
Abstract
Antimalarials were among the first, and today are among the most widely used, anti-infective agents. The fundamental pharmacodynamic endpoint for antimalarials is quite simple: elimination of this eukaryotic protozoal pathogen from its host; numerous surrogates for this have been developed. Antimalarial therapy is confounded by several key factors including the coexistence of multiple pharmacologically distinct Plasmodium life cycle forms in the human host; limited resources for discovery, development, and deployment of new drugs; and a high requirement for safety due to the enormous patient population and use for chemoprophylaxis of healthy travelers. Further, for any particular drug, myriad influences impact the pharmacological endpoint, including rapidity of the onset of action, potency, ‘static vs. ‘cidal activity, susceptibility to parasite resistance, immune status of the host, and the suitability of prevailing pharmacokinetics. Classic and recently described pharmacodynamic endpoints in preclinical models are presented, as are new insights into the pharmacokinetic drivers of antimalarial pharmacodynamics. The efficacy and safety of existing drugs are surveyed, and some novel experimental agents are discussed.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
- Malaria
- Plasmodium
- Antimalarial
- Pharmacokinetics
- Pharmacodynamics
- PK/PD
- Parasite reduction ratio
- Resistance
- Combination therapy
1 Malaria
Malaria is a mosquito-borne infectious disease that afflicts hundreds of millions and kills nearly a million children every year in Africa. Recent efforts have brought encouraging progress toward eliminating this major public health challenge, via a combination of vector control, use of insecticide-impregnated bednets, improved diagnostics, and chemotherapy campaigns. Nevertheless, the lack of a vaccine and widespread drug resistance make the need for effective and safe new antimalarial agents compelling. Of the Plasmodium species pathogenic to humans, falciparum is most aggressive, causing life-threatening infection and having the greatest propensity for drug resistance [1, 2].
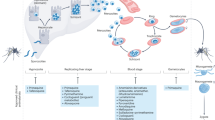
Malaria parasites have a complex life cycle (Fig. 1). From a therapeutic perspective, by far the most important forms are asexually dividing parasites that dwell within erythrocytes. They alone are responsible for the morbidity and mortality of malaria, and in severe infection may number hundreds of millions per milliliter of blood. It is the cyclical release of naked parasites into the bloodstream that gives rise to the classic periodic agues of malaria. P. falciparum’s unique lethality stems from adherence of infected erythrocytes to blood vessel walls, which causes a cytokine response and functional vascular obstruction. In severe cases this results in cerebral malaria, extensive hemolysis, and multiple end-organ failures. The remaining life cycle stages (all of which may coexist simultaneously within the same patient) are responsible for transmission of infection to the mosquito and may cause late reactivation of symptomatic disease. Unfortunately, each life cycle form has a different profile of drug susceptibilities (Fig. 2) [3]; hence multiple drugs may be required for complete eradication of the infection from a patient. In addition to their obvious necessity for the treatment of established infection, antimalarial drugs will also remain essential for the chemoprophylaxis of travelers to malarious regions until a suitable vaccine becomes available.

Life cycle of malaria parasites. Sporozoites inoculated by a mosquito rapidly make their way to the liver and infect hepatocytes. Parasites replicate within the hepatocyte, rupture the cell, and enter the blood stream as merozoites. These merozoites invade erythrocytes, initiating the blood stage of the infection. Erythrocytic parasites amplify 8–32-fold over 48–72 h, rupture the red blood cell, invade new erythrocytes, and restart the cycle. All parasite proliferation within the human is asexual. Some erythrocytic forms differentiate into gametocytes that will infect a biting mosquito, reproduce sexually, and eventually generate sporozoites that complete the life cycle. Some liver stage parasites of Plasmodium vivax and Plasmodium ovale form latent hypnozoites that may remain quiescent for years, before activating and establishing symptomatic disease

Simplified pharmacodynamic specificities of antimalarial drugs. Depicted are the classes of antimalarial drugs, in the context of their major activities against distinct forms of Plasmodium in a human host. Disease-initiating sporozoites are not listed since no drug has meaningful activity against this stage. Group I drugs primarily target the disease-causing asexual blood stages, and form the bulwark of antimalarial therapy. The Group II synergistic combination of atovaquone plus proguanil has reliable action against pathogenic red cell stages as well as the initial liver stage of P. falciparum; it is useful for prophylaxis as well as treatment. Group III primaquine targets regular and latent liver stages and gametocytes, but has no useful activity against asexual blood stages. Interspecies differences and unknown or unreliable activities are not shown
The discovery and development of antimalarial drugs has proven challenging. Plasmodium parasites are host-specific, such that species pathogenic for humans rarely propagate in other animals, and they have proven equally difficult to study in vitro. Obligate intracellular pathogens with an ~80 % AT genome [4], long term in vitro cultivation was not accomplished until 1976, when the preference of P. falciparum for microaerophilic conditions was realized [5]. P. vivax still cannot be maintained in continuous culture. Over time, however, animal models and in vitro assay systems have matured to the point that important parameters such as potency, pharmacokinetic–pharmacodynamic linkage, and proof-of-principle in vivo efficacy can be measured. Though eukaryotic like its human host, Plasmodium has numerous unique and distinguishing features exploitable for drug targeting. These include, for example, a chloroplast-like organelle whose self-contained genome and proteins have clinically useful susceptibility to several conventional antibacterials (e.g., tetracyclines, clindamycin) [6]; efficient hemoglobin digestion and corollary brisk detoxification of byproduct heme by chloroquine-sensitive crystallization [7]; and absolute dependence on pyrimidine biosynthesis disrupted by atovaquone-mediated blockade of mitochondrial electron transport [8]. In recent years availability of the complete genome sequence [4] and use of high throughput screens against libraries of millions of compounds [9–11] have provided much-needed promising new therapeutic leads (Sect. 4.7).
2 Pharmacodynamic Endpoints for Antimalarial Drugs
2.1 In Vivo
For most animal models of malaria neither the host nor the parasite is directly pertinent to human infection, and the time course and characteristics of human disease are not faithfully recapitulated. However, different animal model systems mimic some aspects of the host–parasite interaction and allow for biological and pharmacological investigations . In addition to humans, three major animal models have been utilized for studying antimalarial agents.
2.1.1 Avian
Avian malaria was the initial model of choice for chemotherapeutic development [12]. P. gallinaceum was particularly practical since both the mosquito vector and avian host (ducks, chickens) are easily infected. Although avian red cells are nucleated and the life cycle of P. gallinaceum differs significantly from that of human parasites, the erythrocytic stage is usefully mimicked. Mid-twentieth century development of major drugs chloroquine, primaquine, proguanil, and some antifolates was accomplished using P. gallinaceum-avian malaria. Subsequent discovery of rodent malaria, a system relatively easy to manipulate and with greater similarity to human physiology, resulted in a shift away from avian models.
2.1.2 Rodent
Rodent malaria parasites P. berghei, P. yoelii, and P. chabaudi have proven invaluable for pharmacodynamic screening of candidate compounds [13] and rodent models are now standard in antimalarial drug development [14]. However, no rodent model fully recapitulates the disease profile seen in human malaria, rodent parasite behavior in vivo can differ significantly from that of human-tropic Plasmodium species, and rodents may differ substantially in their handling of antimalarials. Controversy surrounds the use of the P. berghei ANKA-mouse model of cerebral malaria that reproduces some, but not all, pathophysiological aspects of human cerebral malaria [15]. Recent development of a mouse model in which human erythrocytes are maintained by immunosuppression enables pharmacodynamic evaluations against the parasites of greatest interest (asexual blood stages of P. falciparum), but in a mouse background [16].
2.1.3 Nonhuman Primate
Multiple Plasmodium species naturally infect nonhuman primates and mimic important features of human infection, arguably making them the best surrogates for human disease. However, the expense, limited availability, and ethical concerns surrounding use of primates severely limit their practical utility. Notable models include the P. cynomolgi-macaque that mimics human P. vivax infection, and the P. coatneyi- and P. fragile-macaque models that in many aspects resemble human P. falciparum infections [17]. In fact, the P. cynomolgi-macaque model proved critical in the development of primaquine, the only agent known to target latent liver forms (hypnozoites), thus preventing P. vivax and P. ovale relapses. The demonstration that P. knowlesi, normally infective to Old World Monkeys, can cause significant, sometimes lethal, zoonotic disease in humans has made this pathogen a subject of considerable study [18]. While monkey-to-human transmission is more frequent than previously estimated, human-to-human transmission has not been demonstrated. There have been efforts to infect nonhuman primates with human parasites. Some success has been obtained using the Aotus monkey host for P. falciparum, P. vivax, or P. malariae; however, results from these artificial self-curing infections need to be interpreted with caution .
2.1.4 Humans
Experimental malaria in humans has a long and checkered history [19], but thoughtful and safe studies conducted during the World War II era provided invaluable new knowledge on the complex biology (Fig. 1) and pathophysiology of falciparum and vivax malaria, and made possible the rapid development and deployment of amodiaquine, chloroquine, and proguanil to troops fighting in malarious areas of Africa, Europe, and the Pacific. Even today, studying some aspects of antimalarial pharmacodynamics depends heavily on experimental human infections. Prior to large and difficult to control field trials of prophylactic efficacy, small, tightly controlled studies in which well-informed and consenting healthy volunteers are challenged by the bite of malaria-infected mosquitoes or by the inoculation of malaria-infected blood, are standard in assessing drug [20] or vaccine candidates [21] for malaria prophylaxis.
Classical pharmacodynamic endpoints for antimalarial development and use have been clinical—both symptomatic (e.g., time to fever reduction) and microbiological (e.g., time to parasite clearance, 28 day cure rate). In recent years, new metrics have been devised for use in clinical trials. They take cognizance of the fact that unlike most other infections, the pathogenic forms of malaria are confined to erythrocytes in the bloodstream. In severe illness infected cells can number up to 1012, and clinical success of a drug depends on reducing this burden rapidly and completely. From this realization have come clinical measures of rate of killing, rate of recrudescence and parasite reduction ratio (Sect. 2.2.3). In some cases these endpoints are now also being applied to animal and in vitro studies. Demonstrating and counting parasites in a blood smear by simple light microscopy remains the gold standard for measuring antimalarial pharmacodynamic activity. PCR- and antibody-based assays are now also available but logistical and resource limitations restrict their widespread field-deployment.
2.2 In Vitro
For much of the twentieth century, malaria research was restricted to in vivo models since human parasites could not be cultured in vitro. Trager and Jensen’s breakthrough report of the continuous culture of P. falciparum erythrocytic stages in vitro enabled a veritable explosion in malaria research [5]. Unfortunately, this success has not translated to P. vivax, which preferentially infects immature red cells, or to liver stages of the Plasmodium life cycle. Nonetheless, P. falciparum blood stages maintained in vitro form the basis for numerous and diverse pharmacodynamic assays. The following pharmacodynamic endpoints all focus on the asexual erythrocytic parasites, which are responsible for symptomatic disease.
2.2.1 Growth Inhibition
As developed in a micro-titer format, this assay allows high throughput screens and facile measurement of dose–response relationships. Maximum sensitivity is provided by measuring the incorporation of [3H]hypoxanthine into parasite nucleic acid polymers [22]; however, the use of dyes [23] and flow cytometry [24], though less sensitive, avoids the logistical restraints of radioisotopes. Growth inhibition assays do not discriminate between the ‘static or ‘cidal nature of growth inhibition (that is, whether or not the parasite proliferates once drug pressure is lifted). In addition, they provide limited information about the speed of effect—an important parameter for in vivo consideration. Nevertheless, this is a rapid, simple and important first step in screening for antimalarial activity.
2.2.2 Parasiticidal vs. Parasitistatic Activity
A logical follow-up to growth inhibition assay is to determine whether the effect is ‘static, or ‘cidal. In this method parasites are treated with drug, the drug is removed, and individual parasites are cloned out by limiting dilution. Survivors are detectable after a period of 3–6 weeks [25, 26]. The 48 h erythrocytic life cycle and frequent requirement for fresh medium and red cells make these assays cumbersome, time-consuming, costly, and subject to microbial contamination. Less rigorous approaches have been reported [27, 28] but not extensively validated by comparison with the classic method.
2.2.3 Speed of Action and Parasite Reduction Ratio (PRR )
The rapidity of drug action is important in clinical care, and may determine the utilization profile of a drug, particularly in the setting of patients with high parasitemia. Speed of action is measured via the Parasite Reduction Ratio (PRR), the number of parasites present before drug treatment divided by those remaining after treatment. PRR assays are constrained to one life cycle of the parasite; 48 h for P. falciparum. Thus, the higher the PRR, the more rapid is a drug’s effect. PRR studies are usually performed in vivo (in humans or in nonhuman models) wherein high parasitemia before dosing makes measurement of the decline both straightforward and sensitive. Measurement of PRR in vitro has been accomplished, with log PRR values ranging from >8 for artemisinin to <3 for atovaquone [25]. However, in vitro assays rely on limiting dilution cloning and outgrowth, thus taking weeks to yield a result. The standard growth inhibition assay has recently been modified to yield information about speed-of-action of antimalarial drugs [29]. Output of this assay is binary, with compounds being classified as fast-acting or non-fast-acting .
2.2.4 Pharmacokinetic/Pharmacodynamic Linkage
Modifications of an existing hollow fiber cartridge apparatus have very recently made possible studies in which P. falciparum can be exposed in vitro to dynamically changing drug concentrations, akin to those that occur in vivo and distinctly different from the constant drug concentrations usually studied in vitro [30]. This system allows studies of the pharmacokinetics that drive many different antimalarial pharmacodynamics. For example, using known human pharmacokinetics, different dosing regimens can be tested to identify those that provide maximal parasite reduction and/or minimal emergence of resistance. Alternatively, the fundamental governance of drug action by either peak concentration or time of exposure can be discerned by applying a given dose of drug by two artificial (and extremely different) kinetic regimens (Sect. 3.4).
In summary, from a century ago when ducks and chickens were the major vehicle for antimalarial drug development, pharmacodynamic analysis has progressed to the stage where most microbiological endpoints can be assayed in vitro. Complexities of the parasite and the host give rise to a significant number of issues that must be addressed for successful antimalarial development and use, and no single model system or assay is sufficient to address all of them. Multiple approaches remain necessary.
3 Pharmacodynamic Issues
3.1 Life Cycle Stage Specificity
Antimalarial drugs can only be understood, and properly used, in the context of their activity against different forms of the parasite. Plasmodium has a multistage, complex, and dynamic life cycle, even just within the confines of the human host (Fig. 1). Human disease is initiated by the bite of an infected female Anopheline mosquito (the vector), which inoculates sporozoites in the course of taking a blood meal. This form circulates for just a few minutes before infecting hepatocytes, undergoing multiple rounds of asexual reproduction over several weeks, amplifying 10–30,000-fold, rupturing the cell and spilling progeny into the bloodstream. The released merozoites invade erythrocytes, differentiate and replicate asexually 8–32-fold over 48–72 h, before lysing the cell and infecting new erythrocytes. Severely ill patients may harbor up to 1012 erythrocytic parasites. Some erythrocytic forms differentiate into gametocytes, responsible for infecting and undergoing sexual reproduction within the mosquito, eventually to generate salivary gland-resident sporozoites that complete the life cycle. The above biology is common to all pathogenic species of Plasmodium, but vivax and ovale have the additional feature that some liver stage parasites, termed hypnozoites, are latent and may remain quiescent for decades after the mosquito bite, before activating and establishing an erythrocytic cycle [1].
The various life cycle forms within a patient are morphologically, biochemically, and pharmacologically distinct (Fig. 2). The activity of drugs against the various life cycle stages can be used to classify antimalarials into pharmacologically convenient groups [3]. No drug works against sporozoites, and, unfortunately, no drug is active against all forms other than sporozoites. Group I drugs primarily target asexual blood stages. They alone cause morbidity and mortality, hence are the major target of drug therapy. The Group II synergistic combination of atovaquone and proguanil has additional activity against the initial liver stage of P. falciparum. Group III primaquine targets liver stages and gametocytes, but has no useful activity against asexual blood stages. Primaquine’s activity against latent hypnozoites of P. vivax or ovale prevents the late reactivation of symptomatic erythrocytic parasites that characterizes these species. Killing gametocytes prevents transmission of infection to the mosquito and is hence of public health importance.
Choice of appropriate drug is driven, in large part, by the desired outcome. Treatment of, or prophylaxis against, symptomatic malaria is provided by Group I and II agents. Public health campaigns, or regimens striving for complete cure, may include primaquine for its reliable activity against hypnozoites and gametocytes. Prophylaxis can also be obtained by drugs that target liver stage parasites (Groups II and III). Life cycle stage specificity is also important in new drug discovery, the ideal agent being one that has reliable activity against all parasite forms.
3.2 Drug Resistance
The World Health Organization (WHO) defines malaria drug resistance as “the ability of a parasite strain to survive or multiply despite the administration and absorption of a drug given in doses equal to or higher than those usually recommended but within the tolerance of the subject” [31]. Acquisition of resistance, which may be rapid and at a high level, negates the clinical utility of a drug and may jeopardize its entire chemical class. Recognized resistance mechanisms include amplification of, or most commonly point mutation(s) in, a target protein sequence. In recent years experimentally induced resistance to “lead” compounds in drug development has been exploited to obtain invaluable insight into molecular mechanism of action as well as long-term vulnerability to resistance. Interestingly, for leads that generate resistance in the lab, pharmacodynamic utility of the class may be preserved by screening for class members that retain activity against the primary resistance [32]. Additionally, emergence of resistance may be delayed if the molecular mechanism involves multiple molecular targets. As for other anti-infective classes, the most reliable route for precluding resistance in the field is to avoid monotherapy by use of drug combinations.
3.3 Drug Combinations
The pharmacological battle against malaria has laid bare the inadvisability of monotherapy, the end result of which has been emergence and dissemination of resistance to nearly every class of antimalarial drug that has been deployed. Laboratory, and in some cases field, data indicate that antimalarial drug resistance can be delayed, and perhaps avoided, by drug combinations. The earliest and best-studied antimalarial drug combinations stem from Nobel prize-winning studies by Hitchings and Elion on the antibacterial pairing of a sulfonamide inhibitor of dihydropteroate synthase plus a folate reductase inhibitor, both of which interfere with production of essential nutrient tetrahydrofolate [33]. Profound synergism is obtained against malaria parasites by this dual inhibition: when sulfadiazine and pyrimethamine are given in combination (as opposed to singly) the same efficacy is obtained by a 20-fold (or more) reduction in the dose of each drug [34]. (Interestingly, the first-ever inkling of sulfonamide/antifolate synergy against any organism came from Joseph Greenberg’s studies of P. gallinaceum in chicks [35].) The ability to use lesser doses for maximal efficacy reduces cost and the likelihood of host toxicity. Synergism and the well-matched pharmacokinetics of this pair were designed to minimize the emergence of resistance. Two other antimalarial fixed dose combinations have since been marketed. Atovaquone plus proguanil (Sects. 4.4 and 4.5.1) relies on synergistic collapse of the parasite’s transmitochondrial membrane potential [36]. The more empirical pairing of artemether and lumefantrine targets different processes and their pharmacokinetic mismatch (2–3 h vs. 3–6 days) results in long-term persistence of lumefantrine alone, a concern for resistance.
The now-accepted requirement for combination therapy and emergence of several promising new antimalarial leads (Sect. 4.7) have given rise to spirited discussions of the most rational basis for choosing drug pairs [37, 38]. The relative importance of (1) similarities/differences in molecular mechanism of action and in mechanisms of resistance; (2) synergistic, additive or antagonistic interaction; (3) matching half-life; (4) ‘static/‘cidal activity; are all under consideration. While thoughtful consideration and various in silico models can usefully examine these various factors, only experimental work will identify the key determinants of success.
3.4 Pharmacokinetic/Pharmacodynamic Linkage
Ironically, though malaria featured prominently in Paul Ehrlich’s seminal studies toward rational drug discovery [39], and antimalarial drug concentrations were amongst the earliest to be measured in order to understand and guide therapy [40], today the status of pharmacokinetic/pharmacodynamic linkage is notably incomplete. Particularly lacking is an understanding of the fundamental PK governance of antimalarial activity. Extensive study of antibacterials has revealed that pharmacodynamic efficacy is usually driven by a specific pharmacokinetic parameter—C MAX or T MIC (Fig. 3) [41, 42]. Indeed, antibacterials are classified and clinically dosed based this PK/PD link. Recent in vitro studies have demonstrated that such PK/PD governance also pertains for antimalarials. Initial proof of principle work indicates that antimalarials too can be classified as being driven by C MAX or T MIC, independent of their ‘static or ‘cidal action, and that this governance is class-wide [30]. This information may improve empirical dosing regimens [38], and, more importantly, provide new guidance in drug development. For example, an experimental compound that is C MAX-driven may be fully efficacious in vivo, despite having a short-half life. However, short in vivo half-life for a compound that has been shown to be governed by time of exposure would suggest either a NOGO decision or efforts to modify its chemical structure so as to prolong plasma half-life.

Pharmacokinetic parameters of a drug in vivo. Following dosing, drug concentration in blood rises (dashed line) until reaching a peak (C MAX), and then decays at a rate characteristic of the drug and the dosed organism, until all drug is cleared from the system. During this time, drug concentrations spend a certain interval (T MIC) above a predetermined Minimal Inhibitory Concentration (MIC). The Area Under the concentration–time Curve (AUC, diagonal black lines) is an indicator of total drug exposure. The same amount of drug can be dosed via a different regimen using multiple smaller doses (solid line). This regimen yields lower C MAXs but a longer T MIC while maintaining the same AUC (shaded area)
3.5 Safety
Given the enormous number of people afflicted with malaria, the often limited health resources in malaria endemic areas, and the tens of millions of travelers to malarious countries every year who should take chemoprophylaxis (http://www.cdc.gov/malaria/travelers/index.html), it becomes immediately obvious that the requirement for safety is unusually high for antimalarial drugs. This imposing barrier of safety must be kept in mind when selecting candidates for development, and in designing studies to test potential drugs.
3.6 Considerations for New Drug Development
Rational drug development starts with the identification of a suitable molecular target. Screening small molecules in vitro for activity against the cell-free target is followed by testing and development of favored candidates in more complex whole cell assays and animal models. For Plasmodium this process presents several challenges. The eukaryotic nature of Plasmodium, and resultant similarity of basic biochemical mechanisms between the pathogen and its host, immediately narrows the list of unique molecular entities suitable for selective targeting. Intracellular residence of the parasite further complicates drug design. For a lead to be truly efficacious, its molecular target should be accessible and essential during all stages of the parasite life cycle, and preferably be present and required in all species of Plasmodium pathogenic to humans. Finally, there should be a high barrier to resistance, a facet influenced by both the function and redundancy of the target. Rational drug development schema can yield candidates that are tremendously effective in cell-free screens, but a great many of these prove ineffective against erythrocytic parasites in vitro or in vivo. An alternative strategy is to screen against whole cells or animal models; determination of molecular mechanism of action occurs later, if at all, in the process. Most successful antimalarials have been developed through this less-than-rational strategy, indeed chloroquine was discovered and developed using avian and rodent models [43] and its molecular mechanism of action was not described until 50 years later [44]. Artemisinins were co-opted from ancient Chinese remedies [45] and details of their mechanism of efficacy are debated.
The malaria research community has coalesced around the SERCaP—Single Exposure Radical Cure and Prophylaxis —as the ideal for new antimalarial drug development [46]. This sets a very high bar. SERCaP dictates that the treatment regimen be single dose, rapidly efficacious, target all forms of the parasite including latent stages, and have a long-lasting pharmacodynamic effect so as to prevent reinfection. Prudent drug development also demands that the agent be inexpensive, orally bioavailable, and provide a high barrier to resistance. It is improbable that any single molecule will satisfy all of these requirements. Instead, future malaria therapies will likely be based on the combination of multiple agents, each providing a unique spectrum of action. In subsequent sections, we discuss clinically used antimalarial drugs and some experimental agents in development, in context of the aspects described above.
4 Pharmacodynamics of Antimalarial Drugs
4.1 4-Substituted Quinolines
Agents in this class share structural similarities (Fig. 4) and a common molecular mechanism of action; however, resistance is mediated by multiple different mechanisms, not all of which have been characterized.

The substituted quinolines and structurally related compounds . Drugs with substitutions at the 4-position include chloroquine (1), quinine (2), mefloquine (3), and amodiaquine (4). Piperaquine (5) is a bisquinoline while lumefantrine (6) is structurally similar to substituted quinolines. All have potent activity against asexual erythrocytic parasites. Primaquine (7), an 8-substituted quinoline, targets liver stages and gametocytes
4.1.1 Chloroquine
Synthesized by the Germans as Resochin in 1934, and rediscovered by the Allies as SN7618 in 1944, chloroquine (Fig. 4, 1) was for decades the mainstay of antimalarial chemotherapy [43]. Christened chloroquine by E. K. Marshall in 1945, the drug inhibits the essential parasite process of heme detoxification [44]. Plasmodium satisfies most of its amino acid requirements by digesting host cell hemoglobin, releasing free heme in the process. Heme-induced oxidative damage is avoided by nonenzymatic crystallization into the inert polymer hemozoin [7]. Chloroquine concentrates in parasites and inhibits heme crystallization, leading to oxidative damage and death.
While chloroquine-resistant parasites are not readily generated in vitro, the eventual emergence and global dissemination of resistance has rendered chloroquine useless in all but a few locales. Elegant parasite cross-breeding studies pinpointed resistance in P. falciparum to point mutations in PfCRT (P. falciparum chloroquine resistance transporter) [47]. Presence of resistance mutations decreases the accumulation, hence the cytotoxicity, of chloroquine. Curiously, although other 4-substituted quinolines also inhibit heme polymerization [48], and P. vivax may be chloroquine-resistant [31], PfCRT appears not to mediate these resistances [49].
Chloroquine’s tremendous clinical success prior to resistance can be explained by its ability to satisfy many of the pharmacodynamic requirements for an ideal antimalarial. It targets a pathway not present in the human host, is parasiticidal, fast acting, and has a relatively high PRR. Furthermore, the pharmacokinetics of chloroquine are favorable. It persists with a plasma half-life of weeks to months [50]. This long half-life permits the convenient weekly dosing of chloroquine for antimalarial prophylaxis.
Chloroquine toxicity is both dose- and age-related ; doses must be substantially reduced for safe use in children. At doses and regimens required to treat malaria, toxicity in adults is negligible [51]. Rare adverse events include diplopia and dizziness. Cumulative high doses of chloroquine used in anticancer [52] and immunosuppressive therapy [53] may lead to neurotoxicity, cardiotoxicity, and irreversible retinopathy.
4.1.2 Quinine
Quinine (Fig. 4, 2), an alkaloid extracted from the bark of the cinchona tree, was used as an antipyretic by the Quechua in South America. It was transported to Rome by the Jesuits in the early seventeenth century and, in a vivid indication of today’s great need for new antimalarials, this antique natural product remains a drug of choice for treating patients with severe falciparum malaria [54]. Quinine differs from chloroquine in having an ~8 h plasma half-life [50]. This necessitates more frequent dosing, and in severely ill patients, a loading dose [55]. Quinine resistance has been observed in the field, with genetic studies indicating a multifactorial phenotype [56]. Quinine has significant toxicity [51]. Symptoms include the classic cinchonism (disturbances of vision and hearing, headache, nausea), as well as hypoglycemia and hypotension that may be life threatening. This toxicity profile and its relatively short half-life make quinine unsuitable as a prophylaxis agent. Quinidine, an efficacious antiarrhythmic, is a stereoisomer of quinine with potent antimalarial activity. In cases where quinine is unavailable, intravenous quinidine is an acceptable substitute for temporary management of severe malaria [55].
4.1.3 Mefloquine
Mefloquine (Fig. 4, 3) was discovered in a whole-cell screen by the Walter Reed Institute of Medical Research [57], and became an immediate agent of choice for its high activity against drug-resistant P. falciparum [58]. PfCRT does not confer mefloquine-resistance [59]. This fact, combined with a weeks-long half-life [60], enables the use of mefloquine for prophylaxis against chloroquine-resistant malaria. Unfortunately, now-widespread resistance limits mefloquine’s utility. Resistance appears to be multifactorial, and is usually associated with increased expression of multidrug-transporter proteins [59]. Mefloquine toxicity is dose-related, and usually mild at doses used for short-term prophylaxis [51]. The adverse event spectrum expands at higher doses, to include CNS toxicity and neuropsychiatric effects. For this reason, it is not utilized in long-term prophylaxis regimens.
4.1.4 Other 4-Substituted Quinolines and Structural Relatives
Amodiaquine (Fig. 4, 4), an old antimalarial with structural and mechanistic features of chloroquine, has significant activity against chloroquine-resistant Plasmodium [61]. However, its use is disfavored due to an association with hepatotoxicity and agranulocytosis [51]. Piperaquine (Fig. 4, 5) is also active against chloroquine-resistant malaria; its molecular mechanism of action is unclear [62]. It is clinically utilized in combination with dihydroartemisinin. Lumefantrine (Fig. 4, 6), a molecule structurally similar to substituted quinolines, acts against asexual erythrocytic forms of P. falciparum by an unknown molecular mechanism of action [63]. Lumefantrine is FDA-approved and marketed as a fixed-dose combination with artemether (Sect. 4.3) for use against both drug-sensitive and drug-resistant malaria.
4.2 8-Aminoquinol-ines
8-aminoquinoline primaquine (Fig. 4, 7) is the only clinically used antimalarial with reliable activity against initial and latent liver stages and gametocytes (Fig. 2) [64]. Conversely, primaquine has no useful effect on blood stage asexual forms of Plasmodium, and hence it has no place in the acute treatment of symptomatic malaria. Primaquine is the only agent known to eliminate hypnozoites of P. vivax and P. ovale, thus preventing late relapses of these infections. The molecular mechanism of action of primaquine is unclear, but appears to be mediated largely by its metabolites. Primaquine’s vivid toxicities suggest that it acts by generating oxidative species and interfering with redox balance in the pathogen. Primaquine is associated with frequent gastrointestinal intolerance and at high doses causes methemoglobinemia in most people (primaquine use reviewed in [65]). Patients with glucose-6-phosphate dehydrogenase deficiency are particularly susceptible, even at therapeutic doses, to acute, sometimes life threatening, hemolysis and hemolytic anemia [66]. Indeed, primaquine-induced hemolysis led to the discovery of glucose-6-phosphate dehydrogenase deficiency, the first genetic abnormality associated with an enzyme [67].
4.3 The Artemisinins
Artemisinin (Fig. 5, 8) or qinghaosu is the active moiety in Artemesia annua , a plant utilized by Chinese herbalists for over 2000 years [45]. A sesquiterpene lactone endoperoxide, the artemisinins currently form the last line of defense against multidrug-resistant P. falciparum. Their mechanism of action has been the subject of much study. It is generally accepted that the endoperoxide is the active pharmacophore. Iron- or heme-catalyzed cleavage of the oxygen-oxygen bond likely leads to subsequent formation of a carbon-centered radical [68] that in turn alkylates parasite macromolecules. Semisynthetic derivatives artemether (Fig. 5, 9), arteether (Fig. 5, 10), and artesunate (Fig. 5, 11) are more soluble than parent artemisinin, but act in a similar fashion. While these compounds themselves have antimalarial activity, they also act as prodrugs in vivo. All derivatives (8–11) are rapidly converted in vivo to dihydroartemisinin (Fig. 5, 12), itself an antimalarial [69]. The artemisinins have potent and rapid activity against asexual erythrocytic stages of P. falciparum and P. vivax, making them particularly useful in severely ill patients with high parasite burden.

The artemisinins. Members of this family include natural product artemisinin (8), and its semisynthetic derivatives artemether (9), arteether (10), artesunate (11), and dihydroartemisinin (12). These compounds are an obligate component of most clinically efficacious antimalarial drug regimens
A distinguishing characteristic of the artemisinins is their extremely short half-life [70]. Cleared from plasma within minutes, their great activity against a parasite with a complex 48 h life cycle has always been puzzling. However, in vitro PK/PD studies have demonstrated that artemisinin efficacy is C MAX-driven, a mechanism that aligns ideally with its short half-life, providing a satisfactory explanation for the clinical success of such remarkably short-lived drugs [30]. Interestingly, daily dose artemisinin monotherapy, even for 7 days, invariably leads to recrudescence of parasitemia [71]. Explanations for this phenomenon include differential effects on life-cycle stages and/or induction of post-treatment “quiescence” in surviving parasites [72]. Long-lived endoperoxides (Sect. 4.7.4) have been developed in an effort to remedy this problem [73]. In any case, artemisinins are now almost always used in combination regimens.
Efficacy of the artemisinins has recently been threatened by the discovery of a resistance-like phenomenon. While not meeting WHO’s definition of leading to treatment failure, the effect manifests pharmacodynamically as a slower rate of parasite clearance and longer persistence of parasites in vivo [74]. Genetic studies suggest that parasite genotype accounts only partially for the observed clinical phenotype, implying the existence of as-yet-undefined contributions from the human host [75]. Although geographically still restricted, this “resistance” phenotype has begun a slow march out of its initial focus in Southeast Asia. Recent genome-wide association studies on “resistant” parasites [76], as well as attempts to recapitulate resistance in vitro, have focused on polymorphisms in a region of chromosome 13 (encoding a protein containing a kelch 13 propeller domain) that appear to correlate with the phenotype [77]. Recent in vitro work has demonstrated that these kelch 13 polymorphisms are necessary and sufficient to enhance the survival of the parasite ring stage in the face of artemisinin pressure [78]. While the exact molecular mechanism is unclear, transcriptomic analysis suggests that this ‘resistance’ is mediated via an upregulation of the parasite’s unfolded protein response [79]. Artemisinins currently form the last line of defense against drug-resistant malaria and the threat to their efficacy has, appropriately, spurred greater urgency in new antimalarial drug development.
Artemisinins are considered relatively safe, having been used for decades in millions of humans. Recognized toxicities include hemolysis and hypersensitivity reactions . Animal toxicology studies indicate that brain, liver, bone marrow, and fetus may be affected. This adverse event profile has not been unambiguously demonstrated in humans treated with therapeutic doses [51].
4.4 Atovaquone
Atovaquone (Fig. 6, 13) is an analog of coenzyme Q that specifically targets the cytochrome bc1 complex of the mitochondrial respiratory chain. It interferes with mitochondrial functions, including pyrimidine biosynthesis, by inhibiting electron transport and collapsing the mitochondrial transmembrane potential [80]. Potent activity against asexual erythrocytic forms of P. falciparum and P. vivax, and the ability to eliminate P. falciparum liver stage parasites makes this agent especially useful for prophylaxis. Atovaquone is relatively safe—common adverse events include headache, rash, abdominal pain, vomiting, and diarrhea.

Atovaquone, antifolates, and sulfonamides . Atovaquone (13) is a cytochrome bc1 inhibitor that is effective on its own, synergizes with the biguanide proguanil (14), and is clinically always used in combination. Proguanil is a prodrug metabolized in vivo to cycloguanil (15), a dihydrofolate reductase inhibitor. Pyrimethamine (16) also targets dihydrofolate reductase and is used in synergistic combination with sulfadoxine (17), a dihydropteroate synthase-targeting sulfonamide
In its very first clinical trials atovaquone failed spectacularly, and quite unexpectedly, thanks to the rapid emergence of drug-resistant P. falciparum , mediated by point mutations in target cytochrome b (atovaquone reviewed in [36]). Utility of this potent, safe, relatively long-lived candidate was saved by its synergistic combination, demonstrable both in lab and clinic, with veteran antimalarial proguanil (Sect. 4.5.1).
4.5 Antifolates and Sulfonamides
Sulfonamides and antifolates were among the earliest synthetic antimalarial agents, and were developed primarily using avian models of malaria.
4.5.1 Proguanil and Cycloguanil
The biguanide proguanil (Fig. 6, 14) targets Plasmodium in multiple ways. Best known as a prodrug, in vivo it is metabolized by CYP2C19 to cycloguanil (Fig. 6, 15) [81], which inhibits P. falciparum dihydrofolate reductase [82]. However, proguanil is also active in vitro (where it is not converted to cycloguanil) suggesting other direct (and unknown) targets [83]. Pharmacogenetic differences in CYP2C19 can affect the conversion to cycloguanil, and patients with CYP2C19*2–CYP2C19*8 alleles (poor metabolizers) may not fully convert proguanil to cycloguanil [81]. Clinical relevance of the poor metabolizer phenotype with respect to malaria is the subject of debate. Proguanil is remarkably safe at therapeutic doses. Resistance against cycloguanil is conferred by point mutations in Plasmodium dihydrofolate reductase [84], and these arose and spread rapidly in the era of proguanil monotherapy. Clinical use of proguanil is now largely confined to its synergistic combination with atovaquone (Sect. 4.4). While proguanil itself displays no effect on the mitochondrial membrane potential, it greatly enhances atovaquone’s effect [85].
4.5.2 Pyrimethamine
Pyrimethamine (Fig. 6, 16) is a diaminopyrimidine that targets Plasmodium dihydrofolate reductase [82]. The effects of inhibiting folate metabolism manifest late in the replication cycle of asexual erythrocytic forms, making pyrimethamine a slow-acting drug. Pharmacodynamic efficacy of pyrimethamine can be augmented by host immunity, and significantly inhibited by dietary p-aminobenzoic acid or folate. Point mutations in Plasmodium dihydrofolate reductase confer resistance to this drug [86]. Therapeutic doses of pyrimethamine are safe; excessive doses can recapitulate symptoms of folate deficiency [51]. Pyrimethamine is usually dosed in combination with a sulfonamide or sulfone to create a synergistic effect. However, the utility of this combination is limited by widespread drug resistance, as well as by intrinsic toxicity of the sulfonamides.
4.5.3 Sulfonamides and Sulfones
Sulfonamides and sulfones inhibit Plasmodium dihydropteroate synthase [87], an enzyme involved in folate biosynthesis that has no counterpart in humans. These agents are slow acting and readily generate mutations in target protein dihydropteroate synthase [88], which confers class-wide resistance. Clinical utility is confined to coadministration with an antifolate partner drug. The combination of sulfadoxine (Fig. 6, 17) with pyrimethamine is particularly effective since multiple steps of the same biosynthetic pathway are affected. This particular combination was designed to include partners with matched in vivo pharmacokinetics (both drugs are long-lived), thus avoiding functional monotherapy with either agent towards the end of the dosing interval. The use of sulfonamides and sulfonamide-containing combinations is limited by toxicity, including Stevens–Johnson syndrome and exfoliative dermatitis [51]. Additionally, widespread resistance of Plasmodium to both partner drugs has compromised the utility of this drug class and its combinations.
4.6 Antibacterials
Tetracycline (Fig. 7, 18) and doxycycline (Fig. 7, 19) are slow acting agents [61] that target the apicoplast, a chloroplast-like organelle in malaria parasites. Interference with apicoplast function results in delayed death of the parasite: effects of the drug are not manifest until the next replication cycle [89]. This delayed effect makes the tetracyclines unsuitable for treatment of established severe infections; however, they are useful as adjunctive therapy and for short-term prophylaxis. Adverse effects that limit their use include discoloring depositions in bones and teeth, and a tendency to cause photosensitivity [90].

Antibacterials. Tetracycline (18) and doxycycline (19) are slow-acting antimalarials useful for adjunctive therapy and short-term prophylaxis
4.7 Experimental Agents
Today’s global development of new antimalarial drugs is largely coordinated by the Medicines for Malaria Venture (MMV) , a nonprofit public–private foundation. MMV coordinates industrial and academic antimalarial efforts and facilitates progress of drug candidates through the long and complex development pathway [91]. A current snapshot of the global antimalarial development portfolio can be found on the foundation’s website (www.mmv.org). There are multiple new drugs or drug combinations in various stages of development [92], ranging from the discovery phase to post-approval management. The following are agents currently in human trials, with a focus on novel pharmacophores, molecular targets, or pharmacokinetics.
4.7.1 DSM265
Developed by a multinational academic collaboration, DSM265 (Fig. 8, 20) targets dihydroorotate dehydrogenase, a mitochondrial enzyme in P. falciparum essential for pyrimidine biosynthesis and parasite survival [93]. The compound achieves several important benchmarks in that it is potent, selective, active against chloroquine-resistant parasites, bioavailable and metabolically stable. It will likely be deployed in combination therapy with a suitable partner.

Experimental antimalarials. Triazolopyrimidine DSM265 (20) inhibits dihydroorotate dehydrogenase, an essential malarial enzyme. KAE609 (21) targets the PfATP4 (Na+) channel and KAF156 (22) is an imidazolopiperazine active against both blood and liver stages of the parasite life cycle. OZ439 (23) contains the endoperoxide pharmacophore of the artemisinins, but is designed to persist in the blood
4.7.2 KAE609
KAE609 (Fig. 8, 21) is a spiroindolone that targets a P-type ATPase Na(+) channel of P. falciparum [94]. Developed from a natural product screen in an industry–academia collaboration, KAE609 is a novel pharmacophore targeting a subsequently described and hitherto unexplored Plasmodium function, and has successfully completed Phase IIA clinical trials. The relative ease of generating resistance in vitro is somewhat worrisome; however, this efficacious compound will provide much-needed diversity to the antimalarial armamentarium.
4.7.3 KAF 156
An imidazopyrazine derivative discovered and developed through a whole cell screening approach, KAF156 (Fig. 8, 22) targets both liver and blood stages of Plasmodium [95]. This multistage activity, coupled with acceptable pharmacokinetics, suggests that KAF156 has potential to provide both the Radical Cure and the Protection demanded by the SERCaP model.
4.7.4 OZ439
OZ439 (Fig. 8, 23) is member of the synthetic ozonides [96]. Its endoperoxide motif mimics that of the artemisinins, and their mechanism of action is likely in common. The unique feature of OZ439 is its metabolic stability: a terminal half-life of 25–30 h compared to half-lives of just minutes for the artemisinins [73]. Recent in vitro research on artemisinin resistance has yielded the intriguing suggestion that the resistance phenotype may be mediated by a quiescence or dormancy mechanism that simply allows the parasite to outlast the short-lived drug [72]. If so, long-t 1/2 endoperoxide OZ439 will be crucial for combating and perhaps reversing artemisinin resistance in the field.
5 Summary
Malaria is a public health problem of immense proportions. The complex biology of malaria makes drug development and use particularly challenging, a situation exacerbated by drug resistance. The parasite is a eukaryote, which limits the availability of targets unique to the pathogen. Plasmodium species pathogenic to humans will not infect other animals, rendering animal models deficient. Finally, human parasites are difficult, and in some cases impossible, to culture in vitro, limiting development of laboratory assays.
The asexual erythrocytic stages that cause symptomatic illness are the primary targets of treatment. However, prevention and public health strategies necessitate the targeting of liver stages and gametocytes, both of which are pharmacologically distinct from the asexual blood stages.
Initial pharmacodynamic endpoints for antimalarial development were clinical and required human studies. Surrogate animal models and in vitro assays have matured to the point where a rational combination of assays yields adequate information to aid drug development, and these in conjunction with contemporary screening methods have yielded a number of highly promising experimental drug candidates.
Successful antimalarial drugs must fulfil certain criteria. The drug must be potent; provide a single-dose cure; act rapidly; and target all stages of the parasite life cycle. Drug pharmacokinetics in vivo must align with the essential pharmacokinetic–pharmacodynamic linkage. The molecule must possess a high barrier to resistance. And ultimately, the drug must be extremely safe, given expected administration to millions of patients and to healthy uninfected travelers. No single drug currently fulfils all these criteria. Indeed, it is unlikely any agent ever will. Successful antimalarial regimens will require combinations of drugs, rationally selected to provide best activity.
Existing antimalarials have proven successful because they fulfill some of the requirements listed above. New drug development efforts have formalized these benchmarks and progress along the development pathway is now governed by these criteria. While there are several new pharmacophores targeting novel targets, and some old ones aiming for different processes, antimalarial drug development is likely to remain a challenging endeavor for the foreseeable future.
References
Fairhurst RM, Wellems TE (2010) Plasmodium species (Malaria). In: Mandell GL, Bennett JE, Donlin R (eds) Mandel, Douglas and Bennett’s principles and practice of infectious diseases, vol 2, Churchill. Livingstone, Philadelphia, PA, pp 3437–3462
White NJ, Pukrittayakamee S, Hien TT et al (2014) Malaria. Lancet 383(9918):723–735. doi:10.1016/S0140-6736(13)60024-0
Shapiro TA, Goldberg DE (2006) Chemotherapy of protozoal infections: malaria. In: Brunton LL, Lazo JS, Parker KL (eds) Goodman and Gilman’s the pharmacological basis of therapeutics, 11th edn. McGraw-Hill, New York, pp 1021–1047
Gardner MJ, Hall N, Fung E et al (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419(6906):498–511. doi:10.1038/nature01097
Trager W, Jensen JB (1976) Human malaria parasites in continuous culture. Science 193(4254):673–675
Dahl EL, Rosenthal PJ (2008) Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol 24(6):279–284. doi:10.1016/j.pt.2008.03.007
Francis SE, Sullivan DJ Jr, Goldberg DE (1997) Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51:97–123. doi:10.1146/annurev.micro.51.1.97
Painter HJ, Morrisey JM, Mather MW et al (2007) Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446(7131):88–91. doi:10.1038/nature05572
Gamo FJ, Sanz LM, Vidal J et al (2010) Thousands of chemical starting points for antimalarial lead identification. Nature 465(7296):305–310. doi:10.1038/nature09107
Guiguemde WA, Shelat AA, Bouck D et al (2010) Chemical genetics of Plasmodium falciparum. Nature 465(7296):311–315. doi:10.1038/nature09099
Rottmann M, McNamara C, Yeung BK et al (2010) Spiroindolones, a potent compound class for the treatment of malaria. Science 329(5996):1175–1180. doi:10.1126/science.1193225
McGhee RB (1988) Major animal models in malaria research: avian. In: Wernsdorfer WH, McGregor I (eds) Malaria: principles and practice of malariology, vol 2. Churchill Livingstone, Edinburgh, pp 1545–1567
Cox FEG (1988) Major animal models in malaria research: rodent. In: Wernsdorfer WH, McGregor I (eds) Malaria: principles and practice of malariology, vol 2. Churchill Livingstone, Edinburgh, pp 1503–1543
Fidock DA, Rosenthal PJ, Croft SL et al (2004) Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 3(6):509–520. doi:10.1038/nrd1416
Craig AG, Grau GE, Janse C et al (2012) The role of animal models for research on severe malaria. PLoS Pathog 8(2):e1002401. doi:10.1371/journal.ppat.1002401
Angulo-Barturen I, Jimenez-Diaz MB, Mulet T et al (2008) A murine model of falciparum-malaria by in vivo selection of competent strains in non-myelodepleted mice engrafted with human erythrocytes. PLoS One 3(5):e2252. doi:10.1371/journal.pone.0002252
Collins WE (1988) Major animal models in malaria research: simian. In: Wernsdorfer WH, McGregor I (eds) Malaria: principles and practice of malariology, vol 2. Churchill Livingstone, Edinburgh, pp 1473–1501
Cox-Singh J (2012) Zoonotic malaria: Plasmodium knowlesi, an emerging pathogen. Curr Opin Infect Dis 25(5):530–536. doi:10.1097/QCO.0b013e3283558780
Bruce-Chwatt LJ (1967) Clinical trials of anti-malarial drugs. Trans R Soc Trop Med Hyg 61(3):412–426
Shapiro TA, Ranasinha CD, Kumar N et al (1999) Prophylactic activity of atovaquone against Plasmodium falciparum in humans. Am J Trop Med Hyg 60(5):831–836
Seder RA, Chang LJ, Enama ME et al (2013) Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341(6152):1359–1365. doi:10.1126/science.1241800
Desjardins RE, Canfield CJ, Haynes JD et al (1979) Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16(6):710–718
Bennett TN, Paguio M, Gligorijevic B et al (2004) Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob Agents Chemother 48(5):1807–1810
Karl S, Wong RP, St Pierre TG et al (2009) A comparative study of a flow-cytometry-based assessment of in vitro Plasmodium falciparum drug sensitivity. Malar J 8:294. doi:10.1186/1475-2875-8-294
Sanz LM, Crespo B, De-Cozar C et al (2012) P. falciparum in vitro killing rates allow to discriminate between different antimalarial mode-of-action. PLoS One 7(2):e30949. doi:10.1371/journal.pone.0030949
Young RD, Rathod PK (1993) Clonal viability measurements on Plasmodium falciparum to assess in vitro schizonticidal activity of leupeptin, chloroquine, and 5-fluoroorotate. Antimicrob Agents Chemother 37(5):1102–1107
Paguio MF, Bogle KL, Roepe PD (2011) Plasmodium falciparum resistance to cytocidal versus cytostatic effects of chloroquine. Mol Biochem Parasitol 178(1–2):1–6. doi:10.1016/j.molbiopara.2011.03.003
Painter HJ, Morrisey JM, Vaidya AB (2010) Mitochondrial electron transport inhibition and viability of intraerythrocytic Plasmodium falciparum. Antimicrob Agents Chemother 54(12):5281–5287. doi:10.1128/AAC.00937-10
Le Manach C, Scheurer C, Sax S et al (2013) Fast in vitro methods to determine the speed of action and the stage-specificity of anti-malarials in Plasmodium falciparum. Malar J 12:424. doi:10.1186/1475-2875-12-424
Bakshi RP, Nenortas E, Tripathi AK et al (2013) Model system to define pharmacokinetic requirements for antimalarial drug efficacy. Sci Transl Med 5(205):205ra135. doi:10.1126/scitranslmed.3006684
Barrette A, Ringwald P (2010) Global report on antimalarial drug efficacy and drug resistance: 2000–2010. World Health Organization, Switzerland
Zhao X, Xu C, Domagala J et al (1997) DNA topoisomerase targets of the fluoroquinolones: a strategy for avoiding bacterial resistance. Proc Natl Acad Sci U S A 94(25):13991–13996
Hitchings GH (1969) A quarter century of chemotherapy. JAMA 209(9):1339–1340
Richards WHG (1966) Antimalarial activity of sulphonamides and a sulphone, singly and in combination with pyrimethamine, against drug resistant and normal strains of laboratory plasmodia. Nature 212(5069):1494–1495
Greenberg J, Boyd BL, Josephson ES (1948) Synergistic effect of chlorguanide and sulfadiazine against Plasmodium gallinaceum in the chick. J Pharmacol Exp Ther 94(1):60–64
Vaidya A (2001) Atovaquone-proguanil combination. In: Rosenthal PJ (ed) Antimalarial chemotherapy. Humana Press Inc., Totowa, NJ, pp 203–218
Hastings IM, Hodel EM (2014) Pharmacological considerations in the design of anti-malarial drug combination therapies—is matching half-lives enough? Malar J 13(1):62. doi:10.1186/1475-2875-13-62
White NJ (2013) Pharmacokinetic and pharmacodynamic considerations in antimalarial dose optimization. Antimicrob Agents Chemother 57(12):5792–5807. doi:10.1128/AAC.00287-13
Lazarus A (1922) Paul Ehrlich, vol 2. Rikola, Wien
Lipkin IJ, Ramsden W (1918) Nephelometric estimation of quinine in blood and urine. Br Med J 1(2994):560–561
Craig WA (1998) Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26(1):1–10, quiz 11–12
Drusano GL (2004) Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat Rev Microbiol 2(4):289–300. doi:10.1038/nrmicro862
Coatney GR (1963) Pitfalls in a discovery: the chronicle of chloroquine. Am J Trop Med Hyg 12:121–128
Sullivan DJ Jr, Gluzman IY, Russell DG et al (1996) On the molecular mechanism of chloroquine’s antimalarial action. Proc Natl Acad Sci U S A 93(21):11865–11870
Klayman DL (1985) Qinghaosu (artemisinin): an antimalarial drug from China. Science 228(4703):1049–1055
Alonso PA, Djimde A, Magill A et al (2011) A research agenda for malaria eradication: drugs. PLoS Med 8(1):e1000402. doi:10.1371/journal.pmed.1000402
Fidock DA, Nomura T, Talley AK et al (2000) Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6(4):861–871
Sullivan DJ Jr, Matile H, Ridley RG et al (1998) A common mechanism for blockade of heme polymerization by antimalarial quinolines. J Biol Chem 273(47):31103–31107
Nomura T, Carlton JM, Baird JK et al (2001) Evidence for different mechanisms of chloroquine resistance in 2 Plasmodium species that cause human malaria. J Infect Dis 183(11):1653–1661. doi:10.1086/320707
Krishna S, White NJ (1996) Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications. Clin Pharmacokinet 30(4):263–299
Taylor WR, White NJ (2004) Antimalarial drug toxicity: a review. Drug Saf 27(1):25–61
Amaravadi RK, Lippincott-Schwartz J, Yin XM et al (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17(4):654–666. doi:10.1158/1078-0432.CCR-10-2634
Meyer KC, Decker C, Baughman R (2010) Toxicity and monitoring of immunosuppressive therapy used in systemic autoimmune diseases. Clin Chest Med 31(3):565–588. doi:10.1016/j.ccm.2010.05.006
Achan J, Talisuna AO, Erhart A et al (2011) Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J 10:144. doi:10.1186/1475-2875-10-144
Warrell DA (1989) Treatment of severe malaria. J R Soc Med 82(Suppl 17):44–50, discussion 50–51
Foley M, Tilley L (1998) Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther 79(1):55–87
Osdene TS, Russell PB, Rane L (1967) 2,4,7-Triamino-6-ortho-substituted Arylpteridines. A new series of potent antimalarial agents. J Med Chem 10(3):431–434. doi:10.1021/jm00315a031
Trenholme CM, Williams RL, Desjardins RE et al (1975) Mefloquine (WR 142,490) in the treatment of human malaria. Science 190(4216):792–794
Price RN, Uhlemann AC, Brockman A et al (2004) Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364(9432):438–447. doi:10.1016/S0140-6736(04)16767-6
Karbwang J, White NJ (1990) Clinical pharmacokinetics of mefloquine. Clin Pharmacokinet 19(4):264–279. doi:10.2165/00003088-199019040-00002
Rieckmann KH (1971) Determination of the drug sensitivity of Plasmodium falciparum. JAMA 217(5):573–578
Davis TM, Hung TY, Sim IK et al (2005) Piperaquine: a resurgent antimalarial drug. Drugs 65(1):75–87
White NJ, van Vugt M, Ezzet F (1999) Clinical pharmacokinetics and pharmacodynamics and pharmacodynamics of artemether-lumefantrine. Clin Pharmacokinet 37(2):105–125
Arnold J, Alving AS, Hockwald RS et al (1955) The antimalarial action of primaquine against the blood and tissue stages of falciparum malaria (Panama, P-F-6 strain). J Lab Clin Med 46(3):391–397
Hill DR, Baird JK, Parise ME et al (2006) Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg 75(3):402–415
Beutler E (1959) The hemolytic effect of primaquine and related compounds: a review. Blood 14(2):103–139
Alving AS, Carson PE, Flanagan CL et al (1956) Enzymatic deficiency in primaquine-sensitive erythrocytes. Science 124(3220):484–485
Posner GH, Park SB, Gonzalez L et al (1996) Evidence for the importance of high-valent Fe=O and of a diketone in the molecular mechanism of action of antimalarial trioxane analogs of artemisinin. J Am Chem Soc 118:3537–3538
Newton PN, Barnes KI, Smith PJ et al (2006) The pharmacokinetics of intravenous artesunate in adults with severe falciparum malaria. Eur J Clin Pharmacol 62(12):1003–1009. doi:10.1007/s00228-006-0203-2
Benakis A, Paris M, Loutan L et al (1997) Pharmacokinetics of artemisinin and artesunate after oral administration in healthy volunteers. Am J Trop Med Hyg 56(1):17–23
Hassan Alin M, Ashton M, Kihamia CM et al (1996) Multiple dose pharmacokinetics of oral artemisinin and comparison of its efficacy with that of oral artesunate in falciparum malaria patients. Trans R Soc Trop Med Hyg 90(1):61–65
Klonis N, Xie SC, McCaw JM et al (2013) Altered temporal response of malaria parasites determines differential sensitivity to artemisinin. Proc Natl Acad Sci U S A 110(13):5157–5162. doi:10.1073/pnas.1217452110
Moehrle JJ, Duparc S, Siethoff C et al (2013) First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol 75(2):524–537. doi:10.1111/j.1365-2125.2012.04368.x
Dondorp AM, Yeung S, White L et al (2010) Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol 8(4):272–280. doi:10.1038/nrmicro2331
Anderson TJ, Nair S, Nkhoma S et al (2010) High heritability of malaria parasite clearance rate indicates a genetic basis for artemisinin resistance in western Cambodia. J Infect Dis 201(9):1326–1330. doi:10.1086/651562
Takala-Harrison S, Clark TG, Jacob CG et al (2013) Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A 110(1):240–245. doi:10.1073/pnas.1211205110
Ariey F, Witkowski B, Amaratunga C et al (2014) A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505(7481):50–55. doi:10.1038/nature12876
Hudson AT (1993) Atovaquone—a novel broad-spectrum anti-infective drug. Parasitol Today 9(2):66–68
Desta Z, Zhao X, Shin JG et al (2002) Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 41(12):913–958. doi:10.2165/00003088-200241120-00002
Ferone R, Burchall JJ, Hitchings GH (1969) Plasmodium berghei dihydrofolate reductase. Isolation, properties, and inhibition by antifolates. Mol Pharmacol 5(1):49–59
Watkins WM, Sixsmith DG, Chulay JD (1984) The activity of proguanil and its metabolites, cycloguanil and p-chlorophenylbiguanide, against Plasmodium falciparum in vitro. Ann Trop Med Parasitol 78(3):273–278
Foote SJ, Galatis D, Cowman AF (1990) Amino acids in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum involved in cycloguanil resistance differ from those involved in pyrimethamine resistance. Proc Natl Acad Sci U S A 87(8):3014–3017
Srivastava IK, Vaidya AB (1999) A mechanism for the synergistic antimalarial action of atovaquone and proguanil. Antimicrob Agents Chemother 43(6):1334–1339
Peterson DS, Walliker D, Wellems TE (1988) Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci U S A 85(23):9114–9118
Triglia T, Menting JG, Wilson C et al (1997) Mutations in dihydropteroate synthase are responsible for sulfone and sulfonamide resistance in Plasmodium falciparum. Proc Natl Acad Sci U S A 94(25):13944–13949
Wang P, Read M, Sims PF et al (1997) Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization. Mol Microbiol 23(5):979–986
Dahl EL, Shock JL, Shenai BR et al (2006) Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob Agents Chemother 50(9):3124–3131. doi:10.1128/AAC.00394-06
Clendenning WE (1965) Complications of tetracycline therapy. Arch Dermatol 91:628–632
Burrows JN, Burlot E, Campo B et al (2014) Antimalarial drug discovery—the path towards eradication. Parasitology 141(1):128–139. doi:10.1017/S0031182013000826
Flannery EL, Chatterjee AK, Winzeler EA (2013) Antimalarial drug discovery—approaches and progress towards new medicines. Nat Rev Microbiol 11(12):849–862. doi:10.1038/nrmicro3138
Coteron JM, Marco M, Esquivias J et al (2011) Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem 54(15):5540–5561. doi:10.1021/jm200592f
Spillman NJ, Allen RJ, McNamara CW et al (2013) Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 13(2):227–237. doi:10.1016/j.chom.2012.12.006
McNamara CW, Lee MC, Lim CS et al (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504(7479):248–253. doi:10.1038/nature12782
Vennerstrom JL, Arbe-Barnes S, Brun R et al (2004) Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430(7002):900–904. doi:10.1038/nature02779
Acknowledgements
We thank Drs. David Sullivan and Kartiki Desai for their careful reading of the manuscript and thoughtful comments; Dr. Elizabeth Nenortas for proofing chemical structures; and Rachel Shapiro Grasmick for drawing the malaria life cycle. This project was supported by the Johns Hopkins Malaria Research Institute, the Bloomberg Family Foundation, and by NIH grants R01AI095453 and R01AI111962.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Bakshi, R.P., Shapiro, T.A. (2016). Pharmacodynamics of Antimalarial Agents. In: Rotschafer, J., Andes, D., Rodvold, K. (eds) Antibiotic Pharmacodynamics. Methods in Pharmacology and Toxicology. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3323-5_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3323-5_17
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3321-1
Online ISBN: 978-1-4939-3323-5
eBook Packages: Springer Protocols