
Abstract
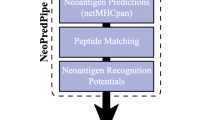
OpenVax is a computational workflow for identifying somatic variants, predicting neoantigens, and selecting the contents of personalized cancer vaccines. It is a Dockerized end-to-end pipeline that takes as input raw tumor/normal sequencing data. It is currently used in three clinical trials (NCT02721043, NCT03223103, and NCT03359239). In this chapter, we describe how to install and use OpenVax, as well as how to interpret the generated results.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
References
González S, Volkova N, Beer P et al (2018) Immuno-oncology from the perspective of somatic evolution. Semin Cancer Biol 52:75–85
Schumacher TN, Scheper W, Kvistborg P (2019) Cancer Neoantigens. Annu Rev Immunol 37:173–200
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
McKenna A, Hanna M, Banks E et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21
Cibulskis K, Lawrence MS, Carter SL et al (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31:213–219
Saunders CT, Wong WSW, Swamy S et al (2012) Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 28:1811–1817
Köster J and Rahmann S (2012) Building and documenting workflows with Python-based Snakemake, In: German Conference on Bioinformatics 2012, Schloss Dagstuhl-Leibniz-Zentrum fuer Informatik
Sherry ST, Ward MH, Kholodov M et al (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311
Forbes SA, Bhamra G, Bamford S et al (2008) The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet 10:11. https://doi.org/10.1002/0471142905.hg1011s57
Kim S, Scheffler K, Halpern AL et al (2018) Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15:591–594
Hoof I, Peters B, Sidney J et al (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13
Karosiene E, Lundegaard C, Lund O et al (2012) NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64:177–186
Peters B, Sette A (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6:132
Kim Y, Sidney J, Pinilla C et al (2009) Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics 10:394
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Kodysh, J., Rubinsteyn, A. (2020). OpenVax: An Open-Source Computational Pipeline for Cancer Neoantigen Prediction. In: Boegel, S. (eds) Bioinformatics for Cancer Immunotherapy. Methods in Molecular Biology, vol 2120. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-0327-7_10
Download citation
DOI: https://doi.org/10.1007/978-1-0716-0327-7_10
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-0326-0
Online ISBN: 978-1-0716-0327-7
eBook Packages: Springer Protocols