notes
-
1.
Store iron stock, preferably in an amber-colored bottle, at 4–10 °C in a refrigerator. However, prolonged storage of auxins is not recommended due to photo-oxidative degradation.
-
2.
Media are generally sterilized by autoclaving at 121 °C with a pressure of 15 (1.06 kg/m2). The time required for sterilization is dependent on the quantity of medium and type of container. A slow exhaust follows sterilization, as quick decompression will cause the medium to boil off the vessels.
-
3.
Media constituents (such as antibiotics) subject to modification by heat can be added to the autoclaved medium following filter-sterilization (passing through pre-autoclaved bacteria-proof membrane filtration unit).
-
4.
Contamination can occur in the stock solutions of vitamins and growth regulators as well as in the media which are inadequately sterilized.
-
5.
When using ready-made powdered media, it is essential that the powder is completely dissolved. Any turbidity of the solution indicates that one or more of the ingredients is not dissolved, and thus not available to the plant cells.
-
6.
Explants are sterilized normally by treating with 3–5 % sodium hypochlorite or 0.05–0.1 % mercuric chloride solution for 5–15 min. When working with explants of a new genotype, it is better to run preliminary screening tests to determine the exposure time tolerated by explants.
-
7.
If the explants turn brown or white, they are over-sterilized and the cells are dead. It is recommended to reduce the length of time or the concentration of sterilizing agent. In contrast, if the surface sterilization treatment does not yield clean cultures then the procedure needs to be more stringent: increase time or concentration of the sterilant.
-
8.
It is recommended to check inoculated cultures for contamination every 2–4 days for at least 10 days. Contaminated cultures should be immediately removed and autoclaved before washing.
-
9.
The standardized protocol for surface sterilization resulted in 60–100 % aseptic culture establishment at initiation stage. However, minor time adjustment may be required in some genotypes. Inclusion of 0.1 % streptomycin was beneficial in some accessions exhibiting bacterial contamination during culture initiation.
-
10.
Propagation methods may require modification depending upon the genotypes/accessions/diverse collections to be conserved, in the Genebank.
-
11.
Amongst various combinations of plant growth regulators tested, MS supplemented with 0.2 mg/L was most suitable (see [2]). The described protocol has been adopted for in vitro establishment and propagation of more than 20 accessions (genotypes) collected from different agro-ecological regions of India. Though there was significant variation with respect to rate of multiplication, a minimum of 11 shoots/explants were obtained in the least responding accession [2].
-
12.
Usually the most critical step of in vitro propagation is the establishment of plantlets in the soil. As the plantlets are propagated in vitro under high relative humidity with little or no photosynthesis, the acclimatization during the transition from culture to soil conditions must be gradual. Watering the plants is very critical as too little water may lead to permanent wilt, while too much water may lead to decay.
-
13.
This protocol has the potential application for the conservation of germplasm using slow growth and cryopreservation techniques [2, 6, 14] and also for bulking up material for secondary metabolite production .
-
14.
Slow growth strategy is based on optimizing subculture duration without risking germplasm loss and genetic stability through stressful treatments. It is important to note that genotype response to growth limiting treatments can be highly variable.
-
15.
Various methods such as change of enclosure, media modification, low temperature incubation etc. have been employed to achieve slow growth in a large number of crops. Amongst several slow growth strategies and different type of explants experimented upon in Bacopa, subculture duration could be enhanced to 14 months with mannitol, 12 months with sucrose , 15 months with minimal media, and 18–24 months with mineral oil overlay, using polypropylene caps as closures, under culture room conditions. Incubation of cultures at low temperature (4 and 10 °C) was not successful.
-
16.
Amongst various storage temperatures, the optimum temperature at which encapsulated shoot tip s, conserved in a cryovial without nutrient medium , remained viable for 6 months was 25 °C. On transfer to medium for regrowth, thus conserved shoot tips, developed into normal shoots. This methodology has the potential for exchange of germplasm, as being free of nutrient medium, problems related to melting of agar and contamination are minimized.
-
17.
This simple single step protocol is suitable for the propagation and conservation of a total of 22 accessions of Bacopa germplasm, on MS + 0.2 mg/L BAP . They have been maintained in the In Vitro Genebank at NBPGR, India, for more than 10 years with subculture duration of 10–16 months, depending upon the accession.
-
18.
One of the limitations of germplasm maintenance in the In Vitro Genebank, especially in the tropical region, has been the contamination of cultures during various stages. Under ideal situation it is best to discard contaminated cultures and start fresh from mother plants. However, in case of non availability of mother plant, as an emergency measure, cultures can be rescued following resterilization of explants using 0.05 or 0.1 % (w/v) mercuric chloride + 0.1 % (w/v) streptomycin for 5–8 min.
-
19.
In the In Vitro Genebank, periodic monitoring of cultures for survival, chlorosis, hyperhydricity and contamination must be done. Subculture of stored cultures should be carried out when 30 % cultures are dead/dried/contaminated. It is recommended not to subculture all cultures of an accession at one go (on the same day) to rule out the possibility of losing the precious germplasm due to contamination/ technical/equipment error/mislabeling.
-
20.
Monitoring genetic stability is an integral part of any in vitro conservation program. In Bacopa, no morphological, molecular or biochemical variation was detected in selected accessions using a few strategies [6]. Hence, the described in vitro conservation strategy can be safely implemented for medium -term conservation of Bacopa germplasm, especially the elite lines till effective long-term conservation protocols are available [13–16].
-
21.
The isolation of DNA from plant cells essentially involves two steps—(1) The lysis of plant cells and solubilisation of DNA and (2) Enzymatic or chemical methods to remove contaminating protein, RNA and other macromolecules. Isolation and purification of DNA is an important step because pure and high molecular weight DNA is essential for the reliability of DNA for molecular biology techniques. Any impurities in the genomic DNA can inhibit enzymatic reactions, electrophoresis and quantification and in turn their downstream applications. For plant cells with a rigid cell wall, the disruption of cells usually requires that the tissue is ground using a pestle and mortar after thoroughly freezing them in liquid nitrogen. The powdered plant tissue is then transferred to an extraction buffer that contains detergent to disrupt the membranes. The extraction buffer also contains a reducing agent (β-mercaptoethanol) and a chelating agent (ethylenediamine tetra acetic acid, EDTA). This helps to inactivate nucleases that are released from the plant cell which can cause serious degradation of the genomic DNA.
-
22.
Dissolve RNase A in 10 mM Tris–Cl, pH 7.5, 15 mM NaCl. Heat at 100 °C for 15 min. Cool to room temperature. Store as aliquots at −20 °C.
-
23.
The RAPD technique is based on the polymerase chain reaction (PCR ). A DNA sequence is exponentially amplified with the help of arbitrary primers and a thermostable DNA polymerase. The reaction involves repeated cycles, each consisting of a denaturation, a primer annealing and an elongation step. In the first step the DNA is made single stranded by increasing the temperature up to 94 °C (denaturing step) in the second step, lower the temperature to about 37 °C results in prime annealing to their target sequences on the template DNA (annealing step). For the third step temperature is chosen where the activity of the thermostable polymerase is optimal, i.e., usually 72 °C. The polymerase now extends the 3′ ends of the DNA-primer hybrids towards the other primer-binding site. Since this happens at both primer-annealing sites on both the DNA strands, the target fragment is completely replicated. Repeating these three step cycles 35–45 times results in the exponential amplification of the target between the 5′ ends of the two primer binding sites. Amplification products are separated by gel electrophoresis and visualized by ethidium bromide staining (Fig. 4).
-
24.
Ethidium bromide is a mutagen and a probable carcinogen. Wear gloves when working with ethidium bromide solutions. Also use care not to contaminate the work area with the solution. UV light is damaging and must be used with caution. UV light causes burns and can damage the eyes.
-
25.
For genetic stability assessment, for each marker system, score the strong amplified fragments for their presence (1), absence (0) and missing data (9). Compute genetic similarity values based on Jaccard’s coefficient using NTSYS-PC version 2.11a. Perform cluster analyses based on similarity matrices for combined scores of RAPD and ISSR using unweighted pair group method with arithmetical averages (UPGMA)—a method used for constructing phylogenetic tree using similarity matrix.
-
26.
ISSRs are semi arbitrary markers amplified by PCR in the presence of one primer complementary to a target microsatellite. Amplification in the presence of non-anchored primers also has been called microsatellite-primed PCR, or MP-PCR. Such amplification does not require genome sequence information and leads to multi-locus and highly polymorphous patterns. Each band corresponds to a DNA sequence delimited by two inverted microsatellites (Fig. 5). Like RAPDs, ISSRs markers are quick and easy to handle, but they seem to have the reproducibility of SSR markers because of the longer length of their primers. ISSR technique differs from the RAPD in the sense that here longer primer sequences are used and hence the annealing temperature is also higher which together provide more stringency to the technique. Consequent to higher stringent conditions, ISSR markers are more reproducible as compared to the RAPD markers.
References
Anonymous (1988) The wealth of India: raw materials, vol 2. Council of Scientific and Industrial Research, New Delhi, India
Sharma N, Satsangi R, Pandey R, Vimala Devi S (2007) In vitro clonal propagation and medium term conservation of Brahmi [Bacopa monnieri (L.) Wettst.]. J Plant Biochem Biotechnol 16:139–144
Sharma N, Vimala Devi S, Satsangi R (2007) Biotechnological approaches in multiplication and conservation of plants of medicinal value. In: Shukla PK, Chaubey OP (eds) Threatened wild medicinal plants: assessment, conservation and management. Anmol Publications Pvt Ltd., New Delhi, India, pp 248–257
Chandel KPS, Shukla G, Sharma N (1996) Biodiversity in medicinal and aromatic plants in India. National Bureau of Plant Genetic Resources, New Delhi, India
Sharma N, Pandey R (2013) Conservation of medicinal plants in tropics. In: Normah MN, Chin HF, Reed BM (eds) Conservation of tropical plant species. Springer, New York, pp 437–487
Sharma N, Satsangi R, Pandey R, Singh R, Kaushik N, Tyagi RK (2012) In vitro conservation of Bacopa monnieri (L.) using mineral oil. Plant Cell Tiss Org Cult 11:291–301
Ceasar AS, Maxwell SL, Prasad KB, Karthigan M, Ignacimuthu S (2010) Highly efficient shoot regeneration of Bacopa monnieri (L.) using a two stage culture procedure and assessment of genetic integrity of micropropagated plants by RAPD. Acta Physiol Plant 32:442–452
Shrivastava N, Rajani M (1999) Multiple shoot regeneration and tissue culture studies on Bacopa monnieri (L.) Pennell. Plant Cell Rep 18:919–923
Tiwari V, Singh BD, Tiwari KN (1998) Shoot regeneration and somatic embryogenesis from different explants of Brahmi [Bacopa monniera (L.) Wettst.]. Plant Cell Rep 17:538–543
Tiwari V, Tiwari KN, Singh BD (2000) Comparative studies of cytokinins on in vitro propagation of Bacopa monniera. Plant Cell Tiss Org Cult 66:9–16
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci U S A 81:8014–8018
Sharma N, Pandey R (2011) Cryopreservation studies on medicinal plants at NBPGR in India. In: Abstract of the 48th Annual Meeting of the Society for Cryobiology (CRYO 2011) at Corvallis, USA, 24–27 July 2011, p 80
Sharma N, Satsangi R, Pandey R (2011) Cryopreservation of shoot tips of Bacopa monnieri (L.) Wettst by vitrification technique. Acta Hortic 908:283–288
Sharma N, Satsangi R, Pandey R (2012) Cryopreservation in medicinal plants—a case study. In: Abstracts of 5th Indian Horticulture Congress 2012—an international meet, PAU, Ludhiana, India, 6–9 Nov 2012, p 418
Sharma N, Pandey R, Tyagi RK (2014) Cryopreservation studies on a commercially important medicinal plant Bacopa monnieri (L.) Wettst. In: Abstracts of freezing biological time—50th anniversary celebration, Annual Scientific Conference & AGM, Society for Low Temperature Biology, at The Royal Botanic Gardens, Kew, London, UK, 8–10 Oct 2014, p 35
Acknowledgements
The authors thank Director, NBPGR for facilities and encouragement. Financial support provided by the Department of Biotechnology, Ministry of Science and Technology, is also gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Sharma, N., Singh, R., Pandey, R. (2016). In Vitro Propagation and Conservation of Bacopa monnieri L.. In: Jain, S. (eds) Protocols for In Vitro Cultures and Secondary Metabolite Analysis of Aromatic and Medicinal Plants, Second Edition. Methods in Molecular Biology, vol 1391. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3332-7_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3332-7_11
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3330-3
Online ISBN: 978-1-4939-3332-7
eBook Packages: Springer Protocols