
Abstract
Whole genome amplification (WGA) is a widely used molecular technique that is becoming increasingly necessary in genetic research on a range of sample types including individual cells, fossilized remains and entire ecosystems. Multiple methods of WGA have been developed, each with specific strengths and weaknesses, but with a common defect in that each method distorts the initial template DNA during the course of amplification. The type, extent, and circumstance of the bias vary with the WGA method and particulars of the template DNA.
In this review, we endeavor to discuss the types of bias introduced, the susceptibility of common WGA techniques to these bias types, and the interdependence between bias and characteristics of the template DNA. Finally, we attempt to illustrate some of the criteria specific to the analytical platform and research application that should be considered to enable combination of the appropriate WGA method, template DNA, sequencing platform, and intended use for optimal results.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
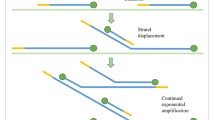

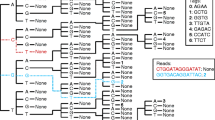
References
Cohen SN, Chang ACY, Boyer HW, Helling RB (1973) Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci 70(11):3240–3244
Jackson DA, Symons RH, Berg P (1972) Biochemical method for inserting new genetic information into DNA of simian virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc Natl Acad Sci 69(10):2904–2909
Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230(4732):1350–1354
Bergen AW, Haque KA, Qi Y, Beerman MB, Garcia-Closas M, Rothman N, Chanock SJ (2005) Comparison of yield and genotyping performance of multiple displacement amplification and OmniPlex whole genome amplified DNA generated from multiple DNA sources. Hum Mutat 26(3):262–270. doi:10.1002/humu.20213
Blainey PC (2013) The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev 37(3):407–427. doi:10.1111/1574-6976.12015
Stepanauskas R (2012) Single cell genomics: an individual look at microbes. Curr Opin Microbiol 15(5):613–620. doi:10.1016/j.mib.2012.09.001
Yilmaz S, Singh AK (2012) Single cell genome sequencing. Curr Opin Biotechnol 23(3):437–443. doi:10.1016/j.copbio.2011.11.018
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M (2011) Tumour evolution inferred by single-cell sequencing. Nature 472(7341):90–94, doi:http://www.nature.com/nature/journal/v472/n7341/abs/10.1038-nature09807-unlocked.html#supplementary-information
Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Höfler G, Eisner F, Sill H, Samonigg H, Pantel K, Riethdorf S, Bauernhofer T, Geigl JB, Speicher MR (2013) Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res 73(10):2965–2975. doi:10.1158/0008-5472.can-12-4140
Shapiro E, Biezuner T, Linnarsson S (2013) Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet 14(9):618–630. doi:10.1038/nrg3542
Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N (1992) Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci U S A 89(13):5847–5851
Telenius H, Carter N, Bebb C, Nordenskjold M, Ponder B, Tunnacliffe A (1992) Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13(3):718–725
Dean F, Hosono S, Fang L, Wu X, Faruqi A, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J (2002) Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A 99(8):5261–5266
Lage J, Leamon J, Pejovic T, Hamann S, Lacey M, Dillon D, Segraves R, Vossbrinck B, Gonzalez A, Pinkel D, Albertson D, Costa J, Lizardi P (2003) Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res 13:294–307
Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC (1998) Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet 19(3):225–232. doi:10.1038/898
Kaper F, Swamy S, Klotzle B, Munchel S, Cottrell J, Bibikova M, Chuang H-Y, Kruglyak S, Ronaghi M, Eberle MA, Fan J-B (2013) Whole-genome haplotyping by dilution, amplification, and sequencing. Proc Natl Acad Sci 110(14):5552–5557. doi:10.1073/pnas.1218696110
Hughes S, Arneson N, Done S, Squire J (2005) The use of whole genome amplification in the study of human disease. Prog Biophys Mol Biol 88(1):173–189, doi: http://dx.doi.org/10.1016/j.pbiomolbio.2004.01.007
Roberts JD, Kunkel TA (1988) Fidelity of a human cell DNA replication complex. Proc Natl Acad Sci 85(19):7064–7068
Wabl M, Burrows PD, von Gabain A, Steinberg C (1985) Hypermutation at the immunoglobulin heavy chain locus in a pre-B-cell line. Proc Natl Acad Sci U S A 82(2):479–482
Kanagawa T (2003) Bias and artifacts in multitemplate polymerase chain reactions (PCR). J Biosci Bioeng 96(4):317–323. doi:10.1016/S1389-1723(03)90130-7
Head IM, Saunders JR, Pickup RW (1998) Microbial evolution, diversity, and ecology: a decade of ribosomal RNA analysis of uncultivated microorganisms. Microb Ecol 35(1):1–21
von Wintzingerode F, Gobel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21(3):213–229
Wagner A, Blackstone N, Cartwright P, Dick M, Misof B, Snow P, Wagner GP, Bartels J, Murtha M, Pendleton J (1994) Surveys of gene families using polymerase chain-reaction – PCR selection and PCR drift. Syst Biol 43(2):250–261. doi:10.2307/2413465
Kurata S, Kanagawa T, Magariyama Y, Takatsu K, Yamada K, Yokomaku T, Kamagata Y (2004) Reevaluation and reduction of a PCR bias caused by reannealing of templates. Appl Environ Microbiol 70:7545–7549
Ishii K, Fukui M (2001) Optimization of annealing temperature to reduce bias caused by a primer mismatch in multitemplate PCR. Appl Environ Microbiol 67(8):3753–3755. doi:10.1128/AEM.67.8.3753-3755.2001
Lueders T, Friedrich MW (2003) Evaluation of PCR amplification bias by terminal restriction fragment length polymorphism analysis of small-subunit rRNA and mcrA genes by using defined template mixtures of methanogenic pure cultures and soil DNA extracts. Appl Environ Microbiol 69(1):320–326
Polz M, Cavanaugh C (1998) Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol 64:3724–3730
Zheng D, Alm EW, Stahl DA, Raskin L (1996) Characterization of universal small-subunit rRNA hybridization probes for quantitative molecular microbial ecology studies. Appl Environ Microbiol 62(12):4504–4513
Arriola E, Lambros M, Jones C, Dexter T, Mackay A, Tan D, Tamber N, Fenwick K, Ashworth A, Dowsett M (2007) Evaluation of Phi29-based whole-genome amplification for microarray-based comparative genomic hybridisation. Lab Invest 87(1):75–83
Bredel M, Bredel C, Juric D, Kim Y, Vogel H, Harsh GR, Recht LD, Pollack JR, Sikic BI (2005) Amplification of whole tumor genomes and gene-by-gene mapping of genomic aberrations from limited sources of fresh-frozen and paraffin-embedded DNA. J Mol Diagn 7(2):171–182. doi:10.1016/S1525-1578(10)60543-0
Suzuki M, Giovannoni S (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630
Hansen MC, Tolker-Nielsen T, Givskov M, Molin S (1998) Biased 16S rDNA PCR amplification caused by interference from DNA flanking the template region. FEMS Microbiol Ecol 26(2):141–149. doi:10.1111/j.1574-6941.1998.tb00500.x
Mathieu-Daude F, Welsh J, Vogt T, McClelland M (1996) DNA rehybridization during PCR: the ‘Cot effect’ and its consequences. Nucleic Acids Res 24(11):2080–2086
Hubé F, Reverdiau P, Iochmann S, Gruel Y (2005) Improved PCR method for amplification of GC-rich DNA sequences. Mol Biotechnol 31(1):81–84. doi:10.1385/MB:31:1:081
Jordan B, Charest A, Dowd J, Blumenstiel J, Yeh Rf R, Osman A, Housman D, Landers J (2002) Genome complexity reduction for SNP genotyping analysis. Proc Natl Acad Sci U S A 99:2942–2947
Woyke T, Tighe D, Mavromatis K, Clum A, Copeland A, Schackwitz W, Lapidus A, Wu D, McCutcheon JP, McDonald BR, Moran NA, Bristow J, Cheng JF (2010) One bacterial cell, one complete genome. PLoS One 5(4):e10314. doi:10.1371/journal.pone.0010314
Wetmur JG, Davidson N (1968) Kinetics of renaturation of DNA. J Mol Biol 31(3):349–370
McDowell DG, Burns NA, Parkes HC (1998) Localised sequence regions possessing high melting temperatures prevent the amplification of a DNA mimic in competitive PCR. Nucleic Acids Res 26(14):3340–3347
Lasken R, Egholm M (2003) Whole genome amplification: abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol 21:531–535
Benita Y, Oosting R, Lok M, Wise M, Humphery-Smith I (2003) Regionalized GC content of template DNA as a predictor of PCR success. Nucleic Acids Res 31:e99
Sahdev S, Saini S, Tiwari P, Saxena S, Singh Saini K (2007) Amplification of GC-rich genes by following a combination strategy of primer design, enhancers and modified PCR cycle conditions. Mol Cell Probes 21(4):303–307, http://dx.doi.org/10.1016/j.mcp.2007.03.004
Paez J, Lin M, Beroukhim R, Lee J, Zhao X, Richter D, Gabriel S, Herman P, Sasaki H, Altshuler D, Li C, Meyerson M, Sellers W (2004) Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res 32:e71
Pinard R, de Winter A, Sarkis GJ, Gerstein MB, Tartaro KR, Plant RN, Egholm M, Rothberg JM, Leamon JH (2006) Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics 7:216. doi:10.1186/1471-2164-7-216
Alsmadi O, Alkayal F, Monies D, Meyer BF (2009) Specific and complete human genome amplification with improved yield achieved by phi29 DNA polymerase and a novel primer at elevated temperature. BMC Res Notes 2:48. doi:10.1186/1756-0500-2-48
Reysenbach AL, Giver LJ, Wickham GS, Pace NR (1992) Differential amplification of rRNA genes by polymerase chain reaction. Appl Environ Microbiol 58(10):3417–3418
Multer GL, Boynton KA (1995) PCR bias in amplification of androgen receptor alleles, a trinucleotide repeat marker used in clonality studies. Nucleic Acids Res 23(8):1411–1418. doi:10.1093/nar/23.8.1411
Sachse K (2004) Specificity and performance of PCR detection assays for microbial pathogens. Mol Biotechnol 26(1):61–80. doi:10.1385/MB:26:1:61
Sipos R, Szekely AJ, Palatinszky M, Revesz S, Marialigeti K, Nikolausz M (2007) Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol Ecol 60(2):341–350. doi:10.1111/j.1574-6941.2007.00283.x
Klein CA, Schmidt-Kittler O, Schardt JA, Pantel K, Speicher MR, Riethmuller G (1999) Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci U S A 96(8):4494–4499
Pirker C, Raidl M, Steiner E, Elbling L, Holzmann K, Spiegl-Kreinecker S, Aubele M, Grasl-Kraupp B, Marosi C, Micksche M, Berger W (2004) Whole genome amplification for CGH analysis: Linker-adapter PCR as the method of choice for difficult and limited samples. Cytometry A 61(1):26–34
Wandeler P, Hoeck PEA, Keller LF (2007) Back to the future: museum specimens in population genetics. Trends Ecol Evol 22(12):634–642, http://dx.doi.org/10.1016/j.tree.2007.08.017
Zimmermann J, Hajibabaei M, Blackburn D, Hanken J, Cantin E, Posfai J, Evans T (2008) DNA damage in preserved specimens and tissue samples: a molecular assessment. Front Zool 5(1):18
Krause J, Dear PH, Pollack JL, Slatkin M, Spriggs H, Barnes I, Lister AM, Ebersberger I, Paabo S, Hofreiter M (2006) Multiplex amplification of the mammoth mitochondrial genome and the evolution of Elephantidae. Nature 439(7077):724–727. doi:10.1038/nature04432
Green RE, Krause J, Ptak SE, Briggs AW, Ronan MT, Simons JF, Du L, Egholm M, Rothberg JM, Paunovic M, Paabo S (2006) Analysis of one million base pairs of Neanderthal DNA. Nature 444(7117):330–336. doi:10.1038/nature05336
Ovchinnikov IV, Gotherstrom A, Romanova GP, Kharitonov VM, Liden K, Goodwin W (2000) Molecular analysis of Neanderthal DNA from the northern Caucasus. Nature 404(6777):490–493. doi:10.1038/35006625
Paabo S (1985) Molecular cloning of Ancient Egyptian mummy DNA. Nature 314(6012):644–645
Taubenberger JK (2006) The origin and virulence of the 1918 “Spanish” influenza virus. Proc Am Philos Soc 150(1):86–112
Taubenberger JK, Reid AH, Krafft AE, Bijwaard KE, Fanning TG (1997) Initial genetic characterization of the 1918 “Spanish” influenza virus. Science 275(5307):1793–1796
Paabo S, Higuchi RG, Wilson AC (1989) Ancient DNA and the polymerase chain reaction. The emerging field of molecular archaeology. J Bio Chem 264(17):9709–9712
Paabo S (1989) Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc Natl Acad Sci U S A 86(6):1939–1943
Dabney J, Meyer M, Paabo S (2013) Ancient DNA damage. Cold Spring Harb Perspect Biol 5(7). doi: 10.1101/cshperspect.a012567
Akane A, Shiono H, Matsubara K, Nakamura H, Hasegawa M, Kagawa M (1993) Purification of forensic specimens for the polymerase chain reaction (PCR) analysis. J Forensic Sci 38(3):691–701
Fisher DL, Holland MM, Mitchell L, Sledzik PS, Wilcox AW, Wadhams M, Weedn VW (1993) Extraction, evaluation, and amplification of DNA from decalcified and undecalcified United States Civil War bone. J Forensic Sci 38(1):60–68
Golenberg EM, Bickel A, Weihs P (1996) Effect of highly fragmented DNA on PCR. Nucleic Acids Res 24(24):5026–5033. doi:10.1093/nar/24.24.5026
Aviel-Ronen S, Qi Zhu C, Coe BP, Liu N, Watson SK, Lam WL, Tsao MS (2006) Large fragment Bst DNA polymerase for whole genome amplification of DNA from formalin-fixed paraffin-embedded tissues. BMC Genomics 7:312. doi:10.1186/1471-2164-7-312
Coombs NJ, Gough AC, Primrose JN (1999) Optimisation of DNA and RNA extraction from archival formalin-fixed tissue. Nucleic Acids Res 27(16):e12
Srinivasan M, Sedmak D, Jewell S (2002) Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol 161(6):1961–1971. doi:10.1016/s0002-9440(10)64472-0
Tokuda Y, Nakamura T, Satonaka K, Maeda S, Doi K, Baba S, Sugiyama T (1990) Fundamental study on the mechanism of DNA degradation in tissues fixed in formaldehyde. J Clin Pathol 43(9):748–751
Williams C, Ponten F, Moberg C, Soderkvist P, Uhlen M, Ponten J, Sitbon G, Lundeberg J (1999) A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol 155(5):1467–1471. doi:10.1016/s0002-9440(10)65461-2
Douglas MP, Rogers SO (1998) DNA damage caused by common cytological fixatives. Mutat Res 401(1-2):77–88
McGhee JD, von Hippel PH (1977) Formaldehyde as a probe of DNA structure. 3. Equilibrium denaturation of DNA and synthetic polynucleotides. Biochemistry 16(15):3267–3276
Beadling C, Neff TL, Heinrich MC, Rhodes K, Thornton M, Leamon J, Andersen M, Corless CL (2013) Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn 15(2):171–176. doi:10.1016/j.jmoldx.2012.09.003
Kroneis T, Geigl JB, El-Heliebi A, Auer M, Ulz P, Schwarzbraun T, Dohr G, Sedlmayr P (2011) Combined molecular genetic and cytogenetic analysis from single cells after isothermal whole-genome amplification. Clin Chem 57(7):1032–1041
Bodelier PLE, Kamst M, Meima-Franke M, Stralis-Pavese N, Bodrossy L (2009) Whole-community genome amplification (WCGA) leads to compositional bias in methane-oxidizing communities as assessed by pmoA-based microarray analyses and QPCR. Environ Microbiol Rep 1(5):434–441. doi:10.1111/j.1758-2229.2009.00066.x
Dean F, Nelson J, Giesler T, Lasken R (2001) Rapid amplification of plasmid and phage DNA using Phi29 polymerase and a multiply-pimed rolling circle amplification. Genome Res 11:1095–1099
Abulencia C, Wyborski D, Garcia J, Podar M, Chen W, Chang S, Chang H, Watson D, Brodie E, Hazen T, Keller M (2006) Environmental whole-genome amplification to access microbial populations in contaminated sediments. Appl Environ Microbiol 72:3291–3301
Lehmann EL (1999) Elements of large-sample theory, Springer texts in statistics. Springer, New York, NY
Mutter GL, Boynton KA (1995) PCR bias in amplification of androgen receptor alleles, a trinucleotide repeat marker used in clonality studies. Nucleic Acids Res 23(8):1411–1418
Walsh PS, Erlich HA, Higuchi R (1992) Preferential PCR amplification of alleles: mechanisms and solutions. PCR Methods Appl 1(4):241–250
Schnell S, Mendoza C (1997) Theoretical description of the polymerase chain reaction. J Theor Biol 188(3):313–318. doi:10.1006/jtbi.1997.0473
Schnell S, Mendoza C (1997) Enzymological considerations for a theoretical description of the quantitative competitive polymerase chain reaction (QC-PCR). J Theor Biol 184(4):433–440
Rutledge R, Stewart D (2008) A kinetic-based sigmoidal model for the polymerase chain reaction and its application to high-capacity absolute quantitative real-time PCR. BMC Biotechnol 8(1):47
Jagers P, Klebaner F (2003) Random variation and concentration effects in PCR. J Theor Biol 224(3):299–304
Peccoud J, Jacob C (1996) Theoretical uncertainty of measurements using quantitative polymerase chain reaction. Biophys J 71(1):101–108. doi:10.1016/S0006-3495(96)79205-6
Lantz O, Bendelac A (1994) An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med 180(3):1097–1106
Karrer EE, Lincoln JE, Hogenhout S, Bennett AB, Bostock RM, Martineau B, Lucas WJ, Gilchrist DG, Alexander D (1995) In situ isolation of mRNA from individual plant cells: creation of cell-specific cDNA libraries. Proc Natl Acad Sci U S A 92(9):3814–3818
Gill P, Puch-Solis R, Curran J (2009) The low-template-DNA (stochastic) threshold—Its determination relative to risk analysis for national DNA databases. Forensic Sci Int Genet 3(2):104–111, http://dx.doi.org/10.1016/j.fsigen.2008.11.009
Gill P, Sparkes R, Kimpton C (1997) Development of guidelines to designate alleles using an STR multiplex system. Forensic Sci Int 89(3):185–197
Whitaker JP, Cotton EA, Gill P (2001) A comparison of the characteristics of profiles produced with the AMPFlSTR SGM Plus multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci Int 123(2-3):215–223
Chandler DP, Fredrickson JK, Brockman FJ (1997) Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol Ecol 6(5):475–482. doi:10.1046/j.1365-294X.1997.00205.x
Piyamongkol W, Bermúdez MG, Harper JC, Wells D (2003) Detailed investigation of factors influencing amplification efficiency and allele drop‐out in single cell PCR: implications for preimplantation genetic diagnosis. Mol Hum Reprod 9(7):411–420. doi:10.1093/molehr/gag051
Wells D, Sherlock J, Handyside A, Delhanty J (1999) Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridisation. Nucleic Acids Res 27:1214–1218
Paunio T, Reima I, Syvanen A (1996) Preimplantation diagnosis by whole-genome amplification, PCR amplification, and solid-phase minisequencing of blastomere DNA. Clin Chem 42:1382–1390
Ng G, Roberts I, Coleman N (2005) Evaluation of 3 methods of whole-genome amplification for subsequent metaphase comparative genomic hybridization. Diagn Mol Pathol 14(4):203–212
Ishoey T, Woyke T, Stepanauskas R, Novotny M, Lasken RS (2008) Genomic sequencing of single microbial cells from environmental samples. Curr Opin Microbiol 11(3):198–204, http://dx.doi.org/10.1016/j.mib.2008.05.006
Woyke T, Xie G, Copeland A, Gonzalez JM, Han C, Kiss H, Saw JH, Senin P, Yang C, Chatterji S, Cheng JF, Eisen JA, Sieracki ME, Stepanauskas R (2009) Assembling the marine metagenome, one cell at a time. PLoS One 4(4):e5299. doi:10.1371/journal.pone.0005299
Rodrigue S, Malmstrom RR, Berlin AM, Birren BW, Henn MR, Chisholm SW (2009) Whole genome amplification and de novo assembly of single bacterial cells. PLoS One 4(9):e6864. doi:10.1371/journal.pone.0006864
Raghunathan A, Ferguson H, Bornarth C, Song W, Driscoll M, Lasken R (2005) Genomic DNA amplification from a single bacterium. Appl Environ Microbiol 71:3342–3347
Handyside A, Robinson M, Simpson R, Omar M, Shaw M, Grudzinskas J, Rutherford A (2004) Isothermal whole genome amplification from single and small numbers of cells: a new era for preimplantation genetic diagnosis of inherited disease. Mol Hum Reprod 10(10):767–772
Tzvetkov MV, Becker C, Kulle B, Nurnberg P, Brockmoller J, Wojnowski L (2005) Genome-wide single-nucleotide polymorphism arrays demonstrate high fidelity of multiple displacement-based whole-genome amplification. Electrophoresis 26(3):710–715. doi:10.1002/elps.200410121
Esteban JA, Salas M, Blanco L (1993) Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J Biol Chem 268(4):2719–2726
Kucera RB, Nichols NM (2001) DNA-dependent DNA polymerases, Current protocols in molecular biology. John Wiley, Hoboken, NJ. doi:10.1002/0471142727.mb0305s84
Blanco L, Bernad A, Lazaro J, Martin G, Garmendia C, Salas M (1989) Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication. J Biol Chem 264(15):8935–8940
Cline J, Braman JC, Hogrefe HH (1996) PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 24(18):3546–3551
Hogrefe HH, Cline J, Lovejoy AE, Nielson KB (2001) DNA polymerases from hyperthermophiles. Methods Enzymol 334:91–116
Kwok S, Chang SY, Sninsky JJ, Wang A (1994) A guide to the design and use of mismatched and degenerate primers. PCR Methods Appl 3(4):S39–S47
Cha RS, Thilly WG (1993) Specificity, efficiency, and fidelity of PCR. PCR Methods Appl 3(3):S18–S29
Brelsford A, Collin H, Perrin N, Fumagalli L (2012) Nonspecific PCR amplification by high-fidelity polymerases: implications for next-generation sequencing of AFLP markers. Mol Ecol Resour 12(1):123–127. doi:10.1111/j.1755-0998.2011.03063.x
Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF (2005) PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol 71(12):8966–8969. doi:10.1128/AEM.71.12.8966-8969.2005
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6(12), e27310. doi:10.1371/journal.pone.0027310
Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12(2):R18. doi:10.1186/gb-2011-12-2-r18
Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, Magrini VJ, Richt RJ, Sander SN, Stewart DA, Stromberg M, Tsung EF, Wylie T, Schedl T, Wilson RK, Mardis ER (2008) Whole-genome sequencing and variant discovery in C. elegans. Nat Methods 5(2):183–188. doi:10.1038/nmeth.1179
Lasken RS, Stockwell TB (2007) Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol 7:19. doi:10.1186/1472-6750-7-19
Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW, Church GM (2006) Sequencing genomes from single cells by polymerase cloning. Nat Biotech 24(6):680–686, http://www.nature.com/nbt/journal/v24/n6/suppinfo/nbt1214_S1.html
Jensen MA, Straus N (1993) Effect of PCR conditions on the formation of heteroduplex and single-stranded DNA products in the amplification of bacterial ribosomal DNA spacer regions. PCR Methods Appl 3(3):186–194
Qiu X, Wu L, Huang H, McDonel PE, Palumbo AV, Tiedje JM, Zhou J (2001) Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl Environ Microbiol 67(2):880–887. doi:10.1128/AEM.67.2.880-887.2001
Thompson JR, Marcelino LA, Polz MF (2002) Heteroduplexes in mixed-template amplifications: formation, consequence and elimination by ‘reconditioning PCR’. Nucleic Acids Res 30(9):2083–2088
Wu J, Liu W, Tseng I, Cheng S (2001) Characterization of a 4-methylbenzoate-degrading methanogenic consortium as determined by small-subunit rDNA sequence analysis. J Biosci Bioeng 91(5):449–455
Paabo S, Irwin DM, Wilson AC (1990) DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem 265(8):4718–4721
Shuldiner AR, Nirula A, Roth J (1989) Hybrid DNA artifact from PCR of closely related target sequences. Nucleic Acids Res 17(11):4409
Odelberg SJ, Weiss RB, Hata A, White R (1995) Template-switching during DNA synthesis by Thermus aquaticus DNA polymerase I. Nucleic Acids Res 23(11):2049–2057
Patel R, Lin M, Laney M, Kurn N, Rose S, Ullman EF (1996) Formation of chimeric DNA primer extension products by template switching onto an annealed downstream oligonucleotide. Proc Natl Acad Sci U S A 93(7):2969–2974
Wang GC, Wang Y (1997) Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl Environ Microbiol 63(12):4645–4650
Blainey PC, Quake SR (2011) Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res 39(4), e19. doi:10.1093/nar/gkq1074
Woyke T, Sczyrba A, Lee J, Rinke C, Tighe D, Clingenpeel S, Malmstrom R, Stepanauskas R, Cheng JF (2011) Decontamination of MDA reagents for single cell whole genome amplification. PLoS One 6(10):e26161. doi:10.1371/journal.pone.0026161
Hutchison CA 3rd, Smith HO, Pfannkoch C, Venter JC (2005) Cell-free cloning using phi29 DNA polymerase. Proc Natl Acad Sci U S A 102(48):17332–17336. doi:10.1073/pnas.0508809102
Marcy Y, Ishoey T, Lasken RS, Stockwell TB, Walenz BP, Halpern AL, Beeson KY, Goldberg SM, Quake SR (2007) Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet 3(9):1702–1708. doi:10.1371/journal.pgen.0030155
Wang J, Fan HC, Behr B, Quake SR (2012) Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150(2):402–412. doi:10.1016/j.cell.2012.06.030
Loakes D, Brown DM (1994) 5-Nitroindole as an universal base analogue. Nucleic Acids Res 22(20):4039–4043
Loakes D, Hill F, Brown DM, Salisbury SA (1997) Stability and structure of DNA oligonucleotides containing non-specific base analogues. J Mol Biol 270(3):426–435. doi:10.1006/jmbi.1997.1129
Peng W, Takabayashi H, Ikawa K (2007) Whole genome amplification from single cells in preimplantation genetic diagnosis and prenatal diagnosis. Eur J Obstet Gynecol Reprod Biol 131(1):13–20. doi:10.1016/j.ejogrb.2006.07.027
Thompson CT, Gray JW (1993) Cytogenetic profiling using fluorescence in situ hybridization (FISH) and comparative genomic hybridization (CGH). J Cell Biochem Suppl 17G:139–143
Fiegl M, Tueni C, Schenk T, Jakesz R, Gnant M, Reiner A, Rudas M, Pirc-Danoewinata H, Marosi C, Huber H et al (1995) Interphase cytogenetics reveals a high incidence of aneuploidy and intra-tumour heterogeneity in breast cancer. Br J Cancer 72(1):51–55
Kallioniemi A, Kallioniemi O, Sudar D, Rutovitz D, Gray J, Waldman F, Pinkel D (1992) Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258(5083):818–821
Bannai M, Higuchi K, Akesaka T, Furukawa M, Yamaoka M, Sato K, Tokunaga K (2004) Single-nucleotide-polymorphism genotyping for whole-genome-amplified samples using automated fluorescence correlation spectroscopy. Anal Biochem 327(2):215–221. doi:10.1016/j.ab.2004.01.012
Hosono S, Faruqi A, Dean F, Du Y, Sun Z, Wu X, Du J, Kingsmore S, Egholm M, Lasken R (2003) Unbiased whole-genome amplification directly from clinical samples. Genome Res 13(5):954–964
Lovmar L, Fredriksson M, Liljedahl U, Sigurdsson S, Syvanen A (2003) Quantitative evaluation by minisequencing and microarrays reveals accurate multiplexed SNP genotyping of whole genome amplified DNA. Nucleic Acids Res 31:e129
Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Peramenschikov A, Williams CF, Jeffery SS, Botstein D, Brown PO (1999) Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet 23:41–46
Gray JW, Collins C (2000) Genome changes and gene expression in human solid tumors. Carcinogenesis 21(3):443–452
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437(7057):376–380. doi:10.1038/nature03959
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, Rasolonjatovo IM, Reed MT, Rigatti R, Rodighiero C, Ross MT, Sabot A, Sankar SV, Scally A, Schroth GP, Smith ME, Smith VP, Spiridou A, Torrance PE, Tzonev SS, Vermaas EH, Walter K, Wu X, Zhang L, Alam MD, Anastasi C, Aniebo IC, Bailey DM, Bancarz IR, Banerjee S, Barbour SG, Baybayan PA, Benoit VA, Benson KF, Bevis C, Black PJ, Boodhun A, Brennan JS, Bridgham JA, Brown RC, Brown AA, Buermann DH, Bundu AA, Burrows JC, Carter NP, Castillo N, Chiara ECM, Chang S, Neil Cooley R, Crake NR, Dada OO, Diakoumakos KD, Dominguez-Fernandez B, Earnshaw DJ, Egbujor UC, Elmore DW, Etchin SS, Ewan MR, Fedurco M, Fraser LJ, Fuentes Fajardo KV, Scott Furey W, George D, Gietzen KJ, Goddard CP, Golda GS, Granieri PA, Green DE, Gustafson DL, Hansen NF, Harnish K, Haudenschild CD, Heyer NI, Hims MM, Ho JT, Horgan AM, Hoschler K, Hurwitz S, Ivanov DV, Johnson MQ, James T, Huw Jones TA, Kang GD, Kerelska TH, Kersey AD, Khrebtukova I, Kindwall AP, Kingsbury Z, Kokko-Gonzales PI, Kumar A, Laurent MA, Lawley CT, Lee SE, Lee X, Liao AK, Loch JA, Lok M, Luo S, Mammen RM, Martin JW, McCauley PG, McNitt P, Mehta P, Moon KW, Mullens JW, Newington T, Ning Z, Ling Ng B, Novo SM, O'Neill MJ, Osborne MA, Osnowski A, Ostadan O, Paraschos LL, Pickering L, Pike AC, Pike AC, Chris Pinkard D, Pliskin DP, Podhasky J, Quijano VJ, Raczy C, Rae VH, Rawlings SR, Chiva Rodriguez A, Roe PM, Rogers J, Rogert Bacigalupo MC, Romanov N, Romieu A, Roth RK, Rourke NJ, Ruediger ST, Rusman E, Sanches-Kuiper RM, Schenker MR, Seoane JM, Shaw RJ, Shiver MK, Short SW, Sizto NL, Sluis JP, Smith MA, Ernest Sohna Sohna J, Spence EJ, Stevens K, Sutton N, Szajkowski L, Tregidgo CL, Turcatti G, Vandevondele S, Verhovsky Y, Virk SM, Wakelin S, Walcott GC, Wang J, Worsley GJ, Yan J, Yau L, Zuerlein M, Rogers J, Mullikin JC, Hurles ME, McCooke NJ, West JS, Oaks FL, Lundberg PL, Klenerman D, Durbin R, Smith AJ (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 (7218): 53–59. doi:10.1038/nature07517
Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, Sidow A, Fire A, Johnson SM (2008) A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res 18(7):1051–1063. doi:10.1101/gr.076463.108
Adey A, Morrison HG, Asan XX, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, Shendure J (2010) Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11(12):R119. doi:10.1186/gb-2010-11-12-r119
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. doi:10.1186/1471-2164-13-341
ElSharawy A, Warner J, Olson J, Forster M, Schilhabel MB, Link DR, Rose-John S, Schreiber S, Rosenstiel P, Brayer J, Franke A (2012) Accurate variant detection across non-amplified and whole genome amplified DNA using targeted next generation sequencing. BMC Genomics 13:500. doi:10.1186/1471-2164-13-500
Indap AR, Cole R, Runge CL, Marth GT, Olivier M (2013) Variant discovery in targeted resequencing using whole genome amplified DNA. BMC Genomics 14:468. doi:10.1186/1471-2164-14-468
Wood HM, Belvedere O, Conway C, Daly C, Chalkley R, Bickerdike M, McKinley C, Egan P, Ross L, Hayward B, Morgan J, Davidson L, MacLennan K, Ong TK, Papagiannopoulos K, Cook I, Adams DJ, Taylor GR, Rabbitts P (2010) Using next-generation sequencing for high resolution multiplex analysis of copy number variation from nanogram quantities of DNA from formalin-fixed paraffin-embedded specimens. Nucleic Acids Res 38(14):e151. doi:10.1093/nar/gkq510
Murphy SJ, Cheville JC, Zarei S, Johnson SH, Sikkink RA, Kosari F, Feldman AL, Eckloff BW, Karnes RJ, Vasmatzis G (2012) Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA Res 19(5):395–406. doi:10.1093/dnares/dss021
Voet T, Kumar P, Van Loo P, Cooke SL, Marshall J, Lin ML, Zamani Esteki M, Van der Aa N, Mateiu L, McBride DJ, Bignell GR, McLaren S, Teague J, Butler A, Raine K, Stebbings LA, Quail MA, D’Hooghe T, Moreau Y, Futreal PA, Stratton MR, Vermeesch JR, Campbell PJ (2013) Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res 41(12):6119–6138. doi:10.1093/nar/gkt345
Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ (2009) Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods 6(4):291–295. doi:10.1038/nmeth.1311
Mamanova L, Andrews RM, James KD, Sheridan EM, Ellis PD, Langford CF, Ost TW, Collins JE, Turner DJ (2010) FRT-seq: amplification-free, strand-specific transcriptome sequencing. Nat Methods 7(2):130–132. doi:10.1038/nmeth.1417
Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ (2008) A large genome center's improvements to the Illumina sequencing system. Nat Methods 5(12):1005–1010. doi:10.1038/nmeth.1270
Taub MA, Corrada Bravo H, Irizarry RA (2010) Overcoming bias and systematic errors in next generation sequencing data. Genome Med 2(12):87. doi:10.1186/gm208
Chen YC, Liu T, Yu CH, Chiang TY, Hwang CC (2013) Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS One 8(4):e62856. doi:10.1371/journal.pone.0062856
Schwartz S, Oren R, Ast G (2011) Detection and removal of biases in the analysis of next-generation sequencing reads. PLoS One 6(1):e16685. doi:10.1371/journal.pone.0016685
Paliy O, Foy BD (2011) Mathematical modeling of 16S ribosomal DNA amplification reveals optimal conditions for the interrogation of complex microbial communities with phylogenetic microarrays. Bioinformatics 27(15):2134–2140. doi:10.1093/bioinformatics/btr326
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Sabina, J., Leamon, J.H. (2015). Bias in Whole Genome Amplification: Causes and Considerations. In: Kroneis, T. (eds) Whole Genome Amplification. Methods in Molecular Biology, vol 1347. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2990-0_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2990-0_2
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2989-4
Online ISBN: 978-1-4939-2990-0
eBook Packages: Springer Protocols