
Abstract
Oil-degrading bacteria and their communities have been in focus of the research for the past few decades for a number of reasons. First, this allows filling the voids in our knowledge on the major mechanisms facilitating the oil biodegradation, to identify the key organisms playing significant roles in these processes and, furthermore, to learn how to effectively manage their performance in situ to enhance the rates of biodegradation. Historically, of a particular interest for genomics studies were the so-called marine hydrocarbonoclastic bacteria, the petroleum biodegradation specialists with very restricted substrate profiles. Apart from their utility in environmental cleanup, oil-degrading bacteria possess an array of enzymes and pathways of a great potential for further biotechnological applications: biopolymers production, oxidation-reduction reactions, chiral synthesis, biosurfactant production, etc. In this chapter we describe current methods for genome and metagenome sequencing and annotation. Importantly, these are not limited to a particular group of microorganisms and are thus almost universally applicable. We focused exclusively on the methods and tools that everyone could use on a non-commercial basis. Due to the availability of numerous alternative methods and approaches, we have arbitrarily chosen reliable protocols that can be used by a common biologist without a great deal of computational biology background.
Author contributed equally with all other contributors.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
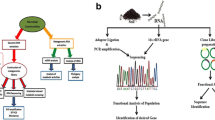

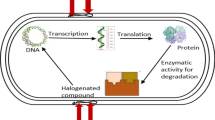
References
Schneiker S, Martins dos Santos VAP, Bartels D, Bekel T, Brecht M, Buhrmester J, Chernikova TN, Denaro R, Ferrer M, Gertler C, Goesmann A, Golyshina OV, Kaminski F, Khachane AN, Lang S, Linke B, McHardy AC, Meyer F, Nechitaylo T, Pühler A, Regenhardt D, Rupp O, Sabirova JS, Selbitschka W, Yakimov MM, Timmis KN, Vorhölter F-J, Weidner S, Kaiser O, Golyshin PN (2006) Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat Biotechnol 24:997–1004. doi:10.1038/nbt1232
Frey KG, Herrera-Galeano JE, Redden CL, Luu TV, Servetas SL, Mateczun AJ, Mokashi VP, Bishop-Lilly KA (2014) Comparison of three next-generation sequencing platforms for metagenomic sequencing and identification of pathogens in blood. BMC Genomics 15:96. doi:10.1186/1471-2164-15-96
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ (2012) Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol 30:434–439. doi:10.1038/nbt.2198
Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P (2007) Prediction of effective genome size inmetagenomic samples. Genome Biol 8:R10. doi:10.1186/gb-2007-8-1-r10
Tamames J, de la Peña S, de Lorenzo V (2012) COVER: a priori estimation of coverage for metagenomic sequencing. Environ Microbiol Rep 4:335–341. doi:10.1111/j.1758-2229.2012.00338.x
Daley T, Smith AD (2013) Predicting the molecular complexity of sequencing libraries. Nat Methods 10:325–327. doi:10.1038/nmeth.2375
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi:10.1093/bioinformatics/btr026
Schmieder R, Edwards R (2011) Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6:e17288. doi:10.1371/journal.pone.0017288
Kelley DR, Schatz MC, Salzberg SL (2010) Quake: quality-aware detection and correction of sequencing errors. Genome Biol 11:R116. doi:10.1186/gb-2010-11-11-r116
Salmela L, Schröder J (2011) Correcting errors in short reads by multiple alignments. Bioinformatics 27:1455–1461. doi:10.1093/bioinformatics/btr170
Chevreux B, Wetter T, Suhai S (1999) Genome sequence assembly using trace signals and additional sequence information In: Computer science and biology: Proceedings of the German conference on bioinformatics. http://www.bioinfo.de/isb/gcb99/talks/chevreux/main.html
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. doi:10.1038/nature03959
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi:10.1101/gr.074492.107
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27:578–579. doi:10.1093/bioinformatics/btq683
Nadalin F, Vezzi F, Policriti A (2012) GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13(Suppl 1):S8. doi:10.1186/1471-2105-13-S14-S8
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi:10.1093/bioinformatics/btp698
Yang X, Chockalingam SP, Aluru S (2013) A survey of error-correction methods for next-generation sequencing. Brief Bioinform 14:56–66. doi:10.1093/bib/bbs015
Nagarajan N, Pop M (2013) Sequence assembly demystified. Nat Rev Genet 14:157–167. doi:10.1038/nrg3367
El-Metwally S, Hamza T, Zakaria M, Helmy M (2013) Next-generation sequence assembly: four stages of data processing and computational challenges. PLoS Comput Biol 9:e1003345. doi:10.1371/journal.pcbi.1003345
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Schatz MC, Phillippy AM, Shneiderman B, Salzberg SL (2007) Hawkeye: an interactive visual analytics tool for genome assemblies. Genome Biol 8:R34. doi:10.1186/gb-2007-8-3-r34
Narzisi G, Mishra B (2011) Scoring-and-unfolding trimmed tree assembler: concepts, constructs and comparisons. Bioinformatics 27:153–160. doi:10.1093/bioinformatics/btq646
Vezzi F, Narzisi G, Mishra B (2012) Reevaluating assembly evaluations with feature response curves: GAGE and assemblathons. PLoS One 7:e52210. doi:10.1371/journal.pone.0052210
Sernova NV, Gelfand MS (2008) Identification of replication origins in prokaryotic genomes. Brief Bioinform 9:376–391. doi:10.1093/bib/bbn031
Zhang R, Zhang C-T (2005) Identification of replication origins in archaeal genomes based on the Z-curve method. Archaea 1:335–346
Luo H, Zhang C-T, Gao F (2014) Ori-Finder 2, an integrated tool to predict replication origins in the archaeal genomes. Front Microbiol 5:482. doi:10.3389/fmicb.2014.00482
Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi:10.1093/nar/gkm160
Cros M-J, de Monte A, Mariette J, Bardou P, Grenier-Boley B, Gautheret D, Touzet H, Gaspin C (2011) RNAspace.org: an integrated environment for the prediction, annotation, and analysis of ncRNA. RNA 17:1947–1956. doi:10.1261/rna.2844911
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41:D226–D232. doi:10.1093/nar/gks1005
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improveddetection of transfer RNA genes in genomicsequence. Nucleic Acids Res 25:955–964
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33:W686–W689. doi:10.1093/nar/gki366
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi:10.1186/1471-2164-9-75
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA (2008) The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi:10.1186/1471-2105-9-386
Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA (2010) RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res 38:D111–D118. doi:10.1093/nar/gkp894
Novichkov PS, Rodionov DA, Stavrovskaya ED, Novichkova ES, Kazakov AE, Gelfand MS, Arkin AP, Mironov AA, Dubchak I (2010) RegPredict: an integrated system for regulon inference in prokaryotes by comparative genomics approach. Nucleic Acids Res 38:W299–W307. doi:10.1093/nar/gkq531
Cipriano MJ, Novichkov PN, Kazakov AE, Rodionov DA, Arkin AP, Gelfand MS, Dubchak I (2013) RegTransBase – a database of regulatory sequences and interactions based on literature: a resource for investigating transcriptional regulation in prokaryotes. BMC Genomics 14:213. doi:10.1186/1471-2164-14-213
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. doi:10.1016/S0022-2836(05)80360-2
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Zhang Z, Schäffer AA, Miller W, Madden TL, Lipman DJ, Koonin EV, Altschul SF (1998) Protein sequence similarity searches using patterns as seeds. Nucleic Acids Res 26:3986–3990
Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL (2012) Domain enhanced lookup time accelerated BLAST. Biol Direct 7:12. doi:10.1186/1745-6150-7-12
Saier MH, Reddy VS, Tamang DG, Västermark A (2014) The transporter classification database. Nucleic Acids Res 42:D251–D258. doi:10.1093/nar/gkt1097
Rawlings ND, Waller M, Barrett AJ, Bateman A (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 42:D503–D509. doi:10.1093/nar/gkt953
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451. doi:10.1093/nar/gks479
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi:10.1093/nar/gkr367
Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics Chapter 2: Unit 2.3. doi:10.1002/0471250953.bi0203s00
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi:10.1093/nar/gkh340
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. doi:10.1006/jmbi.2000.4042
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi:10.1093/molbev/mst010
Wallace IM, O’Sullivan O, Higgins DG, Notredame C (2006) M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res 34:1692–1699. doi:10.1093/nar/gkl091
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi:10.1080/10635150701472164
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Cavalli-Sforza LL, Edwards AW (1967) Phylogenetic analysis. Models and estimation procedures. Am J Hum Genet 19:233–257
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Felsenstein J (2012) Confidence limits on phylogenies: an approach using the bootstrap. Evolution (N Y) 39:783–791
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Huang Y, Niu B, Gao Y, Fu L, Li W (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682. doi:10.1093/bioinformatics/btq003
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi:10.1038/nmeth.1701
Möller S, Croning MD, Apweiler R (2001) Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 17:646–653
Käll L, Krogh A, Sonnhammer ELL (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338:1027–1036. doi:10.1016/j.jmb.2004.03.016
Bendtsen JD, Nielsen H, Widdick D, Palmer T, Brunak S (2005) Prediction of twin-arginine signal peptides. BMC Bioinformatics 6:167. doi:10.1186/1471-2105-6-167
Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S (2004) Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel 17:349–356. doi:10.1093/protein/gzh037
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41:D808–D815. doi:10.1093/nar/gks1094
Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD (2014) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 42:D459–D471. doi:10.1093/nar/gkt1103
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199–D205. doi:10.1093/nar/gkt1076
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi:10.1093/nar/gku340
Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. doi:10.1093/nar/gkh468
Biegert A, Mayer C, Remmert M, Söding J, Lupas AN (2006) The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res 34:W335–W339. doi:10.1093/nar/gkl217
Hildebrand A, Remmert M, Biegert A, Söding J (2009) Fast and accurate automatic structure prediction with HHpred. Proteins 77(Suppl 9):128–132. doi:10.1002/prot.22499
Acknowledgements
This research was supported by the European Community Projects MAGICPAH (FP7-KBBE-2009-245226), ULIXES (FP7-KBBE-2010-266473) KILLSPILL (FP7-KBBE-2012-312139); MicroB3 (OCEAN.2011-2- 287589) and the Royal Society UK-Russia Travel Grant IE130218. The work of IK was supported by RFBR Grant # 13-04-0215715 and RSF Grant # 14-24-00165.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this protocol
Cite this protocol
Toshchakov, S.V., Kublanov, I.V., Messina, E., Yakimov, M.M., Golyshin, P.N. (2015). Genomic Analysis of Pure Cultures and Communities. In: McGenity, T., Timmis, K., Nogales , B. (eds) Hydrocarbon and Lipid Microbiology Protocols. Springer Protocols Handbooks. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8623_2015_126
Download citation
DOI: https://doi.org/10.1007/8623_2015_126
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-50449-9
Online ISBN: 978-3-662-50450-5
eBook Packages: Springer Protocols