
Abstract
Background
Gibbons (Hylobatidae) are the most diverse group of living apes. They exist as geographically-contiguous species which diverged more rapidly than did their close relatives, the great apes (Hominidae). Of the four extant gibbon genera, the evolutionary histories of two polyspecific genera, Hylobates and Nomascus, have been the particular focus of research but the DNA sequence data used was largely derived from the maternally inherited mitochondrial DNA (mtDNA) locus.
Results
To investigate the evolutionary relationships and divergence processes of gibbon species, particularly those of the Hylobates genus, we produced and analyzed a total of 11.5 kb DNA of sequence at 14 biparentally inherited autosomal loci. We find that on average gibbon genera have a high average sequence diversity but a lower degree of genetic differentiation as compared to great ape genera. Our multilocus species tree features H. pileatus in a basal position and a grouping of the four Sundaic island species (H. agilis, H. klossii, H. moloch and H. muelleri). We conducted pairwise comparisons based on an isolation-with-migration (IM) model and detect signals of asymmetric gene flow between H. lar and H. moloch, between H. agilis and H. muelleri, and between N. leucogenys and N. siki.
Conclusions
Our multilocus analyses provide inferences of gibbon evolutionary histories complementary to those based on single gene data. The results of IM analyses suggest that the divergence processes of gibbons may be accompanied by gene flow. Future studies using analyses of multi-population model with samples of known provenance for Hylobates and Nomascus species would expand the understanding of histories of gene flow during divergences for these two gibbon genera.
Similar content being viewed by others
Background
Gibbons are a family (Hylobatidae) of ape species endemic to the rainforests of the mainland and islands of Southeast Asia, including the Malay Peninsula, Sumatra, Borneo, Java and Mentawai Islands (Figure 1). They are the closest relatives of the great ape family (Hominidae) to which humans belong, and offer an interesting opportunity for comparisons. Like present-day great apes, the extant gibbons comprise four genera, which feature strikingly different numbers of chromosomes and extensive rearrangement of chromosomes [1–6]. Some 14 to 19 gibbon species have been recognized and classified into the genera Hylobates, Hoolock, Nomascus, and Symphalangus, indicating that gibbons consist of many more species than the seven species comprising the current day great apes [1, 7–13]. In contrast to the geographically-discontinuous distribution of current-day great apes, gibbons now or recently live in close geographic proximity to one another. Moreover, the divergence of the four extant great ape genera (Homo, Gorilla, Pan and Pongo) apparently occurred over a time span of more than five million years [14–16], whereas the radiation of the four gibbon genera may have occurred within less than two million years [10, 17]. Due to their close relationship to great apes, high taxonomic diversity and rapid diversification, gibbons have increasingly been the subject of molecular genetic analyses (e.g. [10, 17–23]).

Analyses of chromosomal number and structure and sequences of mitochondrial DNA (mtDNA) and nuclear loci consistently find that all four gibbon genera are monophyletic [3, 10, 17–21, 24–26], although the exact phylogenetic relationships among the genera are still debated. Different phylogenetic trees have been proposed, and a basal position of Nomascus was indicated based on sequences of mtDNA control region and cytochrome b gene [10, 20, 23] but this result conflicts with the basal placement of the genus Hoolock in studies based on sequences of mtDNA ND3-ND4 genes [24], the concatenated sequences of mtDNA, Y-linked and X-linked loci [17], sequences of autosomal and X chromosomal regions [22] and chromosomal analysis [3], as well as with the basal position of the Symphalangus genus in an Alu-based phylogeny of gibbons [27]. Moreover, the evolutionary relationships within the two polyspecific genera, Hylobates and Nomascus, have been the particular focus of research using sequence data from the mtDNA and nuclear loci (e.g. [10, 12, 17–19, 25, 28, 29]). MtDNA cytochrome b gene sequence analyses suggested that the divergences among Nomascus species accompanied a successive migration from north to south in which the two northernmost species N. hainanus and N. nasutus diverged first, followed by N. concolor and the two groups of southern species (the species N. leucogenys and N. siki and the two southernmost species N. annamensis and N. gabriellae) diverged last (Figure 1B; [10, 12]). In contrast to the pattern found in the genus Nomascus, the biogeographic scenario for the dispersal of Hylobates species is still in question [10, 17, 20, 25]. Nonetheless, the phylogenetic tree inferences from several datasets of mtDNA sequences (Figure 2B; [24, 25]) and the concatenated sequences of the mtDNA, Y-linked and X-linked loci [17] suggest that the two northernmost mainland species (H. lar and H. pileatus) may have branched off earlier than other Hylobates gibbons, although the tree inferences from other sequence datasets of mtDNA cytochrome b gene and Y chromosome placed H. klossii[10], H. moloch[20] or H. muelleri (Figure 2C [19]) as basal species in Hylobates phylogeny Figure 2.

Bayesian phylogenetic trees of gibbons. (A) Species tree inferred based on sequences from 14 autosomal loci of 44 gibbon individuals (this study). (B) Mtgenome tree inferred based on mtgenome sequences of 49 gibbon individuals excluding the control regions (Additional file 1; [18]). (C) Y chromosome tree inferred based on the partitioned concatenated dataset of seven Y chromosomal regions of 26 gibbon individuals [19]. The support values for the nodes according to the Bayesian posterior probability (PP) are shown as the circles (filled circles: PP ≥0.85; grey circles; 0.70 < PP < 0.85; open circles: PP <0.50). The colored boxes indicate species of taxa.
Although sequence data have thus been utilized to elucidate the phylogenetic relationships of Hylobates and Nomascus gibbons, most of these data were derived from single uniparentally inherited loci such as the maternally-inherited mtDNA [10, 12, 18, 20, 24, 25, 29] or the Y chromosome [28]. While mtDNA in particular provides useful information due to its rapid rate of evolution, DNA sequence data from multiple autosomal loci are needed to provide more comprehensive insights into the evolutionary relationships of species [30–32]. Recent advances in sequencing technologies have facilitated the gathering of multilocus sequence data from multiple individuals with extreme efficiency and thereby it has been feasible to acquire such data to explore evolutionary questions on species or population levels, even for non-model species [33, 34]. A number of analytical approaches have been developed that process multilocus data to estimate parameters in a coalescent framework with use of the Bayesian Markov chain Monte Carlo (MCMC) method, which enables multilocus species tree inference in contrast to the trees inferred from single genes or concatenated sequences of multiple genes (e.g. [35–41]). In addition, multilocus data are suitable for the investigation of whether the divergence of closely related species was accompanied by gene flow (e.g. [39, 40, 42–51]).
These coalescent-based approaches provide an opportunity to use multilocus sequence data to re-examine the phylogenetic relationships of Hylobates species and Nomascus species through reconstruction of species trees as well as to investigate the role of gene flow in the evolutionary histories of these gibbons. Among the Hylobates species, three areas of sympatry with natural hybridization have been documented between species through the observations of wild populations: H. agilis and H. lar in the north-western part of peninsular Malaysia, H. lar and H. pileatus in eastern central Thailand and, H. muelleri and H. albibarbis in central Borneo, respectively [52–54]. Also, small areas of sympatry between N. concolor and N. leucogenys in regions of northern Vietnam, northwestern Laos and southern China have been reported and possible hybrid individuals between N. concolor and N. leucogenys have been identified [9, 53, 55]. A recent study of a large amount of nuclear sequence data (16 nuclear loci with a total length of ~40 kb) from a limited number of individuals detected a signal of gene flow between N. gabriellae and N. leucogenys[22]. These findings highlighted the possibility that gene flow may occur between other pairs of co-generic gibbon species during their divergences as well.
In this study, we generated multilocus autosomal DNA sequence data to investigate the evolutionary histories of gibbons. Specifically, we sequenced 14 autosomal noncoding loci, which in previous studies were useful for elucidating evolutionary histories of great apes, from each of 44 gibbon individuals. We examined the levels of genetic variation and differentiation among the gibbon taxa, and further compare levels of genetic variation and patterns of genetic diversity between the gibbon and great ape families. We reconstructed evolutionary relationships among the sampled gibbon taxa using coalescent species tree analysis employed in the program *BEAST and we further applied the isolation-with-migration (IM) model implemented in the program IMa2 to assess the possibility of gene flow between closely related species.
Results and discussion
Levels of genetic diversity in gibbons
We amplified and sequenced a total of ~11.5 kb from 14 autosomal noncoding loci from each of 44 gibbon individuals (Table 1). These individuals represent six Hylobates, four Nomascus and one Symphalangus species (Table 2). We observed a total of 713 polymorphic sites among all gibbons. The nucleotide diversity levels were generally similar among regions with relatively lower diversity levels in locus 4 and locus 10 (Table 1). The average values of π and θw across all loci were calculated for genera and species (Table 2). Of the three sampled genera, we observed the highest diversity level in Hylobates (N = 58, π = 0.61% and θw = 0.76%) and the lowest level in Symphalangus (N = 12, π = 0.17% and θw = 0.19%), consistent with results based on sequence data of mtgenomes and Y chromosomes (Table 2), although comparison to the genus Hoolock cannot be made since these data are lacking from this genus, which is not in captivity in Europe and so not readily sampled by us. However, comparative sequence data from 1140 bp of the mitochondrial cytochrome b gene and from 20 nuclear loci (17 autosomal and three X chromosomal loci, totaling ~60 kb in length), also suggested that Hylobates had highest genetic diversity and Symphalangus was the least diverse among the four existing gibbon genera (Table 2).
We next compared the diversity levels among eight extant genera of the two ape families, Hominidae and Hylobatidae. Among great apes, sequence data from 16–25 autosomal loci, of which 14 were the same ones analyzed in this work on gibbons, found that orangutans (genus Pongo) had the highest diversity level (N = 32, π = 0.36% and θw = 0.35) as compared to the other three genera of great apes (Table 2) [57, 59, 60]. Interestingly, we observed diversity levels in the gibbon genera Nomascus (N = 18, π = 0.36% and θw = 0.42%) and Hylobates (N = 58, π = 0.61% and θw = 0.76%) as high, or even higher than that seen in orangutans. The higher diversity level of Hylobates than that of Pongo also was observed in the sequence data of the 20 nuclear loci (N = 16, π = 0.53% and θw = 0.50%, Table 2) [20]. Although there are just four genera in each of the two ape families, the high taxonomic diversity of 14–19 nominal gibbon species stands in contrast to the mere seven species of great apes [1, 7–12].
Within Hylobates, we observed considerable variation in the diversity levels among six Hylobates species (ranging from 0.06%-0.44% for π and 0.07%-0.45 for θw), and found within H. muelleri (N = 6, π = 0.44% and θw = 0.45%) a level over six times higher than that of H. pileatus (N = 8, π = 0.06% and θw = 0.07%). This finding of relatively higher diversity of H. muelleri, also seen from mtgenome sequences (Table 2), was also evident in a study of 20 nuclear loci employing eight individuals representing five Hylobates species (H. agilis, H. moloch, H. muelleri and H. pileatus) (Table 2) [22]. A recent study of the mtDNA cytochrome b gene, which used fairly comprehensive sampling of extant gibbon species including the recently identified H. albibarbis, provided estimates consistent with those based on mtgenome and nuclear datasets of relatively small sample sizes (Table 2) and supported the inference that H. muelleri may be the most genetically diverse Hylobates species [10]. Among the four sampled Nomascus species, we observed that N. gabriellae and N. leucogenys had similar diversity levels, which concurs with diversity estimates based on sequences of mtDNA cytochrome b and 20 nuclear loci (Table 2).
Sequence divergence and genetic differentiation between gibbon taxa
We also investigated the patterns of inter-genus and interspecies sequence divergence and genetic differentiation between gibbons by calculating the average number of differences per site (πb) and pairwise FST statistics, respectively, between genera (Table 3). Using our sequence data from 14 autosomal loci, we observed levels of sequence divergence and genetic differentiation between our three sampled gibbon genera (mean πb = 1.47% and FST = 0.72) similar to levels observed based on analysis of 20 nuclear loci (17 autosomal and three X chromosomal loci) for the same three genera (mean πb = 1.41% and FST = 0.77) [22]. These two estimates were slightly increased when the Hoolock (the unsampled genus in this study) was included (mean πb = 1.50% and FST = 0.80) [22]. We observed slightly lower sequence divergence between Nomascus and Symphalangus (πb = 1.38%) as compared to between Hylobates and Nomascus (πb = 1.46%) and between Hylobates and Symphalangus (πb = 1.58%). These inter-genus sequence divergence estimates are similar to those observed between African apes of different genera (πb = 1.12%-1.55%) but much lower than the levels between orangutans and any of the four African ape species (gorillas, bonobos, chimpanzees and humans; πb = 3.02%-3.19%) [57]. Moreover, we also observed lower levels of genetic differentiation between the three gibbon genera (FST = 0.65-0.79) as compared to the levels between humans and orangutans (FST = 0.94), between human and gorillas (FST = 0.92) or between human and chimpanzees (FST = 0.89) [57]. In sum, these results are consistent with a relatively low level of genetic differentiation among the gibbon genera as compared to among the great ape genera.
For the differentiation levels between species within the same genus, we found that within the genus Hylobates, the πb values were higher in the species comparisons including H. pileatus (ranging from 0.86% to 0.92%) than those in other species pairs (ranging from 0.50% to 0.67%) (Table 3). Relatively higher FST values were consistently found in species pairs including H. pileatus (ranging from 0.65 to 0.84) compared to those of other species pairs (ranging from 0.28 to 0.64). The higher pairwise πb and FST values estimated between H. pileatus and other Hylobates species were in general agreement with results obtained using data from 20 nuclear loci, in which five Hylobates species (H. agilis, H. moloch, H. muelleri and H. pileatus) were sampled and the highest πb and FST values were observed between H. muelleri and H. pileatus (πb = 0.82%) and between H. agilis and H. pileatus (FST = 0.66), respectively [22]. The relatively high levels of divergence and genetic differentiation between H. pileatus and other Hylobates species are consistent with phylogenetic analyses suggesting that H. pileatus diverged initially from the others during Hylobates evolution (Figure 2A, B; [17, 24, 25]). Moreover, although analyses using sequences of 20 nuclear loci found no evidence for genetic differentiation between H. moloch and H. muelleri (FST = 0.00) [22], our data from 14 autosomal loci showed a level of genetic differentiation between these two species (FST = 0.33) similar to the level between eastern and western chimpanzees (FST = 0.32) [57]. Within the genus Nomascus, the levels of divergence and genetic differentiation between our four sampled Nomascus species (πb = 0.30%-0.46% and FST = 0.30-0.56) (Table 3) were similar to those between bonobos and three chimpanzee subspecies (πb = 0.31%-0.32% and FST = 0.49-0.68) [57].
Patterns of gibbon divergence
To elucidate the divergence processes in gibbon evolutionary histories, we analyzed our multi-locus sequence data using two coalescent-based approaches: the reconstruction of a species tree and the isolation-with-migration (IM) model for population/species divergence. We first inferred a species tree for the sampled gibbon species using the coalescent-based Bayesian MCMC method implemented in the program *BEAST [35, 61, 62] and the sequence data of 14 autosomal loci. We found that the monophyly of the three sampled gibbon genera previously suggested was well supported [3, 10, 17, 19–21, 24–26] (Figure 2). Within the genus Hylobates, our species tree of biparentally inherited multilocus data suggests that the species H. pileatus is the most basal taxon and the four species with geographic distribution restricted to the Sundaic inlands (H. agilis, H. klossii, H. moloch and H. muelleri; Figure 1A) cluster together (Bayesian posterior probability, PP =0.85) (Figure 2A). These results are largely consistent with those in the single locus mtDNA analyses (Figure 2B; [18, 24, 25]) as well in the tree generated using a concatenated dataset of mtDNA, Y-linked and X-linked loci [17]. However, our data here are insufficient to resolve the relationships among the four Sundaic species while the mtgenome tree showed closer relationships of H. agilis-H. muelleri and H. klossii-H. moloch (Figure 2B). Because of a higher mutation rate, a smaller effective population and consequently a shorter coalescence time than typical autosomal loci, the uniparentally inherited mtDNA is known to be conducive to resolving phylogenetic relationships of recently diverging taxa [31, 32]. The divergence of the six Hylobates species analyzed here was estimated to occur over a short interval of about one million years [10, 17] and thus an even shorter time for the divergence of four Sundaic species could be expected. Compared to the biparentally inherited autosomal loci, the mtgenome sequence data provided higher resolution for disentangling the phylogenetic relationships of the four Sundaic species.
We next used a program (IMa2) based on the isolation-with-migration model [49–51, 63] to estimate multiple demographic parameters (including divergence time, migration rate and effective population sizes of derived populations and their ancestral populations) using MCMC simulation. Since the number of loci in our dataset (14) was insufficient for including all five sampled Hylobates species in a single IMa2 analysis, we conducted pairwise comparisons for these species as well as for the four Nomascus species studied. Species pairs were chosen for analysis based upon their close phylogenetic relationships or geographical proximity and consequent potential hybridization between them. Namely, IMa2 analyses were run for comparisons of seven Hylobates species pairs and for comparisons of three Nomascus species pairs (Table 4). The marginal posterior probability distributions of divergence time parameters showed clear peaks and bounds within the prior distribution for all pairwise comparisons except for N. gabriellae × N. siki which was tailed at the upper limit of the parameter prior space and hence returned an unreliable 95% highest posterior density (HPD) interval (Figure 3A). For Hylobates species, we found that the divergence time estimates were fairly consistent with the branching patterns of the species tree: (((H. agilis, H. muelleri, H. moloch, H. klossii), H. lar), H. pileatus) (Figures 2A and 3A). In the seven comparisons involving Hylobates, we found the oldest divergence time estimates between H. lar and H. pileatus (3 MYA) (Table 4 and Figure 2A). Following that, relatively younger time estimates were observed in pairwise comparisons between H. lar and the three species H. agilis, H. moloch and H. muelleri (2.2-2.5 MYA, H. lar × H. agilis, H. lar × H. moloch and H. lar × H. muelleri), which were older than the estimates of comparisons among the three species (1.5-2.1 MYA, H. moloch × H. agilis, H. muelleri × H. agilis, and H. moloch × H. muelleri) clustered together in the species tree (Table 4 and Figure 2A). Likewise, the younger estimate of divergence between N. leucogenys and N. siki (1.4 MYA) as compared to those between N. leucogenys and N. gabriellae and between N. gabriellae and N. siki (1.7-1.8 MYA) was consistent with branching patterns showing the prior divergence of N. gabriellae and the later divergence of the clade containing N. leucogenys and N. siki in the species tree (Table 4 and Figure 2A).

Marginal posterior probability distribution for parameters in IMa2 pairwise comparison analyses. Curves are shown for (A) estimates of divergence time parameter (in years) and (B) estimates of population migration rate (2NM) in each pairwise comparison analyses of Hylobates or Nomascus species.
Our analyses suggest that H. muelleri has the largest and H. pileatus the smallest effective population sizes among the five analyzed Hylobates species (202,300 and 11,100, respectively) (Table 4). Given the relatively larger inferred effective population sizes of H. muelleri in all pairwise comparison analyses and the smaller population size of H. pileatus when compared to those of their respective ancestral population sizes (Table 4), we would suggest that the species H. muelleri has experienced population expansion while the H. pileatus population has decreased in size since the divergence. Similarly, N. leucogenys also appears to have expanded as the current effective population size is larger than those of ancestral populations in both comparisons N. leucogenys × N. gabriellae and N. leucogenys × N. siki (Table 4).
Unidirectional gene flow between gibbon species
In addition to the estimations of divergence time and effective population sizes, the IMa2 analyses also provide inferences on the extent and patterns of gene flow in the divergence processes of species. Migration parameters (m) in the IM model can be transformed to obtain the estimates of population migration rate (2NM) which is the effective number of gene migrations received by a population per generation [49, 64, 65]. We found population migration rates significantly different from zero (2NM = 0.065-0.261, P < 0.05) in the comparisons of H. lar × H. agilis, H. lar × H. moloch, H. lar × H. muelleri, H. moloch × H. agilis, and H. muelleri × H. agilis (Table 4 and Figure 3B). However, after applying Bonferroni correction for multiple testing, significantly nonzero population migration rates were only found in the comparisons H. lar × H. moloch and H. muelleri × H. agilis, which indicated gene flow from H. moloch to H. lar (2NM = 0.074, P < 0.0035) and from H. muelleri to H. agilis (2NM = 0.261, P < 0.0035) (Table 4). We also detected significantly nonzero population migration rate for the gene flow from N. leucogenys to N. siki (2NM = 0.364, P < 0.008) (Table 4). Also notable is the asymmetry in gene flow, as we only found significant signals of gene flow in one direction but not in opposite direction in the comparisons where significantly nonzero migration rates were observed (Table 4 and Figure 3B).
Among the Hylobates, most species are currently separated from each other by bodies of water (e.g. the Java Sea and Karimata Strait between H. lar and H. moloch and the Karimata Strait between H. agilis and H. muelleri). Given these barriers, it is necessary that any signal of gene flow found between such species cannot be the consequence of recent contact. Rather, lower sea levels and changes in the distributions of gibbon populations in the past may have allowed some interactions among these Hylobates taxa [10, 66, 67]. Furthermore, it is possible that the signals of gene flow we detected between Hylobates species may underestimate the history of gene flow during Hylobates divergence. Our dataset was insufficient for attempting IMa2 analyses in the multi-population model which can reveal historical gene flow involving ancestral populations [49, 64]. Consequently, we could only assess gene flow between two derived populations and any gene flow between the derived populations and the ancestral populations was not addressed in our pairwise comparison analyses. The importance of this consideration was shown in an analysis of bonobos and three chimpanzee subspecies in which asymmetric gene flow was detected in a pairwise analysis between allopatric central and western chimpanzees while further three- and four-population analyses suggested that there had actually been gene flow between the western chimpanzees and the ancestral population of central and eastern chimpanzees [64]. Accordingly, that the gene flow from the western to the central chimpanzees identified in the two-population analysis likely reflects the history of gene flow from the western chimpanzees into the ancestor of central and eastern chimpanzees identified in multi-population analyses [64]. In the case of Hylobates species, the four Sundaic species (H. agilis, H. klossii, H. moloch and H. muelleri) shared the same ancestral population according to phylogenetic analyses (Figure 2A, B; [17, 24, 25]) and the gene flow suggested by our IMa2 analyses of the comparisons among H. lar and three of these species (H. agilis, H. moloch and H. muelleri) could reflect historical gene flow between H. lar and the ancestral population of the Sundaic species. Our analyses did not allow us to date the time of potential gene flow between species. However, mtDNA studies have supported the monophylies of Hylobates species, while Y-chromosome studies are less clear on the possibility of sharing of Y-haplotypes between groups (Figure 2B, C; [10, 19, 24, 25]). Thus, it is possible that any female-mediated gene flow in Hylobates may have occurred deep enough in the past to allow for mitochondrial lineage sorting, or that any more recent gene flow has been male-mediated.
Among Nomascus species, N. leucogenys and N. siki, who share more similarities of genetics, morphology and acoustics to each other than to other Nomasacus species [12, 53, 68], exist in adjacent distribution areas (Figure 1B), which might allow some gene exchanges between these two species in their contact zones. We detected a significant signal of gene flow between N. leucogenys and N. siki (Table 4 and Figure 3B), but cannot assess whether this may be a result of gene exchanges occurring during their divergence processes or a consequence of relatively recent secondary contact after speciation. In the case of secondary contact after speciation, the exchanged alleles may have not spread over the ranges of the two species. Analyses with geographically selective sampling, where individuals would be sampled far from and in/near the contact zones, may be helpful to distinguish between the scenarios of divergence with gene flow and secondary contact, as the signal of gene flow could be reduced or eliminated when excluding particular individuals from or near contact zones (e.g. [40, 45]). Our sampling of N. leucogenys and N. siki is limited and the provenances of these individuals are unclear. The contemporary geographic distributions of Nomascus species have been recently revised via vocal and genetic analyses of individuals with known geographic origins and especially those from areas of potential species boundaries, but because these noninvasively collected samples and tissue samples from museum specimens yield DNA of poor quality, limiting the scope of the genetic analyses to the Nomascus phylogeny based on mtDNA cytochrome gene sequence data [12, 68].
Gene flow between N. leucogenys and N. gabriellae with a migration rate equivalent to a rate about one migrant every two generations has also been suggested [22]. However, although we obtained population migration rate estimates of 2NM = 0.015 for migration from N. gabriellae into N. leucogenys and 2NM = 0.151 from N. leucogenys into N. gabriellae, these two estimates were not significantly different from zero (P > 0.1), indicating no gene flow between these two species whose geographic distributions are currently discontiguous and interrupted by the distributions of N. siki and N. annamensis (Table 4 and Figure 1B).
The detection of asymmetric gene flow between gibbon species is not unexpected but has also been seen for closely related taxa in other primates (e.g. gorillas [59], macaques [45], chimpanzees [64, 69], and baboons [70]) and other animals (review in [65]). For example, more gene flow may take place from eastern to western gorillas (2NM = 0.350) than western to eastern gorillas (2NM = 0.141) [59]. Unidirectional patterns of gene flow apparently occurred between three Mus species [43] as well as between two macaque species [45]. In the case of macaques, the IM analyses with exclusion of loci violating neutrality found that the gene flow from rhesus into cynomologus macaques was estimated as 2NM = 0.493, while gene flow in the other direction was not significantly different from zero [45]. Moreover, the extent of gene flow we detected here between gibbons species was similar to that estimated between other closely related taxa (e.g. gorillas [59] or Mus species [43]). However, the population genetic structure within species might lead to different inferences about the extent of gene flow between species. For example, a signal of gene flow may be underestimated or not be detected if samples are not collected from populations in potential contact zones [45]. The magnitude of gene flow detected between two species would also increase with decreasing distance between the sampled populations of two species [70]. Our sample sizes of each gibbon species are limited and their geographic origins are unclear. Therefore, the extent and patterns of gene flow detected here likely represent a minimum estimate and additional work is needed incorporating extensive sampling of individuals with known geographic provenance.
Conclusions
Our analyses of sequence data of 14 autosomal loci, coupled with two coalescent-based analyses (*BEAST and IMa2), provide inferences of species trees and the extent and patterns of gene flow among gibbon taxa. Our tree (Figure 2A), like those based upon mtDNA sequences or the concatenated sequences of mtDNA, Y-linked and X-linked loci, shows H. pileatus as the basal Hylobates taxon and groups the four Sundaic species (Figure 2B; [17, 24, 25]). We find evidence for unidirectional gene flow between some gibbon species; namely between H. lar and H. moloch, between H. agilis and H. muelleri and between N. leucogenys and N. siki. Further insights will require the use of multi-population analyses investigating historical gene flow involving ancestral populations [49, 64] by use of a larger dataset of more loci as well as a larger set of samples of known geographic origin.
Methods
Gibbon DNA samples and PCR amplification of 14 autosomal loci
We used 44 high-quality genomic DNA samples, including representatives of six Hylobates, four Nomascus and one Symphalangus species. All DNA samples used derive from the long-term sample collections of the authors and were not acquired specifically for this study. These samples were originally collected in the course of routine veterinary examinations of captive gibbons. We performed whole genome amplification (WGA) on all genomic DNA samples using the multiple displacement amplification procedure implemented in the GenomiPhi HY DNA Amplification Kit (GE Healthcare). The WGA products were purified by ethanol precipitation following manufacturer’s instructions. The purified WGA products were quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.) and used as templates for subsequent polymerase chain reactions (PCRs) for the amplification of autosomal loci.
We amplified 14 autosomal loci (Table 1) previously shown to be noncoding and selectively neutral and used in studies on the evolutionary histories of great apes [57–60, 71]. The polymerase chain reaction (PCR) primers used for these 14 loci were described in the previous studies [57, 59], which were designed by using the human and chimpanzees. The PCR amplification reactions were carried out in a volume of 50 μl containing 60–100 ng of purified WGA products, 0.4 mM MgCl2, 0.2 μM of each forward and reverse primer, 200 μM of each dNTP, 10 × SUPER TAQ PCR buffer (containing MgCl2), and 1.5 units of SUPER TAQ (HT Biotechnology, Cambridge, UK) premixed 2:1 with 1 μg/μl TaqStart monoclonal antibody (BD Bioscience Clontech). The PCR condition included following steps: initial denaturation at 94°C for 1 min; 35 cycles of denaturation at 94°C, primer annealing at 57–61°C for 1 min 30 sec, elongation at 72°C, and a final elongation step of 7 min at 72°C. The PCR products were gel-cut and purified using QIAquick Gel Extraction Kit (Qiagen).
Sequencing of 14 autosomal locus amplicons
We used the high-throughput Illumina sequencing platform to sequence 14 autosomal loci for 44 gibbons. A sequencing library containing 14 autosomal locus amplicons was created for each of 44 individuals using a modified Illumina protocol [72] where a PCR reaction was used to add individual-specific indexing oligos to both ends of library molecules. This indexing PCR procedure allowed us to identify and sort read sequences by individuals during data processing. The 44 individual indexed libraries were pooled in equimolar ratio and sequenced on a single lane of the flow cell of an Illumina Genome Analyzer IIx instrument with paired-end sequencing of 76 + 7 cycles according to the manufacturer’s instructions (Illumina). Bases and quality scores were generated with the Ibis base caller [73]. The reads were then processed based on their indexes and the indexed reads were aligned with the Burrows-Wheeler Aligner (BWA) software [74] to the chimpanzee homologues of 14 autosomal locus sequences with default parameters, resulting in the 44 individual BAM files (44 gibbons). Subsequently, the BAM files were processed separately and the reads of potential PCR duplicates were removed using SAMtools [75]. The consensus sequences of 14 autosomal loci of 44 gibbons were generated also by using SAMtools. In sum, for each consensus sequence, each base had a minimum averaged PHRED score of 25 and the alternate alleles of heterozygous sites had a coverage rate of at least 30%. The summary statistics of the reads for each gibbon is provided in Additional file 2 and the consensus sequences have been deposited in Genbank under the accession numbers KC480606-KC481221.
Sequence data analysis
Multiple alignments for the consensus sequences were generated with ClustalW v2.0 [76] and then edited and checked with BioEdit v7.0.5 [77]. Haplotype phases were inferred with PHASE v2.1 [78, 79] for each locus where the program SeqPHASE [80] was used to interconvert FASTA files of the alignments to the formats of PHASE input and output files. The program DnaSP v5.10 [81] was used to calculate two standard diversity indices, π [56] and θw[82], and pairwise FST statistics between genera or species [83]. We also estimated the average number of differences per site between sequences sampled from two different genera or two species (nucleotide diversity between populations, πb). To test the selective neutrality of each locus, we calculated Tajima’s D [84] using DnaSP with 10,000 coalescent simulations, suggesting no signal of a departure from neutral evolution.
Estimation of gene flow
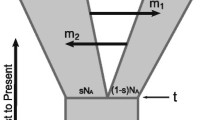
We used program IMa2 [49] to assess the extents of gene flow that may have occurred between Hylobates species and between Nomascus species. Since IMa2 assumes no recombination in each locus analyzed, we tested the possibility of intralocus recombination with methods implemented in the programs Recombination Detection Program (RDP) v3.44 [85]: RDP [86], GENECONV [87], MaxChi [88], Chimaera [89], SiScan [90] and 3Seq [91]. When recombination was detected, only non-recombining blocks of sequences were used in the input datasets. The base positions with gaps/indels in the alignments were removed from IMa2 analyses. The program IMa2 is based on an isolation-with-migration (IM) model and estimates the posterior probability densities using Markov chain Monte Carlo (MCMC) simulation for the parameters scaled by the mutation rate (μ): bi-directional migration rate (m = M/μ, where M is the migration rate per generation per gene copy), population size (θ = 4Neμ, where Ne is effective population size) and divergence time (t = tμ, where t is the time since population splitting) [49–51, 63]. The parameter estimates of population size and divergence time resulting from the IMa2 analyses were converted to the estimates of effective population size (Ne) in individuals and divergence time (t) in years. The migration rate parameters can be transformed into the estimates of population migration rate (i.e. 2NM = 4Neμ × M/μ/2), the effective number of migrant gene copies per generation [49, 64]. For these conversions, we used a time of splitting between gibbon and great apes of approximately 19.5 million years [15, 92–94] and an assumed generation time of 15 years. The life histories of gibbons have been suggested to resemble those of great apes rather than same-sized monkeys [95]. The female age of first reproduction has been estimated for wild populations of H. lar at 11.06 years, which was only slightly younger than that of great ape females, and its interbirth interval has been estimated as 41 months, longer than those of same-sized monkeys (e.g. Macaca or Cercocebus) [95]. Therefore, we assumed a generation time of 15 years for gibbons. We estimated the mutation rate per year for each locus using the average divergence of sequences (Dxy) [56] between gibbon and chimpanzee (Table 1) with the divergence time (T) of 19.5 million years between them (Dxy = 2Tμ approximately under a neutral evolution model). The average mutation rate (mutation/site/year) of the 14 loci was calculated as 0.94 × 10-9, which is similar to the commonly-used per-site genome-wide mutation rates of human and other great apes [14, 96–98].
Due to the limited number of loci and large number of gibbon species, we were unable to include all Hylobates species in a single IMa2 analysis and thus we conducted the IMa2 analyses with pairwise comparisons between Hylobates species as well as between Nomascus species. Species for which we had sequenced only one individual were excluded from the analyses (i.e. H. klossii and N. concolor). The species pairs were selected for analysis based on: (1) evidence for close phylogenetic relationships or (2) the presence of contact zones or suggestions of potential hybridization. Accordingly, we conducted seven comparisons for the Hylobates spp. and three comparisons for the Nomascus spp. (Table 4). Preliminary runs were performed to estimate the settings of uniform priors (upper bound on the uniform prior distribution) for the parameters, the necessary duration of runs and the heating terms of Metropolis-coupled chains required for well-mixed Markov chains. Once optimal priors and heating schemes were devised from initial runs, 20 independent Markov chains (−hn20 -ha0.96 -hb0.9) and six independent runs with different starting seeds (adjusting only the starting seed) were performed for each analysis. We saved 20,000 genealogies per run after a sufficient burn-in period (1,000,000 burn-in steps). Stationarity was reached already during the burn-in period. The adequate convergence of the MCMC simulation was assessed by (i) inspection of autocorrelation values over the course the run and effective sample sizes (ESS); (ii) inspection of the parameter trend plots; and (iii) checking that the parameter estimates calculated using genealogies sampled in the first and second halves of the run were highly similar. At least 100,000 sampled genealogies pooled from independent MCMC runs were used to calculate marginal posterior probability density estimates for the parameters using “L-mode” in IMa2, and LLR test (likelihood ratio test) statistics for assessing whether the estimated migration rates are significantly different from zero with a mix chi-squared distribution [49, 63].
Bayesian inference of species tree
We conducted a coalescent-based method for species tree reconstruction employed in the program *BEAST [35] using chimpanzee sequences as the outgroup. This method jointly estimates the posterior distributions of species tree and a set of gene trees from multi-locus sequence data and is implemented in the BEAST software package v1.7.2 [61, 62]. The best-fit substitution models were assessed using the Akaike information criterion (AIC) by Model-Generator v0.85 [99] and were set independently for each locus partition. In addition, the clock and tree models were unlinked for all locus partitions. Four independent BEAST runs of 100,000,000 generations (100 million) were carried out with lognormal relaxed clock model and Yule speciation process in tree prior, sampling every 5,000 generations. Convergence was assessed in Tracer 1.5 [100] and the burn-in period was set as 4,000 trees. We combined the log output files from four individual BEAST runs using LogCombiner 1.7.2 [62] and the ESS values for all parameters were above 200. The maximum-clade-credibility tree was generated using TreeAnnotator 1.7.2 [62] and visualized using FigTree 1.3.1 [101].
Abbreviations
- IM:
-
Isolation with migration
- mtDNA:
-
Mitochondrial DNA
- MCMC:
-
Markov chain Monte Carlo
- mtgenome:
-
Mitochondrial genome
- HPD:
-
Highest posterior density
- MYA:
-
Million years ago
- WGA:
-
Whole genome amplification
- PCR:
-
Polymerase chain reaction
- BWA:
-
Burrows-Wheeler Aligner
- ESS:
-
Effective sample sizes
- AIC:
-
Akaike information criterion.
References
Groves C: Primate taxonomy. 2001, Washington: Smithsonian Institution Press
Jauch A, Wienberg J, Stanyon R, Arnold N, Tofanelli S, Ishida T, Cremer T: Reconstruction of genomic rearrangements in great apes and gibbons by chromosome painting. Proc Natl Acad Sci U S A. 1992, 89: 8611-8615. 10.1073/pnas.89.18.8611.
Müller S, Hollatz M, Wienberg J: Chromosomal phylogeny and evolution of gibbons (Hylobatidae). Hum Genet. 2003, 113: 493-501. 10.1007/s00439-003-0997-2.
Van Tuinen P, Ledbetter DH: Cytogenetic comparison and phylogeny of three species of Hylobatidae. Am J Phys Anthropol. 1983, 61: 453-466. 10.1002/ajpa.1330610408.
Capozzi O, Carbone L, Stanyon RR, Marra A, Yang F, Whelan CW, de Jong PJ, Rocchi M, Archidiacono N: A comprehensive molecular cytogenetic analysis of chromosome rearrangements in gibbons. Genome Res. 2012, 22: 2520-2528. 10.1101/gr.138651.112.
Misceo D, Capozzi O, Roberto R, Dell’oglio MP, Rocchi M, Stanyon R, Archidiacono N: Tracking the complex flow of chromosome rearrangements from the Hominoidea Ancestor to extant Hylobates and Nomascus Gibbons by high-resolution synteny mapping. Genome Res. 2008, 18: 1530-1537. 10.1101/gr.078295.108.
IUCN Red List. 2012, http://www.iucnredlist.org/,
Mootnick A, Groves C: A new generic name for the hoolock gibbon (Hylobatidae). Int J Primatol. 2005, 26: 971-976. 10.1007/s10764-005-5332-4.
Mootnick AR, Fan PF: A comparative study of crested gibbons (Nomascus). Am J Primatol. 2011, 73: 135-154. 10.1002/ajp.20880.
Thinh VN, Mootnick AR, Geissmann T, Li M, Ziegler T, Agil M, Moisson P, Nadler T, Walter L, Roos C: Mitochondrial evidence for multiple radiations in the evolutionary history of small apes. BMC Evol Biol. 2010, 10: 74-10.1186/1471-2148-10-74.
Thinh VN, Mootnick AR, Thanh VN, Nadler T, Roos C: A new species of crested gibbon, from the central Annamite mountain range. Vietn J Primatol. 2010, 4: 1-12.
Thinh VN, Rawson B, Hallam C, Kenyon M, Nadler T, Walter L, Roos C: Phylogeny and distribution of crested gibbons (genus Nomascus) based on mitochondrial cytochrome b gene sequence data. Am J Primatol. 2010, 72: 1047-1054. 10.1002/ajp.20861.
Chivers DJ, Roos C, Groves CP, Rawson BDW: Gibbons (Hylobatidae). Handbook of the mammals of the world Vol 3 Primates. Edited by: Mittermeier RA, DE W. Barcelona: Lynx Edicions, in press
Hobolth A, Dutheil JY, Hawks J, Schierup MH, Mailund T: Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 2011, 21: 349-356. 10.1101/gr.114751.110.
Locke DP, Hillier LW, Warren WC, Worley KC, Nazareth LV, Muzny DM, Yang SP, Wang Z, Chinwalla AT, Minx P, et al: Comparative and demographic analysis of orang-utan genomes. Nature. 2011, 469: 529-533. 10.1038/nature09687.
Wilkinson RD, Steiper ME, Soligo C, Martin RD, Yang Z, Tavare S: Dating primate divergences through an integrated analysis of palaeontological and molecular data. Syst Biol. 2011, 60: 16-31. 10.1093/sysbio/syq054.
Israfil H, Zehr SM, Mootnick AR, Ruvolo M, Steiper ME: Unresolved molecular phylogenies of gibbons and siamangs (Family: Hylobatidae) based on mitochondrial, Y-linked, and X-linked loci indicate a rapid Miocene radiation or sudden vicariance event. Mol Phylogenet Evol. 2011, 58: 447-455. 10.1016/j.ympev.2010.11.005.
Chan YC, Roos C, Inoue-Murayama M, Inoue E, Shih CC, Pei KJ, Vigilant L: Mitochondrial genome sequences effectively reveal the phylogeny of Hylobates gibbons. PLoS One. 2010, 5: e14419-10.1371/journal.pone.0014419.
Chan YC, Roos C, Inoue-Murayama M, Inoue E, Shih CC, Vigilant L: A comparative analysis of Y chromosome and mtDNA phylogenies of the Hylobates gibbons. BMC Evol Biol. 2012, 12: 150-10.1186/1471-2148-12-150.
Chatterjee HJ: Phylogeny and biogeography of gibbons: a dispersal-vicariance analysis. Int J Primatol. 2006, 27: 699-712. 10.1007/s10764-006-9044-1.
Hayashi S, Hayasaka K, Takenaka O, Horai S: Molecular phylogeny of gibbons inferred from mitochondrial-DNA sequences - preliminary report. J Mol Evol. 1995, 41: 359-365. 10.1007/BF01215183.
Kim SK, Carbone L, Becquet C, Mootnick AR, Li DJ, de Jong PJ, Wall JD: Patterns of genetic variation within and between gibbon species. Mol Biol Evol. 2011, 28: 2211-2218. 10.1093/molbev/msr033.
Roos C, Geissmann T: Molecular phylogeny of the major hylobatid divisions. Mol Phylogenet Evol. 2001, 19: 486-494. 10.1006/mpev.2001.0939.
Takacs Z, Morales JC, Geissmann T, Melnick DJ: A complete species-level phylogeny of the Hylobatidae based on mitochondrial ND3-ND4 gene sequences. Mol Phylogenet Evol. 2005, 36: 456-467. 10.1016/j.ympev.2005.03.032.
Whittaker DJ, Morales JC, Melnick DJ: Resolution of the Hylobates phylogeny: congruence of mitochondrial D-loop sequences with molecular, behavioral, and morphological data sets. Mol Phylogenet Evol. 2007, 45: 620-628. 10.1016/j.ympev.2007.08.009.
Garza JC, Woodruff DS: A phylogenetic study of the gibbons (Hylobates) using DNA obtained noninvasively from hair. Mol Phylogenet Evol. 1992, 1: 202-210. 10.1016/1055-7903(92)90016-A.
Meyer TJ, McLain AT, Oldenburg JM, Faulk C, Bourgeois MG, Conlin EM, Mootnick AR, de Jong PJ, Roos C, Carbone L, Batzer MA: An Alu-based phylogeny of gibbons (Hylobatidae). Mol Biol Evol. 2012, 29: 3441-3450. 10.1093/molbev/mss149.
Hong KW, Huh JW, Kim DS, Ha HS, Kim HS: Molecular relationship of hylobates based on Alu elements of the Y chromosome. Korean J Genet. 2007, 29: 379-387.
Monda K, Simmons RE, Kressirer P, Su B, Woodruff DS: Mitochondrial DNA hypervariable region-1 sequence variation and phylogeny of the concolor gibbons, Nomascus. Am J Primatol. 2007, 69: 1285-1306. 10.1002/ajp.20439.
Brown WM, George M, Wilson AC: Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A. 1979, 76: 1967-1971. 10.1073/pnas.76.4.1967.
Moore WS: Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995, 49: 718-726. 10.2307/2410325.
Zink RM, Barrowclough GF: Mitochondrial DNA under siege in avian phylogeography. Mol Ecol. 2008, 17: 2107-2121. 10.1111/j.1365-294X.2008.03737.x.
Ekblom R, Galindo J: Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011, 107: 1-15. 10.1038/hdy.2010.152.
Rokas A, Abbot P: Harnessing genomics for evolutionary insights. Trends Ecol Evol. 2009, 24: 192-200. 10.1016/j.tree.2008.11.004.
Heled J, Drummond AJ: Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010, 27: 570-580. 10.1093/molbev/msp274.
Liu L, Pearl DK: Species trees from gene trees: reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst Biol. 2007, 56: 504-514. 10.1080/10635150701429982.
Liu L, Pearl DK, Brumfield RT, Edwards SV: Estimating species trees using multiple-allele DNA sequence data. Evolution. 2008, 62: 2080-2091. 10.1111/j.1558-5646.2008.00414.x.
Liu L: BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics. 2008, 24: 2542-2543. 10.1093/bioinformatics/btn484.
Lee YH, Lin CP: Pleistocene speciation with and without gene flow in Euphaea damselflies of subtropical and tropical East Asian islands. Mol Ecol. 2012, 21: 3739-3756. 10.1111/j.1365-294X.2012.05654.x.
Jackson ND, Austin CC: Inferring the evolutionary history of divergence despite gene flow in a lizard species, Scincella lateralis (Scincidae), composed of cryptic lineages. Biol J Linn Soc Lond. 2012, 107: 192-209. 10.1111/j.1095-8312.2012.01929.x.
Amaral AR, Jackson JA, Moller LM, Beheregaray LB, Manuela Coelho M: Species tree of a recent radiation: the subfamily Delphininae (Cetacea, Mammalia). Mol Phylogenet Evol. 2012, 64: 243-253. 10.1016/j.ympev.2012.04.004.
Carling MD, Lovette IJ, Brumfield RT: Historical divergence and gene flow: coalescent analyses of mitochondrial, autosomal and sex-linked loci in Passerina buntings. Evolution. 2010, 64: 1762-1772. 10.1111/j.1558-5646.2010.00954.x.
Geraldes A, Basset P, Gibson B, Smith KL, Harr B, Yu HT, Bulatova N, Ziv Y, Nachman MW: Inferring the history of speciation in house mice from autosomal, X-linked, Y-linked and mitochondrial genes. Mol Ecol. 2008, 17: 5349-5363. 10.1111/j.1365-294X.2008.04005.x.
Lim HC, Sheldon FH: Multilocus analysis of the evolutionary dynamics of rainforest bird populations in Southeast Asia. Mol Ecol. 2011, 20: 3414-3438. 10.1111/j.1365-294X.2011.05190.x.
Stevison LS, Kohn MH: Divergence population genetic analysis of hybridization between rhesus and cynomolgus macaques. Mol Ecol. 2009, 18: 2457-2475. 10.1111/j.1365-294X.2009.04212.x.
Tavares ES, de Kroon GHJ, Baker AJ: Phylogenetic and coalescent analysis of three loci suggest that the Water Rail is divisible into two species, Rallus aquaticus and R. indicus. BMC Evol Biol. 2010, 10: 226-10.1186/1471-2148-10-226.
Walstrom VW, Klicka J, Spellman GM: Speciation in the White-breasted Nuthatch (Sitta carolinensis): a multilocus perspective. Mol Ecol. 2012, 21: 907-920. 10.1111/j.1365-294X.2011.05384.x.
Becquet C, Przeworski M: A new approach to estimate parameters of speciation models with application to apes. Genome Res. 2007, 17: 1505-1519. 10.1101/gr.6409707.
Hey J: Isolation with migration models for more than two populations. Mol Biol Evol. 2010, 27: 905-920. 10.1093/molbev/msp296.
Hey J, Nielsen R: Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004, 167: 747-760. 10.1534/genetics.103.024182.
Hey J, Nielsen R: Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc Natl Acad Sci U S A. 2007, 104: 2785-2790. 10.1073/pnas.0611164104.
Brockelman WY, Gittins SP: Natural hybridization in the Hylobates lar species group: implications for speciation in gibbons. The lesser apes Evolutionary and behavioural biology. Edited by: Brockelman WY, Preuschoft H, Chivers DJ, Creel N. 1984, Edinburgh: Edinburgh University Press, 498-532.
Geissmann T: Gibbon systematics and species identification. Int Zoo News. 1995, 42: 65-77.
Marshall J, Sugardjito J: Gibbon systematics. Comparative primate biology Volume 1 Systematics, evolution and anatomy. Edited by: Swindler DR, Erwin J. 1986, New York: Alan R. Liss, 137-185.
Dao VT: On the north indochinese gibbons (Hylobates concolor) (Primates, Hylobatidae) in North Vietnam. J Hum Evol. 1983, 12: 367-372. 10.1016/S0047-2484(83)80164-X.
Nei M: Molecular evolutionary genetics. 1987, New York: Columbia University Press
Fischer A, Pollack J, Thalmann O, Nickel B, Paabo S: Demographic history and genetic differentiation in apes. Curr Biol. 2006, 16: 1133-1138. 10.1016/j.cub.2006.04.033.
Voight BF, Adams AM, Frisse LA, Qian Y, Hudson RR, Di Rienzo A: Interrogating multiple aspects of variation in a full resequencing data set to infer human population size changes. Proc Natl Acad Sci U S A. 2005, 102: 18508-18513. 10.1073/pnas.0507325102.
Thalmann O, Fischer A, Lankester F, Paabo S, Vigilant L: The complex evolutionary history of gorillas: insights from genomic data. Mol Biol Evol. 2007, 24: 146-158.
Fischer A, Wiebe V, Paabo S, Przeworski M: Evidence for a complex demographic history of chimpanzees. Mol Biol Evol. 2004, 21: 799-808. 10.1093/molbev/msh083.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Drummond AJ, Suchard MA, Xie D, Rambaut A: Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012, 29: 1969-1973. 10.1093/molbev/mss075.
Nielsen R, Wakeley J: Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001, 158: 885-896.
Hey J: The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol Biol Evol. 2010, 27: 921-933. 10.1093/molbev/msp298.
Pinho C, Hey J: Divergence with Gene Flow: Models and Data. Annu Rev Ecol Evol Syst. 2010, 41: 215-230. 10.1146/annurev-ecolsys-102209-144644.
Woodruff DS: Biogeography and conservation in Southeast Asia: how 2.7 million years of repeated environmental fluctuations affect today’s patterns and the future of the remaining refugial-phase biodiversity. Biodivers Conserv. 2010, 19: 919-941. 10.1007/s10531-010-9783-3.
Woodruff DS, Turner LM: The Indochinese-Sundaic zoogeographic transition: a description and analysis of terrestrial mammal species distributions. J Biogeography. 2009, 36: 803-821. 10.1111/j.1365-2699.2008.02071.x.
Thinh VN, Hallam C, Roos C, Hammerschmidt K: Concordance between vocal and genetic diversity in crested gibbons. BMC Evol Biol. 2011, 11: 36-10.1186/1471-2148-11-36.
Won YJ, Hey J: Divergence population genetics of chimpanzees. Mol Biol Evol. 2005, 22: 297-307.
Charpentier MJ, Fontaine MC, Cherel E, Renoult JP, Jenkins T, Benoit L, Barthes N, Alberts SC, Tung J: Genetic structure in a dynamic baboon hybrid zone corroborates behavioural observations in a hybrid population. Mol Ecol. 2012, 21: 715-731. 10.1111/j.1365-294X.2011.05302.x.
Frisse L, Hudson RR, Bartoszewicz A, Wall JD, Donfack J, Di Rienzo A: Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium levels. Am J Hum Genet. 2001, 69: 831-843. 10.1086/323612.
Meyer M, Kircher M: Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010, 2010: pdb prot5448-2010
Kircher M, Stenzel U, Kelso J: Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 2009, 10: R83-10.1186/gb-2009-10-8-r83.
Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The sequence alignment/Map format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23: 2947-2948. 10.1093/bioinformatics/btm404.
Hall TA: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999, 41: 95-98.
Stephens M, Donnelly P: A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003, 73: 1162-1169. 10.1086/379378.
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001, 68: 978-989. 10.1086/319501.
Flot JF: SEQPHASE: a web tool for interconverting phase input/output files and fasta sequence alignments. Mol Ecol Resour. 2010, 10: 162-166. 10.1111/j.1755-0998.2009.02732.x.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Watterson GA: On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975, 7: 256-276. 10.1016/0040-5809(75)90020-9.
Hudson RR, Slatkin M, Maddison WP: Estimation of levels of gene flow from DNA sequence data. Genetics. 1992, 132: 583-589.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P: RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010, 26: 2462-2463. 10.1093/bioinformatics/btq467.
Martin D, Rybicki E: RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000, 16: 562-563. 10.1093/bioinformatics/16.6.562.
Padidam M, Sawyer S, Fauquet CM: Possible emergence of new geminiviruses by frequent recombination. Virology. 1999, 265: 218-225. 10.1006/viro.1999.0056.
Smith JM: Analyzing the mosaic structure of genes. J Mol Evol. 1992, 34: 126-129.
Posada D, Crandall KA: Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci U S A. 2001, 98: 13757-13762. 10.1073/pnas.241370698.
Gibbs MJ, Armstrong JS, Gibbs AJ: Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 2000, 16: 573-582. 10.1093/bioinformatics/16.7.573.
Boni MF, Posada D, Feldman MW: An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics. 2007, 176: 1035-1047.
Matsudaira K, Ishida T: Phylogenetic relationships and divergence dates of the whole mitochondrial genome sequences among three gibbon genera. Mol Phylogenet Evol. 2010, 55: 454-459. 10.1016/j.ympev.2010.01.032.
Matsui A, Rakotondraparany F, Munechika I, Hasegawa M, Horai S: Molecular phylogeny and evolution of prosimians based on complete sequences of mitochondrial DNAs. Gene. 2009, 441: 53-66. 10.1016/j.gene.2008.08.024.
Schrago CG, Russo CA: Timing the origin of New World monkeys. Mol Biol Evol. 2003, 20: 1620-1625. 10.1093/molbev/msg172.
Reichard UH, Barelli C: Life history and reproductive strategies of Khao Yai Hylobates lar: Implications for social evolution in apes. Int J Primatol. 2008, 29: 823-844. 10.1007/s10764-008-9285-2.
Dutheil JY, Ganapathy G, Hobolth A, Mailund T, Uyenoyama MK, Schierup MH: Ancestral population genomics: the coalescent hidden Markov model approach. Genetics. 2009, 183: 259-274. 10.1534/genetics.109.103010.
Prufer K, Munch K, Hellmann I, Akagi K, Miller JR, Walenz B, Koren S, Sutton G, Kodira C, Winer R, et al: The bonobo genome compared with the chimpanzee and human genomes. Nature. 2012, 486: 527-531.
Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, et al: Insights into hominid evolution from the gorilla genome sequence. Nature. 2012, 483: 169-175. 10.1038/nature10842.
Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO: Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006, 6: 29-10.1186/1471-2148-6-29.
Rambaut A, Drummond AJ: Tracer v1.5. 2004, http://tree.bio.ed.ac.uk/software/tracer/,
Rambaut A: FigTree v1.3.1: Tree figure drawing tool. http://tree.bio.ed.ac.uk/software/figtree/,
Acknowledgements
We are grateful to the following colleagues, zoos and institutions for providing valuable gibbon materials: the late Osamu Takenaka of Kyoto University, Akiko Takenaka of Nagoya Bunri University, Bambang Suryobroto of Bogor Agricultural University, Suchinda Malaivijitnond of Chulalongkorn University, Bristol Zoo, Duisburg Zoo, Krefeld Zoo, Leipzig Zoo, Nuremberg Zoo, Rostock Zoo, Twycross Zoo, London Zoo, Wuppertal Zoo, Chiang Mai Zoo, Dusit Zoo, Ragunan Zoo, Taipei Zoo, Primate Research Institute of Kyoto University, Japan Monkey Centre, and Pingtung Rescue Center of Endangered Wild Animals. This work was supported by a grant from the Leakey Foundation (L.V.), a fellowship of the Deutscher Akademischer Austausch Dienst (Y.-C.C.) and the Max Planck Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Y-CC carried out the experimental work of the study, analyzed the data and wrote the manuscript. CR and EI contributed samples and helped to draft the manuscript, and MI-M, KJ-CP and C-CS contributed samples. LV conceived of the study and wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12862_2013_2320_MOESM1_ESM.doc
Additional file 1: Reconstruction of the mtgenome phylogeny tree. Bayesian analysis of gibbon phylogenetic relationships based on the mtgenome sequences, excluding the control regions, from 49 individuals. (DOC 32 KB)
12862_2013_2320_MOESM2_ESM.doc
Additional file 2: Tables S1 to S2: This file includes details regarding gibbon samples used in the present study and summary statistics of the reads for each gibbon. (DOC 102 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chan, YC., Roos, C., Inoue-Murayama, M. et al. Inferring the evolutionary histories of divergences in Hylobates and Nomascus gibbons through multilocus sequence data. BMC Evol Biol 13, 82 (2013). https://doi.org/10.1186/1471-2148-13-82
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-13-82