
Abstract
Stem cell models of Alzheimer’s disease provide an opportunity to study the mechanisms underlying disease pathology at a resolution that is not possible in animal models. Furthermore, the ability to reprogram patient somatic cells to a pluripotent state ensures that the disease can be investigated in the correct genetic context. Here, we describe the directed differentiation of human pluripotent cells to cortical progenitors by recapitulating key developmental signaling events in vitro. Over a timeframe that mirrors human development, these progenitors give rise to functional lower and upper layer neurons. We also describe biochemical and imaging based methods to analyse key APP and Tau phenotypes in neurons generated from pluripotent stem cells from individuals with either monogenic familial Alzheimer’s disease or Down’s syndrome.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Disease modeling
- Alzheimer’s disease
- Stem cell
- APP
- Abeta peptides
- Tau
- Cortical differentiation
- Neurodegeneration
- Down’s syndrome
1 Introduction
Human cellular models of Alzheimer’s disease (AD) have the potential to complement existing animal models for carrying out mechanistic studies of AD initiation and progression. There is also considerable interest in using such models for high throughput and high content analyses, including chemical and genetic screens. Ideally, human stem cell models of AD should use the cell types affected by the disease and develop disease-relevant pathologies. For practical purposes, such models would undergo disease initiation in a reproducible manner and over a relatively short timescale.
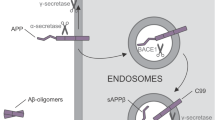
Combining cellular reprogramming to generate patient-specific pluripotent stem cells (PSCs), with our understanding of neural development has enabled researchers to generate specific neuronal cell types in vitro, including excitatory and inhibitory cortical neurons [1–3]. When generated from PSCs derived from individuals affected by genetic forms of AD, including monogenic familial AD and trisomy 21/Down’s syndrome, neurons develop many of the classical hallmarks of AD initiation. These include altered APP processing to generate changes in Abeta peptide production, Abeta aggregation, increases in Tau phosphorylation and the cellular localization of Tau [4–6].
In this chapter, we describe methods to generate human excitatory, glutamatergic cerebral cortex neurons from PSCs. This approach for making cerebral cortex neurons works equally well for embryonic stem cells and patient derived induced pluripotent stem cells [7]. A key aspect of this process is the need to allow time for the neurons generated to undergo functional maturation, as reflected in their firing properties: this is typically of the order of 2 months for their firing properties to resemble those of mature neurons. We also describe techniques to monitor the efficiency of neuronal production by immunofluorescence and confocal microscopy, and to analyse the development of specific AD phenotypes, with an emphasis on AD initiation, reflected in changes in APP processing.
With respect to human stem cells generated from individuals with genetic forms of AD and other dementias, a number of lines have been published and well characterized, including from individuals with Down’s syndrome, familial AD (Psen1 mutations, APP duplication, APP mutations) and frontotemporal dementia, as detailed in Table 1. Recommended culture times of stem cells with specific AD-like pathologies are presented in Table 2.
2 Materials
Main media and solutions. For preparation, tissue culture grade media/solutions and distilled water are used.
-
1.
STO medium for cultivation of mouse embryonic fibroblasts (MEFs): Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % (v/v) fetal bovine serum, 1 mM l-glutamine, 50 U/mL penicillin and 50 mg/mL streptomycin. Store at 4 °C and use within 2 weeks.
-
2.
hPSC medium for cultivation of human pluripotent stem cells: DMEM/mixture F-12-GlutaMAX medium (DMEM/F-12 GlutaMAX) supplemented with 20 % (v/v) knockout serum replacement media (KnockOut SR, KSR), 10 ng/mL FGF2 (fibroblast growth factor basic), 100 μM non-essential amino acids, 100 μM 2-mercaptoethanol, 50 U/mL penicillin and 50 mg/mL streptomycin. Store at 4 °C and use within 2 weeks.
-
3.
MEF conditioned medium: Collected after incubating MEFs in hPSC medium without FGF2 overnight. Pass through a 0.22 μM pore filter and store at −20 °C for up to 2 months. When required, thaw medium and add 10 ng/mL FGF2. Discard thawed aliquots after 5 days.
-
4.
PBS. Tissue culture grade phosphate-buffered saline (PBS).
-
5.
Dispase solution, Dispase II (neutral protease) resuspended in tissue culture grade phosphate-buffered saline (PBS) to 10 mg/mL at 37 °C before sterilisation through at 0.22 μm filter. Store at −20 °C for up to 2 months. Preventing or reducing clumping of human and mouse cells.
-
6.
hPSC freezing medium: Defined fetal bovine serum, 10 % (v/v) dimethyl sulfoxide (DMSO) and 10 μM Y-27632 (Rho-kinase inhibitor with benefits in hPSC expansion). Make fresh and do not store.
-
7.
N2 stem cell culture medium (N2 medium): DMEM/F-12 GlutaMAX with 1× N2 Supplement, 5 μg/mL insulin, 1 mM l-glutamine, 100 μM non-essential amino acids (NEAA), 100 μM 2-mercaptoethanol, 1 mM sodium pyruvate, 50 U/mL penicillin and 50 mg/mL streptomycin. Store at 4 °C and use within 3 weeks.
-
8.
B27 medium: Neurobasal with 1× B27 Supplement, 200 mM l-glutamine, 50 U/mL penicillin and 50 mg/mL streptomycin. Store at 4 °C and use within 3 weeks. Basal media for neural cell culture.
-
9.
N2B27 medium: A 1:1 mixture of N2 medium and B27 medium, store at 4 °C and use within 2 weeks.
-
10.
Neural induction medium: N2B27 with 1 μM Dorsomorphin and 10 μM SB431542 (inhibitors of bone morphogenic protein (BMP) signaling coordinating development; stimulating proliferation and differentiation). Store at 4 °C and use within 1 week.
-
11.
Accutase cell dissociation reagent. Detachment solution.
-
12.
Laminin stock solution. Cell attachment, coating solution.
-
13.
Neural freezing medium: N2B27 with 10 % (v/v) DMSO and 20 ng/mL FGF2. Make fresh and do not store.
-
14.
Fixative: 4 % paraformaldehyde (PFA) in 1× PBS.
-
15.
Tris buffered saline (TBS; 10×): 1.5 M NaCl, 0.1 M Tris–HCl, pH 7.4.
-
16.
TBS-Tx: TBS (1×) with 0.3 % Triton X-100.
-
17.
Blocking solution: 5 % normal donkey serum in TBS-Tx.
3 Methods
Ongoing cultures should be handled in a sterile laminar flow tissue culture hood. All media should be warmed to 37 °C in a water bath before use. All incubation steps should be carried out at 37 °C in 5 % CO2 and all centrifugation steps should be performed at room temperature.
3.1 Plating Mouse Embryonic Fibroblasts
-
1.
Add 1 mL of 0.1 % gelatin in distilled water to each well of two 6-well plates. Incubate at 37 °C for 10 min.
-
2.
Partially thaw a vial of frozen irradiated mouse embryonic fibroblasts (MEFs) in a water bath at 37 °C and transfer to 10 mL of STO medium.
-
3.
Centrifuge for 3 min at 180 × g and resuspend the cells in 1.2 mL of STO medium.
-
4.
Aspirate the gelatin solution from plates and replace with 2 mL of STO medium per well.
-
5.
Add 100 μL of the MEF cell suspension to each well.
-
6.
Gently rock the dishes forward, back and side-to-side to disperse the cells evenly.
-
7.
Culture the MEFs for at least 6 h before the addition of stem cells.
3.2 Culturing Human Pluripotent Cells
-
1.
Aspirate STO medium from MEF culture and wash each well in 2 mL of tissue culture grade phosphate buffered serum (PBS).
-
2.
Aspirate PBS and replace it with 2 mL of hPSC medium without FGF2.
-
3.
Partially thaw a vial of human pluripotent stem cells (PSC) in a water bath and transfer to 10 mL of hPSC medium (see Note 1 ).
-
4.
Centrifuge for 2 min at 180 × g. Aspirate the supernatant.
-
5.
Gently resuspend the colonies in 200 μL of hPSC medium before transferring them to one well of a 6 well plate.
-
6.
Add 10 ng/mL FGF2 and 10 μM Y-27632 solution to the wells containing PSCs, collect MEF conditioned medium from the remaining wells. Incubate the cells overnight.
-
7.
Maintain PSC colonies by daily replacement of media with hPSC containing 10 μM FGF2 until colonies become visible to the naked eye (~1 mm in diameter) (see Note 2 ).
3.3 Passaging PSCs
-
1.
Prepare fresh mouse embryonic fibroblasts (MEFs) as described in Subheading 3.1.
-
2.
Remove STO from MEFs, wash once with PBS and add 2 mL of hPSC medium containing 10 μM FGF2 to each well of the 6 well plate. Return MEFs in hPSC medium to incubator.
-
3.
Add 200 μL of dispase solution to each well of PSCs and incubate for 20–40 min (see Note 3 ).
-
4.
Remove STO from MEFs, wash once with PBS and add 2 mL of hPSC medium containing 10 μM FGF2 to each well of the 6 well plate. Return MEFs in hPSC medium to incubator.
-
5.
Gently rock the plate to ensure detachment of PSC colonies from MEFs and differentiated cells.
-
6.
Transfer colonies to 10 mL of PBS and centrifuge for 2 min at 180 × g. Aspirate solution and repeat PBS washes a further three times.
-
7.
To passage cells and continue the culture, resuspend PSCs in 600 μL of hPSC medium and gently break up colonies to approximately 50–100 cells with P200 pipette (see Note 4 ).
-
8.
Transfer 100 μL of PSC suspension to each well of a 6 well plate containing hPSC medium with 10 μM FGF2 (1:6 passage) and maintain as described in Subheading 3.2, step 7 (see Note 5 ).
-
9.
Alternatively, colonies can be resuspended without being broken up in hPSC freezing medium after step 8 and transferred to cryovials.
-
10.
Cells for frozen storage should be immediately placed at −80 °C in a cell freezing box and transferred to liquid nitrogen after 24 h.
3.4 Neural Induction
-
1.
Coat a 12 well plate with an appropriate substrate to allow feeder-free stem cell culture. Dependent on colony density, 5 wells of PSCs will typically yield enough cells for 3–6 wells of neural induction.
-
2.
Follow steps 3, 5 and 6 of Subheading 3.3, pool together the 5 wells of PSCs to be used for induction and keep the cells to be passaged separate.
-
3.
Passage 1 well of the 6 well plate as described in Subheading 3.3, steps 7 and 8 to continue the PSC culture.
-
4.
Add 500 μL of Accutase to the PSC pellet for induction, gently agitate the cells and incubate in a water bath for 5 min.
-
5.
Gently use a P1000 pipette to create a single cell suspension of PSCs by slowly pipetting the solution 4–6 times.
-
6.
Add 5 mL of hPSC medium to the suspension to inactivate the Accutase and centrifuge at 260 × g for 3 min.
-
7.
Resuspend the cells in 1 mL of MEF conditioned medium supplemented with 10 ng/mL FGF2 and 10 μM Y-27632.
-
8.
Determine the number of cells using a standard haemocytometer or automated cell counter. Each well of neural induction should contain 7 × 105 viable cells per 1 mL of suspension.
-
9.
Dilute the cell suspension to the required volume with MEF conditioned medium supplemented with 10 ng/mL FGF2 and 10 μM Y-27632.
-
10.
Transfer 1 mL of PSC suspension per well. Incubate overnight.
-
11.
The following day, aspirate medium from cells and replace with 1 mL of MEF conditioned medium supplemented with 10 ng/mL FGF2 and culture overnight.
-
12.
Wash PSC wells with 1 mL of PBS and check that they are 100 % confluent (see Note 6 ).
-
13.
Aspirate the PBS and replace with 1 mL of neural induction medium.
-
14.
Monitor cells and replace the media everyday for the next 11 days to give a total of 12 days in induction medium (see Note 7 ).
3.5 Transferring Neuroepithelial Sheet to Laminin Substrate
-
1.
Preparation of laminin coated well plates. Dilute laminin stock to a final concentration of 10 μg/mL in PBS and add 1 mL per well of a 6 well plate. Usually, one well of neural induction is transferred to one well of a 6 well plate. Incubate the plate for at least 4 h before use.
-
2.
Aspirate the laminin solution and replace with 2 mL of neural induction medium, return plate to the incubator.
-
3.
Add 100 μL of dispase to each well of neural induction and incubate until the neuroepithelial sheet detaches from the substrate, approximately 5 min.
-
4.
Transfer the sheet as complete as possible to 10 mL of N2B27 medium and centrifuge at 180 × g for 2 min.
-
5.
Perform two further 10 mL N2B27 washes to remove any residual dispase.
-
6.
Add 200 μL of neural induction medium to each cell pellet and very gently break up the neuroepithelial sheet into aggregates of approximately 500 cells with a P100 pipette (see Note 8 ).
-
7.
Transfer the cell aggregate suspension to neural induction medium in laminin coated plates and incubate overnight.
-
8.
The next day, if the aggregates have attached to the laminin coating, change the medium to N2B27 with 20 ng/mL FGF2.
-
9.
If the aggregates have not attached, centrifuge the cell suspension at 180 × g for 2 min. Resuspend in 200 μL of N2B27 and transfer into 2 mL of N2B27 with 20 ng/mL FGF2 on freshly coated laminin plates.
-
10.
Change N2B27 with 20 ng/mL FGF2 medium every 2 days for a total of 4 days. Monitor the cells for the appearance of polarised neuroepithelial rosettes, which confirms a successful neural induction (see Note 9 ).
-
11.
After 4 days of culture in N2B27 with 20 ng/mL FGF2, withdraw the FGF2 medium and maintain the neural progenitors by changing N2B27 medium every 2 days.
3.6 Passaging Neural Progenitor Cells
-
1.
Between days 16 and 20 after neural induction, progenitors should be passaged in order to expand the culture and remove differentiated or non-neuronal cells. Typically neural progenitors are passaged 1:2 to expand the culture, depending on confluency.
-
2.
Prepare laminin coated 6 well plates as described in Subheading 3.5, step 1. Aspirate the laminin solution and replace with 2 mL of N2B27 medium before returning to the incubator.
-
3.
Add 200 μL of dispase to each well of neural progenitors and incubate for 5 min (see Note 10 ).
-
4.
Transfer cells to 10 mL of tissue culture grade PBS and centrifuge at 180 × g for 2 min.
-
5.
Repeat PBS washes a total of three times to remove any residual dispase.
-
6.
Add 200 μL of N2B27 to the progenitors and gently break them into aggregates of approximately 500 cells using a P1000 pipette.
-
7.
Transfer 100 μL of the cell suspension to N2B27 in laminin coated plates, gently disperse the aggregates by rocking the plate before returning to the incubator overnight.
-
8.
The next day, aspirate the media and replace with N2B27. Continue to change N2B27 medium every 2 days.
-
9.
Repeat steps 3–8 a maximum of three times between days 16 and 26 after neural induction to enrich and expand the neural progenitor culture. Substantial neurogenesis should occur at the edge of rosettes during this period.
-
10.
At day 26, progenitor aggregates should be passaged using Accutase to create a single cell suspension. Depending on confluency, passages can be performed 1:1 to maintain cell density or up to 1:4 to expand the culture.
-
11.
Aspirate N2B27 medium and wash the cells once with 2 mL of tissue culture grade PBS.
-
12.
Aspirate PBS and replace with 750 μL of Accutase per well. Incubate for 5 min.
-
13.
Gently use a P1000 pipette to create a single cell suspension before transferring to 10 mL of N2B27.
-
14.
Centrifuge at 180 × g for 3 min.
-
15.
Resuspend the cells in 200 μL of N2B27 per well, transfer 200 μL of the suspension to 2 mL of N2B27 in a laminin coated 6 well plate.
-
16.
Maintain the culture by replacing N2B27 medium every 2 days.
-
17.
Repeat steps 11–15 several times between days 26 and 38 after neural induction to expand the culture.
3.7 Freezing and Thawing Neural Progenitors
-
1.
Neural progenitors can be frozen for long-term storage as single cells at any point after day 26 by resuspending the cell pellet generated in Subheading 3.6, step 14 in neural freezing medium and transferring to cryovials.
-
2.
Immediately place cryovials in a cell freezing box and store at −80 °C for 24 h before transferring to liquid nitrogen for long-term storage.
-
3.
To resume a progenitor culture from a frozen stock, prepare laminin coated plates as described in Subheading 3.5, step 1.
-
4.
Partially thaw a cryovial of neural progenitors in a water bath and transfer to 10 mL of N2B27 medium.
-
5.
Centrifuge at 260 × g for 3 min.
-
6.
Resuspend the cells in 2 mL of N2B27 with 20 ng/mL FGF2, transfer the suspension to one well of a laminin coated 6 well plate and return the plate to the incubator overnight.
-
7.
The next day, remove the media from cells and replace with N2B27 without FGF2.
-
8.
Continue with the protocol from Subheading 3.6, step 16.
3.8 Final Plating of PSC Derived Cortical Neurons
-
1.
Allow thawed neurons to recover for at least 48 h before final plating.
-
2.
To prepare cell culture dishes for final progenitor plating, pre-coat each 35 mm dish with 400 μL of poly-ornithine and incubate overnight. The following day remove the poly-ornithine and add 400 μL of 20 μg/mL laminin in PBS and incubate for at least 4 h before applying cells (see Notes 11 and 12 ).
-
3.
Between day 33 and day 38 after neural induction progenitor cells can be passaged as previously described in Subheading 3.6, steps 11–14 and resuspended in 1 mL of N2B27 (see Note 13 ).
-
4.
Progenitors should be counted and plated at a density of 100,000 cells per cm2 (i.e. 350,000 cells per 35 mm dish).
-
5.
Replace N2B27 medium every 2 days to maintain the neural culture.
-
6.
Add laminin into the medium every 10 days at a final concentration of 10 μg/mL.
3.9 Immunofluorescent Analysis of PSC Derived Cortical Neurons
-
1.
Remove N2B27 media and wash cells once with PBS.
-
2.
Add 4 % paraformaldehyde to each well and incubate at room temperature for 10 min (see Note 14 ).
-
3.
Wash each well three times with 1× TBS.
-
4.
To permeabilise the cells, apply TBS-Tx to each well and leave on a rocking platform for half an hour at room temperature (see Note 15 ).
-
5.
Apply blocking solution to each well and incubate at room temperature for 1 h.
-
6.
Dilute primary antibodies (Table 3) to the appropriate concentration in blocking solution (e.g. manufacturer’s instructions).
Table 3 Recommended primary antibodies for identifying Alzheimer’s phenotypes -
7.
Add antibody solution to each well and incubate on a rocking platform overnight at 4 °C.
-
8.
Wash each well three times in 1× TBS, followed by an additional three times in TBS-Tx.
-
9.
Dilute secondary antibodies appropriately in blocking solution.
-
10.
Apply secondary antibodies and incubate for 1 h at room temperature protected from light.
-
11.
Wash six times in 1× TBS (see Note 16 ).
-
12.
Mount and image on a confocal microscope.
3.10 Measuring Abeta Peptide Production by Stem Cell-Derived Neurons
-
1.
Harvest medium from neuronal cultures every 48 h and replace with fresh N2B27 media.
-
2.
Collect media from each well into a separate 1.5 mL tube.
-
3.
Centrifuge at 1,200 × g for 3 min to pellet cellular debris.
-
4.
Remove supernatant and store in protein low-bind 1.5 mL tubes at −80 °C until needed. Avoid freeze-thaw cycles.
-
5.
Follow the protocol provided with your analytical method of choice (see Note 17 ).
4 Notes
-
1.
PSCs should be partially thawed and immediately transferred to hPSC medium to minimize the toxic effects of DMSO in the freezing medium.
-
2.
Colonies should be checked daily for non-specific differentiation. This is often seen as a loss of defined colony boundaries and a change in local cell density at these edges.
-
3.
Dispase preferentially dissociates PSC colonies from MEFs and differentiated cells. However, all cells will detach from the substrate if digestion is prolonged or the plate is agitated.
-
4.
Single cell colonies of PSCs are challenging to propagate, whereas larger colonies tend to merge and undergo some differentiation. Therefore, breaking up colonies to the correct size and their even distribution in the well is critical to maintain a PSC culture.
-
5.
A 1:6 passage ratio is typically performed to maintain a PSC culture, however cells should be split according to their confluency. This is particularly important when thawing a frozen culture, where 1:2 or 1:3 passages are more appropriate.
-
6.
Cells must be 100 % confluent before neural induction. Gaps in the monolayer will result in differentiation to non-cortical cell types, typically neural crest.
-
7.
During the course of induction the PSCs should become tightly packed and reduce their nuclear volume as they are specified to become neural progenitors.
-
8.
Neuroepithelial cells must be kept as aggregates to ensure that they retain their progenitor identity. Progenitors plated at a low local density will exit the cell cycle and become neurons.
-
9.
FGF2 promotes the proliferation of neural progenitors, however cells should not be treated for longer than 4 days as this can posteriorise the tissue.
-
10.
Dispase has a clear preference for dissociating neural progenitors rather than differentiated cells. This allows for purification of the culture over several passages.
-
11.
Neurons for comparison should be plated in the same type of dishes and the identical well size, as both of these factors can heavily influence the amount of free Abeta peptide in the media.
-
12.
Cortical cultures should be plated onto plastic dishes rather than glass cover slips in preparation for fixation and immunostaining, as this provides a better substrate for long term adhesion.
-
13.
Cortical cultures should not be passaged beyond day 40 as neurons are particularly fragile and survival rates following passaging are low.
-
14.
To improve the detection of surface antigens, cells can be fixed in pre-cooled 100 % methanol at −20 °C for 20 min prior to fixation in PFA. Triton can be omitted from later steps if using this method.
-
15.
Put the rocker at very low speed for washes as neurons are easily dislodged from culture plates following extended periods in culture.
-
16.
Add DAPI fluorescent stain (binding to A-T rich DNA regions) to one of the final wash steps if it is not present in the mounting media of choice (fluorescent labeling of nuclei).
-
17.
A number of platforms are available to determine the concentration of peptides/proteins of interest within the cell culture medium. Due to their sensitivity, dynamic range and convenience our preferred system is the multiplex MesoScale Discovery (MSD) assays.
References
Shi Y, Kirwan P, Smith J et al (2012) Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 15:477–486, S1
Maroof AM, Keros S, Tyson JA et al (2013) Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12:559–572
Nicholas CR, Chen J, Tang Y et al (2013) Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12:573–586
Israel MA, Yuan SH, Bardy C et al (2012) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482:216–220
Shi Y, Kirwan P, Smith J et al (2013) A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med 4:124ra29
Qiang L, Fujita R, Yamashita T et al (2011) Directed conversion of Alzheimer’s disease patient skin fibroblasts into functional neurons. Cell 146:359–371
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Park IH, Arora N, Huo H et al (2008) Disease-specific induced pluripotent stem cells. Cell 134:877–886
Yagi T, Ito D, Okada Y, Akamatsu W et al (2011) Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet 20:4530–4539
Kondo T, Asai M, Tsukita K et al (2013) Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12:487–496
Acknowledgements
Work in the lab of FJL is supported by the Wellcome Trust, Alzheimer’s Research UK, the Medical Research Council and the EU Innovative Medicines Initiative StemBANCC. NS is supported by the Woolf Fisher Trust. FJL is a Wellcome Trust Senior Investigator.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Saurat, N.G., Livesey, F.J., Moore, S. (2016). Cortical Differentiation of Human Pluripotent Cells for In Vitro Modeling of Alzheimer’s Disease. In: Castrillo, J., Oliver, S. (eds) Systems Biology of Alzheimer's Disease. Methods in Molecular Biology, vol 1303. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2627-5_16
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2627-5_16
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2626-8
Online ISBN: 978-1-4939-2627-5
eBook Packages: Springer Protocols