
Abstract
Pectinolytic enzymes produced by a large variety of organisms are well characterized concerning their physiological and pathological activities during modification or degradation of the complex plant cell wall. The exponential growth in structural information of these enzymes over past decades has rendered insights into functionally relevant residues, active sites and molecular basis of the enzymatic mechanism, which in turn, endorses its usage in industrial applications. This review highlights a comprehensive and up to date summary of structural information and the structure–function correlation of pectinolytic enzymes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
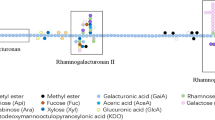
Structural proteins and polysaccharides—predominantly cellulose, hemicellulose, and pectin, contribute to the structural integrity of the plant cell walls (Jacob 2009). The structural analysis of primary and secondary cell walls of plant shows the chemical compositions of these polysaccharides differ significantly between plant species. Based on the pectin and hemicellulose content the primary cell walls are usually divided into type I and type II walls. Type I walls in dicots, non-graminaceous monocots, and gymnosperms contain xyloglucan (20% w/w) as major hemicellulose and 20–30% (w/w) pectin. Type II walls in grasses are rich in xylans (20–30% w/w) and contain 2–10% (w/w) pectin. In both types, the cellulose content is 30–40% (w/w) (Held et al. 2015; O’Neill and York 2003; Rytioja et al. 2014). The pectate network is divided into two regions: ‘smooth’ regions consist of homogalacturonans (HG) and ‘hairy’ regions which mainly consist of highly branched rhamnogalacturonans (RG) and xylogalacturonans (XGA) (Mohnen et al. 2008; Vincken et al. 2003). In HG, d-galacturonic acid (GalA) residues are joined together by α-1,4-glycosidic bonds and partially methylated at O-6 or acetylated at O-2 or O-3 (Atmodjo et al. 2013; Mohnen 2008; Normand et al. 2010). Depending on the pattern of substitution and side-chain composition, RG is further classified as RG-I and RG-II. RG-I is a polymer with a repeating dimer of d-GalA and l-rhamnose, to which l-arabinose and d-galactose are attached at O-4 of l-rhamnose. Some of GalA in RG-I at the O-2 or O-3 may also be acetyl-esterified (Caffall and Mohnen 2009). RG-II has a backbone of short stretches of d-GalA residues with four distinct side chains comprising of 12 different glycosyl residues attached to O-2 or O-3 of the main chain (Rytioja et al. 2014; Yapo 2011). XGA consists of a galacturonic acid backbone with xylose attached to the O-3 through β-1,3-glycosidic bond and can be methyl esterified (Harholt et al. 2010). XGAs from different sources show different linkage pattern; apple and potato comprise 1,4-linked xylose residues (Zandleven et al. 2006), while in soybean, 1,4- and 1,2-linked xylose residues can be observed (Nakamura et al. 2002). XGA from pea pectin contains xylose linked with 1,2- and 1,3-glycosidic bonds (Le Goff et al. 2001; Wong 2008). Although the comprehension of an exact methodology of connection of different pectic polysaccharides is elusive, a schematic model is shown in Fig. 1 (Mohnen 2008).

Schematic representation of pectin showing homogalacturonan (HG), rhamnogalacturonan-I (RG-I), rhamnogalacturonan-II (RG-II) and xylogalacturonan (XGA) linked to each other (Mohnen 2008)
Pectinolytic enzymes, pectic enzymes, or pectinases are the umbrella term that encompasses enzymes for modification and degradation of the complex pectate network. Based on the cleavage site, substrate specificity, and degradation mechanism, pectinases belong to three classes: (a) glycoside hydrolases (GHs) consisting of polygalacturonase (PG; endo-PG, EC 3.2.1.15; and exo-PG, EC 3.2.1.67), exo-polygalacturonosidase (EPGD; EC 3.2.1.82), rhamnogalacturonase or RG-hydrolase (RGH; EC 3.2.1.171), RG-galacturonohydrolase (RG-GH; EC 3.2.1.173), RG-rhamnohydrolase (RG-RH; EC 3.2.1.174), endo-xylogalacturonan hydrolase (endo-XGH; EC 3.2.1.-); (b) polysaccharide lyases (PLs) comprising of pectin lyase (PNL; EC 4.2.2.10), pectate lyase (PGL; EC 4.2.2.2), exo-pectate lyase (exo-PGL; EC 4.2.2.9), rhamnogalacturonan lyase (RGL; RG endo-lyase, EC 4.2.2.23; and RG exo-lyase, EC 4.2.2.24), oligogalacturonate lyase (OGL; now also classified as pectate lyase family 22; EC 4.2.2.6); and (c) carbohydrate esterases (CEs) consisting of pectin methylesterase (PME; EC 3.1.1.11), pectin acetylesterase (PAE; EC 3.1.1.6), rhamnogalacturonan acetylesterase (RGAE; EC 3.1.1.86) (Azadi et al. 1995; Gou et al. 2012; Hatanaka and Ozawa 1971; Jensen et al. 2010; Martens-Uzunova et al. 2006; Mutter et al. 1994, 1998; Ochiai et al. 2009) (Table 1).
So far, the Carbohydrate-Active enZYme database (CAZy) (Lombard et al. 2014) lists 156, 29 and 16 families of GHs, PLs, and CEs, respectively. Based on the sequence homologies, all pectin degrading hydrolases including PGs, EPGD, RGH, RG-GH, RG-RH, and XGH are classified into family GH28. Pectin and pectate lyases are categorized into PL1, PL2, PL3, PL9, PL10, and PL22, whereas RGLs belong to three families, PL4, PL11, and PL26. PAEs belong to CE12 and CE13 of the carbohydrate esterase family. Similarly, PMEs and RGAEs are categorized as members of family CE8 and CE12, respectively.
These enzymes are widely reported in plants, fungi, bacteria, and many yeasts (Garg et al. 2016). Pectinase from phytopathogenic fungi and bacteria contributes to pathogenicity or virulence (Herron et al. 2000; Lionetti et al. 2012; Liu et al. 2017; Wu et al. 2018). During fruit ripening/softening, it changes cell wall organization and reduces firmness (Brummell and Harpster 2001; Paniagua et al. 2017; Wang et al. 2000, 2018). PME involvement has been reported in cell elongation (Derbyshire et al. 2007; Pelletier et al. 2010), pollen grain germination in Arabidopsis (Leroux et al. 2015) and pollen tube growth (Bosch et al. 2005; Tian et al. 2006; Yue et al. 2018). The industrial applications of these enzymes are also substantially explored (Ahlawat et al. 2008; Amin et al. 2019; Kashyap et al. 2001; Khan et al. 2013; Lee et al. 2006; Murthy and Naidu 2011; Najafian et al. 2009; Sandri et al. 2011; Singh et al. 2019). The main emphasis of this review is on analysis of structural and mechanistic features of these enzymes relating to their catalytic mechanism.
Mode of action of pectinase
Pectinases degrade the complex pectic network through depolymerization (hydrolysis/β-elimination) and de-esterification reactions. Depolymerizing enzymes from the GH28 family hydrolyze glycosidic bonds through an inverting mechanism (Zandleven et al. 2005). HG-degrading enzymes (endo-PGs, exo-PGs, and EPGDs) cleave the α-1,4-glycosidic bond between two d-GalA units (Bonnin et al. 2014), whereas RG-degrading enzymes target specifically on the RG-I backbone (Silva et al. 2016). RGHs, attack randomly in an endo-fashion on α-1,2-glycosidic bonds between d-GalA and l-rhamnose (van den Brink and de Vries 2011). Contrary to that, RG-GHs and RG-RHs (exo-acting enzymes) act at the non-reducing end of the RG-I backbone (Silva et al. 2016). XGHs are endo-acting enzymes which degrade glycosidic bond between two β-xylose-substituted GalA-backbone (Zandleven et al. 2005). The other group of depolymerases, PNLs, PGLs, and RGLs, degrade the network via the β-elimination mechanism. Pectate and pectin lyases cleave α-1,4-glycosidic linkage resulting in the formation of 4,5-unsaturated oligogalacturonates (Yip and Withers 2006). PGLs prefer non-methylated or low-esterified substrates and require Ca2+ for catalysis, except those from PL2 family which preferentially utilize Mn2+ ions (Abbott et al. 2013). In contrast, PNLs prefer methyl esterified substrate and do not require Ca2+ ions (Rytioja et al. 2014). RGLs degrade the backbone of RG-I by breaking down the α-1,4-glycosidic bond resulting in the formation of oligomers with unsaturated d-galactopyranosyluronic acid at the non-reducing end (Silva et al. 2016). Pectin esterases (PMEs, PAEs, and RGAEs) remove methyl and acetyl groups from the pectin backbone. PMEs hydrolyze methyl ester bond at C-6 through a double-displacement mechanism of methyl esterified GalA releasing methanol and protons (Jolie et al. 2010; Pelloux et al. 2007). PAEs and RGAEs catalyze the removal of O-2 and/or O-3 acetyl group from HG or RG-I. Deacetylation in RG-I provides access to other RG-I modifying enzymes (lyases and hydrolases) for further degradation of the complex network (Silva et al. 2016). The overall catalytic mechanism is illustrated in Fig. 2.

Overview of reaction mechanism a homogalacturonan (HG) with methyl and acetyl esterification and xylogalacturonan (XGA), enzymes that cleave HG and XGA are polygalacturonase (PG), pectin/pectate lyase (PNL/PGL), pectin methylesterase (PME), pectin acetylesterase (PAE), xylogalacturonan hydrolase (XGH), b rhamnogalacturonan (RG) and modifying enzymes are RG-hydrolase (RGH), RG-lyase (RGL), and RG-acetylesterase (RGAE). Site of action is indicated by arrows
Structural features and enzymatic mechanism
Three-dimensional structures enable understanding of the functional significance and interaction properties at the molecular level. Yoder et al. (1993a) reported the first crystallographic structure of pectinase, Erwinia chrysanthemi pectate lyase C (PelC) and since then, many structures from plant pathogens and other sources have been resolved (Table 2; references cited therein). The X-ray analysis reveals that most of the members of pectinase family share a right-handed β-helix topology. Others like PGLs in PL2 and PL10, RGLs in PL4 and PL11 and RGAE, adopt (α,α)7-barrel, (α,α)3-barrel, β-sandwich + β-sheets, β-propeller and α/β/α-sandwich structures, respectively (Lombard et al. 2010; Mølgaard et al. 2000). Recently identified exo-RGL of PL26 family shows β-Sandwich + (α/α)n barrel fold (Kunishige et al. 2018). The major differences in topology are seen in the number of turns, the conformation of loops, and the terminal regions. On the basis of their shared topological structure, members of the pectinase family can be classified into six classes, namely (a) right-handed β-helix, (b) (α/α)n-barrel, (c) β-sandwich + β-sheets, (d) β-sandwich + (α/α)n-barrel, (e) β-propeller, and (f) α/β/α-sandwich (Table 3, Fig. 3).

Representative fold structures of each pectinase family. GH28 (exo-PG, PDB ID: 3JUR; endo-PG, PDB ID: 2IQ7; RGH, PDB ID: 1RMG; endo-XGH, PDB ID: 4C2L), PL1 (PDB ID: 2QXZ), PL3 (PDB ID: 1EE6), PL9 (PDB ID:1RU4), CE8 (PDB ID: 1QJV), PL10 (PDB ID: 1GXM), PL2 (PDB ID: 2V8I), PL4 (PDB ID: 1NKG), PL26 (PDB ID: 5XQ3), PL11 (PDB ID: 2ZUX), PL22 (PDB ID: 3PE7), CE12 (PDB ID: 1DEO)
Right-handed β-helix class
Enzymes of the GH28 family, three families of pectin and pectate lyases (PL1, PL3, PL9), and PMEs from the CE8 family share the right-handed β-helix fold. Pectate lyase C (PelC) from Erwinia chrysanthemi was the first structure to describe the right-handed β-helix fold (Yoder et al. 1993a). Subsequently, similar structural fold has been reported in structures of, PelC (Herron et al. 2003; Yoder and Jurnak 1995a, b), PelC complex with plant cell wall (Scavetta et al. 1999), pectate lyase E (PelE) (Lietzke et al. 1994, 1996; Yoder et al. 1993b), pectate lyase A (PelA) (Dehdashti et al. 2003; Thomas et al. 2002), pectate lyase L (Pel9A) (Jenkins et al. 2004), pectate lyase (PelI) in complex with substrate (Creze et al. 2008) from Erwinia chrysanthemi; pectate lyase complex with Ca2+ from Bacillus subtilis (BsPel) (Pickersgill et al. 1994), pectate lyase (Pel-15) and Bsp165PelA from Bacillus sp. strain KSM-P15 and strain N16-5, respectively (Akita et al. 2001; Zheng et al. 2012), pectate lyase from Acidovorax citrulli (AcPel) (Tang et al. 2013); Aspergillus niger pectin lyase A (PL1A) (Mayans et al. 1997) and pectin lyase B (PL1B) (Vitali et al. 1998); Erwinia carotovora endo-PG (PehA) (Pickersgill et al. 1998), endo-PG II (van Santen et al. 1999) and endo-PG I (van Pouderoyen et al. 2003) from Aspergillus niger, Aspergillus aculeatus PG (Cho et al. 2001), Fusarium moniliforme PG (FmPG) (Federici et al. 2001), Stereum purpureum endo-PG I (Shimizu et al. 2002), Yersinia enterocolitica exo-PG (YeGH28) (Abbott and Boraston 2007b), PG from Colletotrichum lupini (CluPG1) (Bonivento et al. 2008); exo-PG from Thermotoga maritime (Pijning et al. 2009); RGHA from Aspergillus aculeatus (Petersen et al. 1997); endo-XGH from Aspergillus tubingensis (XghA) (Rozeboom et al. 2013); PME from Erwinia chrysanthemi (Fries et al. 2007; Jenkins et al. 2001), Daucus carota (Johansson et al. 2002), Solanum lycopersicum (Di Matteo et al. 2005), Yersinia enterocolitica (Boraston and Abbott 2012), Sitophilus oryzae (rice weevils; first animal PME) (Teller et al. 2014) and Aspergillus niger (Kent et al. 2016).
In general, the central core region consists of 7–13 complete β-helical turns. The sequential arrangement of β-strands results in the formation of three parallel β-sheets (PB1, PB2, and PB3) connected by loops. An antiparallel β-sandwich arrangement can be seen between PB1 and PB2, and PB3 lies almost perpendicular to PB2 (Sharma et al. 2013). Contrary to the structural fold observed in lyase, few members of GH28 enzymes have additional β-sheets as seen in Aspergillus niger endo-PG II, Aspergillus aculeatus RGHA, and Aspergillus tubingensis endo-XGH. Despite similar right-handed β-helix topology, the reaction mechanisms within this enzyme class are completely different. PGs and RGHs cleave the substrate by acid/base-catalyzed hydrolysis (single displacement), PMEs use double displacement mechanism, whereas lyases (PNLs, PGLs) cleave by β-elimination.
Kester et al. (1996) suggested four blocks of residues (NxD, G/QDD, HG, and RxK) conserved in all endo- and exo-PGs and which are clustered at subsites − 1 and + 1. These conserved patches on the surface of the PB1 and nearby loops form the catalytic site (Pickersgill et al. 1998). Site-directed mutagenesis studies in endo-PG II from Aspergillus niger have revealed the involvement of aspartate residues in the catalytic function (Armand et al. 2000; Pagès et al. 2000). All enzymes from GH28 family act via an inverting mechanism (Abbott and Boraston 2007b). In inverting enzymes, the reaction proceeds through a single displacement mechanism. Two acidic amino acid residues acting as general acid and base, respectively, are involved in the reaction process (Shallom et al. 2005). Comparative sequence alignment of known structures of PGs, RGH, and XGH shows seven conserved amino acid residues (Supplementary Figure, S1). Among them, three catalytic aspartates are functionally conserved (D202, D223, and D224; sequence numbering according to endo-PG from Erwinia carotovora (PehA), PDB ID: 1BHE) (Fig. 4a) except in RGH from Aspergillus aculeatus where D224 (198 in RGH) is substituted by glutamate (E). One out of the three conserved aspartates protonates the scissile glycosidic oxygen, while the other two activate the nucleophilic water molecule. In Aspergillus niger endo-PG II (PDB ID: 1CZF), van Santen et al. (1999)suggested that D180 and D202 activate the water molecule and D201 acts as a proton donor. A generalized reaction mechanism based on endo-PG II is shown in Fig. 4b. In Aspergillus tubingensis endo-XGH (XghA) (PDB ID: 4C2L), the active site contains catalytic residues D207, D228, and D229 (Rozeboom et al. 2013). Other conserved residues N205 and Y322 are required for substrate binding. The substrate binding cleft is wider (~ 15.3 Å) in XghA as compared to other GH28 endo-PGs (ranges from 6.4 to 11.0 Å) (Rozeboom et al. 2013). Similar arrangements have also been observed in the other GH28 enzymes (Table 3). The structural firmness of the β-helix is stabilized by disulfide bridges. In fungal endo-PGs and RGH, four disulfide bridges are found to be conserved. N- and C-terminal disulfide bonds ensure the capping of β-helix, while other two disulfides located in the middle of the β-helix confirm correct local folding around the active site. On the other hand, bacterial endo-PG (PehA from Erwinia carotovora) has only two disulfides (C41-C62, C115-C125; numbering in PehA) (Pickersgill et al. 1998) and endo-XGH from Aspergillus tubingensis (XghA) has shown six disulfide bridges: C37-C60, C85-C88, C230-C247, C321-C329, C32-C369 and C389-C400 (numbering in XghA) (Rozeboom et al. 2013).

Right-handed β-helix fold a overall structural fold of Erwinia carotovora endo-PG with catalytic residues (D202, D223, and D224) (PDB ID: 1BHE); b generalized reaction mechanism for inverting family 28 GHs (based on endo-PG II). D201 acts as proton donor, while D180 and D202 activate the hydrolytic water molecule (van Santen et al. 1999)
Structural comparison of PMEs in CE8 family shows similarities in folding topology and reaction mechanisms. Sequence analysis of known structures reveals five functionally important conserved sequence motifs with few replacements: GxYxE, QAVAL, QDTL, DFIFG, and LGRPW (Markovič and Janeček 2004; Zega and D’Ovidio 2016) (Supplementary Figure, S2). PMEs catalyze reactions through a double displacement mechanism which includes the formation of glycosyl-enzyme intermediate and subsequent hydrolysis. The process requires two aspartates as nucleophile and acid/base catalyst. A catalytic triad, glutamine-aspartate-aspartate, is strictly conserved in all PME active site. In Erwinia chrysanthemi PME (PDB ID: 2NSP), the reaction mechanism requires Q177, D178, and D199. As proposed by Fries et al., D199 performs a nucleophilic attack on carboxymethyl carbonyl carbon supposedly forming a tetrahedral intermediate. The negative charge on the carbonyl oxygen of the intermediate is stabilized by Q177 and D178. D178 acts as a proton donor and assists the release of methanol. Later, D178 acts as a base and accepts proton from water molecule restoring the active site (Fries et al. 2007) (Fig. 5). Similar reaction mechanisms have also been observed in plant PMEs, carrot PME (PDB ID: 1GQ8) (Johansson et al. 2002) and tomato PME (PDB ID: 1XG2) (Di Matteo et al. 2005). Polarized by the presence of R225 (R221 in tomato PME), D157 (D153 in tomato PME) acts as the nucleophile and attacks the carboxymethyl group forming an intermediate. The negative charge of the intermediate is stabilized by Q113 and Q135 (Q109 and Q131 in tomato PME). D136 (D132 in tomato PME), first acts as an acid result in the release of methanol and subsequently as a base to draw proton from the water molecule. Apart of the catalytic residues, many aromatic residues (F80, Y135, F156, Y218, W223, W248 in tomato PME; F84, Y139, F160, Y222, W227, F250, W252 in carrot PME) are assumed to be involved in substrate binding (Di Matteo et al. 2005; Johansson et al. 2002). When plant PMEs compared with bacterial PME, the overall folding topologies found to be very similar, except longer loops in the bacterial PME and an additional helix in C-termini of plant PME. Structural analysis of Sitophilus oryzae (rice weevils) PME (RwPME; PDB ID: 4PMH) also suggests the similar arrangement of catalytic residues (Q199, D200, and D226) and enzymatic mechanism (Teller et al. 2014).

The right-handed β-helix fold a overall structural fold of Erwinia chrysanthemi PME with hexasaccharide (PDB ID: 2NSP) and Daucus carota PME (PDB ID: 1GQ8); b reaction mechanism of PME (as proposed by Fries et al. 2007)
Unlike PGs and PMEs, lyases (PNLs, PGLs) degrade pectin by a β-elimination which involves neutralization of carboxyl group at C-5, proton abstraction at C-5, and removal of glycosidic linkage (Garron and Cygler 2010). Members of family PL1 (only PGLs), PL3, and PL9 shows Ca2+-assisted β-elimination. Ca2+ ion is essential for catalysis as it helps to neutralize the acidic substrate. PGL structures have two kinds of Ca2+ ion: (a) primary Ca2+ ion, in the absence of substrate it binds to the enzyme, as seen in many PGLs structures: BsPel from Bacillus subtilis (PDB ID: 1BN8), Erwinia chrysanthemi PelC (PDB ID: 1O88), Pel-15 from Bacillus sp. KSM-P15 (PDB ID: 1EE6), Bsp165PelA (PDB ID: 3VMV), and Bsp47Pel (PDB ID: 1VBL; to be published); and (b) additional Ca2+ ions (two or three), which coordinate the enzyme and substrate (Tang et al. 2013). The active site of PGL from family PL1 shows the involvement of a group of conserved acidic residues in Ca2+ coordination (D184, D223, and D227; BsPel numbering) (Fig. 6a). Within the extracellular PGLs family, D184 and D227 are conserved, whereas D223 is conservatively substituted with glutamate (E). Usually, arginine or lysine and water molecule act as a catalytic base and acid, respectively (Fig. 6e). The available structural data suggest that arginine is strictly conserved among the PL1 family, whereas the catalytic base in family PL3 and PL9 is lysine.

The right-handed β-helix fold in lyases (PGLs and PNLs) a overall structural fold of Bacillus subtilis PGL (PDB ID: 1BN8) with calcium-binding site (D184, D223, D227). The catalytic base is R279; b structural fold of Erwinia chrysanthemi PGL (PDB ID: 1RU4) with calcium-binding site (D209, D233, D234, D237). The putative catalytic base is K273; c structural fold of Aspergillus niger PNLs (PDB ID: 1IDK and PDB ID: 1QCX) with catalytic residues; d structural fold of Bacillus sp. KSM-P15 PGL (PDB ID: 1EE6) with calcium-binding site (D63, E83, D84). The putative catalytic base is K107; e general reaction mechanism of PGL (calcium assisted β-elimination) where arginine or lysine acts as a catalytic base (in representation, calcium ions are shown as red balls)
Analysis of Caldicellulosiruptor bescii PGL (PDB ID: 4Z06) of PL3 family exhibits an antiperiplanar trans-elimination reaction mechanism. K108 abstracts a proton from the C5 atom and a water molecule protonates the O4 atom of the substrate completing the elimination process (Alahuhta et al. 2015). Acidic residue E84, E39, and D107 are involved in calcium coordination, whereas Q111 and R133 play a role in substrate binding (Alahuhta et al. 2015). Similarly, in Bacillus sp. KSM-P15 PGL (PDB ID: 1EE6) the calcium-binding site is formed by D63, E83, D84, and the putative catalytic base is K107 (Fig. 6d). In PL9 family, PGL from Erwinia chrysanthemi (PDB ID: 1RU4), four aspartate residues (D209, D233, D234, and D237) coordinate calcium-binding site (Jenkins et al. 2004) (Fig. 6b). However, not all enzymes of the PL1 family require Ca2+ ion for the catalytic mechanism. PNLs from Aspergillus niger (PNLA, PDB ID: 1IDK and PNLB, PDB ID: 1QCX) eliminate the need of Ca2+ ion. In contrast to PGLs, aromatic residues (D154, R176, and R236) dominate the binding site of PL1A and PL1B. The catalytic arginine residue (R236) plays a key role in catalysis, while D176 in a similar position to catalytic Ca2+ ion in PGLs stabilizes the negative charge of the substrate and aids proper orientation of R236 (Mayans et al. 1997; Vitali et al. 1998).
(α/α)n barrel class
PGLs from PL2 and PL10 family display (α/α)n barrel topology. The formation of such structural domain is due to the repetition of a pair of antiparallel α-helices (Garron and Cygler 2010). PGLs of PL2 and PL10 family have seven repeats, (α/α)7 and three repeats, (α/α)3, respectively (Fig. 7a). The crystal structure of PL2 family PGLs from Yersinia enterocolitica (YePL2A) (PDB ID: 2V8I) (Abbott and Boraston 2007a), and Vibrio vulnificus (VvPL2) (PDB ID: 5A29) (McLean et al. 2015) showed that Mn2+ is the preferred transition metal for catalysis over Ca2+. The metal-binding site is coordinated by two histidine and one glutamate (H109, H172, E130 in YePL2A, and H129, H192, E150 in VvPL2). In YePL2A, R171 at subsite + 1 assumed to play the role of the catalytic base while water molecule acts as the proton donor and R272 is involved in substrate recognition. Similarly, in VvPL2, R191 and R304 acts as the catalytic base and stabilizing residue, respectively (McLean et al. 2015).

The (α/α)n fold a structural arrangement of PGL from Yersinia enterocolitica (PDB ID: 2V8I) and Azospirillum irakense (PDB ID: 1R76); b Ca2+ assisted reaction mechanism of PL10 enzymes where catalytic residue R307 abstracts proton (Novoa de Armas et al. 2004)
In PL10 family, PGL from Cellvibrio japonicus (Pel10Acm) (PDB ID: 1GXM) (Charnock et al. 2002) and Azospirillum irakense (PelA) (PDB ID: 1R76) (Novoa de Armas et al. 2004) shows the same structural topology with three repeats; (α/α)3. Both Pel10Acm and PelA comprise of two domains and a deep pocket between them hosts substrate binding and catalytic site (Novoa de Armas et al. 2004). In Pel10Acm, analysis of the catalytic center reveals the presence of residues D389, N390, D451, R524, Y526, E527, E535, R596, R610, R625, G628, and S630 (Novoa de Armas et al. 2004; Walker and Ryan 2003). Similarly, in PelA, residue D171, N172, D236, R307, F309, E310, E318, R378, R392, R407, G410, and A412 are present in the active site (Novoa de Armas et al. 2004). In Pel10Acm, the carboxylate group of the substrate at − 1 subsite forms an ionic bond with R596 and a coordinate bond with the Ca2+ ion. Simultaneously, the Ca2+ ion makes coordinate interaction with carboxylate oxygen of D451 as well as + 1 subsite sugar carboxylate (Novoa de Armas et al. 2004). Substitution of D451 leads to loss of enzymatic activity (Charnock et al. 2002). Arginine as a potential catalytic base is located at the + 1 subsite (R524 in Pel10Acm and R307 in PelA) (Fig. 7b) (Novoa de Armas et al. 2004). Although the core topology between PL10 and PL1 is different, superposition of enzymes of both families shows structurally conserved active center. These conserved identical catalytic residues in the active site indicate the convergent evolution of catalytic mechanism (Charnock et al. 2002). In both cases, a conserved acidic residue is essential for stabilization of Ca2+ ion (D451in Pel10Acm and E166 in PelC).
β-Sandwich + β-sheet class
RGLs from Aspergillus aculeatus in the PL4 family (PDB ID: 1NKG) display β-sandwich fold with three structural domains (Fig. 8a). Two antiparallel β-sheets, each comprising eight β-strands form domain I, with two disulfide bonds C30-C73 and C164-C173. Domain II displays topology similar to the fibronectin type III and domain III adopts β-sandwich fold like carbohydrate-binding modules (McDonough et al. 2004). Domain I hosts catalytic (K150, H210) as well as substrate binding residues (R107, R111), whereas R451 and R455 in domain III support the formation of the substrate-binding groove. The correct orientation of domains I and III is mediated by domain II. Mutational studies of RGL with bound substrate (PDB ID: 2XHN) also reveals the involvement of K150 and H210 in catalysis (Jensen et al. 2010). The carboxyl group of the substrate is protonated at the + 1 subsite by D139. K150 plays the role of proton abstractor, while H210 acts as a proton donor (Fig. 8b, c). Unlike PGLs from several PL families, in which the reaction proceeds through the metal-assisted manner, RGLs in PL4 family differ in the mechanism by eliminating the requirement of the Ca2+ ion.

The β-sandwich + β-sheet fold a domain arrangement of Aspergillus aculeatus RGL (PDB ID: 2XHN); b catalytic residues on domain I; c reaction mechanism of RGL where K150 acts as proton abstractor and H210 acts as proton donor (Jensen et al. 2010)
β-Sandwich + (α/α)n barrel class
Recently, the three-dimensional structure of Penicillium chrysogenum exo-RGL (PcRGLX) (PDB ID: 5XQ3) of PL26 family shows a unique structural arrangement (Fig. 9a). Domain I and II display β-sandwich fold similar as PL4 endo-RGL of Aspergillus aculeatus and domain III exhibits an (α/α)6-barrel structure like PL2 PGL. Domain I consists of two three-stranded antiparallel β-sheets stabilized by a disulfide bond C24-C113 and domain II possesses β-sandwich fold comprising two antiparallel β-sheets having eighteen β-strands surrounded by five α-helices. A serine-rich loop can be seen at the interface between domains I and II. Domain III has an antiparallel arrangement of six lateral and six inner helices and hosts a Ca2+-binding site enclosed by D562, N585, H616, D621, and H639 (Fig. 9b). An l-shaped cleft active site provides a compact framework for substrate binding specifically galactosyl side chains. Mutagenesis studies suggest the involvement of residues Y458, D460, R634, H635, Q646, R648, and H782 in the catalytic mechanism (Kunishige et al. 2018). At the + 1 subsite, the hydrogen bond between R634 and the carboxyl group of GalA advocates the role of R634 as a neutralizer. The catalytic mechanism proceeds through β-elimination but differs significantly from PL11 Bacillus subtilis RGL (YesX) by eliminating the requirement of divalent cations. PcRGLX shares similar structural folds to l-rhamnose-α-1,4-d-glucuronate lyase of PL27 (Munoz–Munoz et al. 2017), although they show 7% sequence identity.

The β-Sandwich + (α/α)n barrel fold a structural arrangement of exo-RGL from Penicillium chrysogenum (PDB ID: 5XQ3), the red box shows serine-rich loop (S117-I130) located between domains I and II; b Ca2+ ion (shown as red ball) is coordinated by D562, N585, H616, D621, H639 and located in domain III
β-Propeller class
RGLs from PL11 and OGL from PL22 family adopt β-propeller fold. So far, the crystal structures of PL11 family RGLs (endo-acting YesW and exo-acting YesX) from Bacillus subtilis (PDB ID: 2Z8S and 2ZUX) (Ochiai et al. 2007, 2009), and RGI lyase WT from Bacillus licheniformis (PDB ID: 4CAG) (Silva et al. 2014) have been determined. Both YesW and YesX share sequence identity of 67.8% and exhibits similar structure comprising N-terminal β-sheet domain and eight-bladed β-propeller domain (A-H blades) (Fig. 10a) (Ochiai et al. 2007, 2009). Except for blades A, G in YesW and blade A in YesX, each blade contains four antiparallel β-strands. Besides, all blades except for D in YesW and D, G in YesX contain one or two Ca2+ ions. Substrate binding site in both YesW and YesX is located within the pocket of β-propeller. Catalytic residues such as R452, T534, K535, and Y595 in YesW (R419, T518, K519, and Y579 in YesX) are responsible for substrate recognition. During the catalysis, the negative charge of the carboxyl group is neutralized by positively charged R452 and K535 (R419 and K519 in YesX) and Y595 (Y579 in YesX) is important for stacking interaction. Also, residues such as D153, N596 (in site 1) and H363, H399, D401, and E422 (in site 2) are involved in calcium binding within the active site. One Ca2+ ion is assumed to be involved in substrate binding, while the other nine Ca2+ ions help in stabilizing the β-propeller fold (Ochiai et al. 2007). Despite the same structural fold, an extended loop of nine residues (PPGNDGMSY) in YesX determines the difference in substrate specificity and mode of action between YesX and YesW (Ochiai et al. 2009).

The β-propeller fold a overall structural fold of Bacillus subtilis RGL constituting eight blades (A–H) stabilized by Ca2+ ions (PDB ID: 2ZUX); b structural fold of Bacillus subtilis RGL showing seven blades (A–G) (PDB ID: 3PE7); in both representation, calcium ions are shown as red balls and manganese ion as purple ball
OGL from Yersinia enterocolitica (YeOGL) (PDB ID: 3PE7) and Vibrio parahaemolyticus (VpOGL) (PDB ID: 3C5 M, unpublished data) in PL22 family adopts a seven-bladed β-propeller fold. Each propeller consists of a repeating four-stranded antiparallel β-strands. In the structure of YeOGL, Mn2+ ion is located in the active site, which is stabilized by H287, H353, H355, and Q350 (Fig. 10b). Residue H242 acts as the Brønsted base, which is highly conserved across OGL family, and residues such as H211 and R217 are involved in substrate stabilization (Abbott et al. 2010).
(α/β/α)-Sandwich class
RGAE from CE12 family exhibits α/β/α sandwich fold. The structure of Aspergillus aculeatus RGAE (AaRGAE) (PDB ID: 1DEO) displays an arrangement of five parallel β-strands surrounded by α-helices (Mølgaard et al. 2000). The 3-layered structural fold is stabilized by two disulfide bonds C88-C96 and C214-C232. At the active site, residue S9 forms a hydrogen bond with the adjacent H195 and D192 establishing a catalytic triad S9-D192-H195 (Fig. 11). The stability of β-turn is maintained by aspartate residue (D8) which forms a hydrogen bond between its side-chain O atom and the Nu + 1 amide group (Mølgaard et al. 2000; Mølgaard and Larsen 2002). Homology modeling of RGAE from Bacillus subtilis (BsYesT) (Martínez-Martínez et al. 2008) and Bacillus halodurans (BhRGAE) (Navarro-Fernández et al. 2008) using the AaRGAE as the template shows similar structural features. As per SUPERFAMILY library (Gough et al. 2001), AaRGAE along with hypothetical protein YXIM_BACsu from Bacillus subtilis (PDB ID: 2O14, unpublished data) (Bolvig et al. 2003), putative protein YesY and YesT from Bacillus subtilis (Martínez-Martínez et al. 2008), esterase from Streptomyces scabies (SsEst) (PDB ID: 1ESC) (Wei et al. 1995), Bos taurus Platelet-activating factor acetylhydrolase (Ho et al. 1997), haemagglutinin-esterase-fusion protein (HEF) of influenza C virus (PDB ID: 1FLC) (Rosenthal et al. 1998), Hypothetical lipase protein alr1529 from Nostoc sp. (PDB ID: 1Z8H, unpublished data), Bacillus sp. KCCM10143 cephalosporin C deacetylase (CCD) (Choi et al. 2000), Erwinia chrysanthemi PAE (PaeY) (Mølgaard et al. 2000) are classified as members of SGNH-hydrolase family. The characteristic features that distinguish SGNH-hydrolase family from α/β hydrolase family are: central five-stranded parallel β-sheets, conserved residue blocks (GDS, G, GXND, and DXXHP), nucleophilic serine at C-terminal of β-strand1, close proximity of aspartate and histidine, absence of nucleophilic elbow motif (Martínez-Martínez et al. 2008; Mølgaard et al. 2000).

The (α/β/α)-Sandwich fold a schematic representation of overall structure of Aspergillus aculeatus RGAE (PDB ID: 1DEO); b active site of RGAE showing three catalytic residues, S9, H195, and D192
Concluding remarks and prospectives
The emergence of structural information leads to a better comprehension of structure–function correlation. Based on structural analysis, pectinase can be classified as right-handed β-helix, (α/α)n-barrel, β-sandwich + β-sheets, β-sandwich + (α/α)n-barrel, β-propeller, and α/β/α-sandwich. These enzymes display high selectivity towards specific bonds and work via hydrolysis/β-elimination and de-esterification reaction mechanism. Functionally conserved patches, NxD, G/QDD, HG, and RxK, determine the catalytic activity among GH28 family, except few residues in RGH. PGs, RGH, and endo-XGH exhibit a single displacement inverting reaction mechanism involving two aspartate residues at the active site. PME catalyzes reactions through double-displacement mechanisms which also involves two catalytic aspartates rather than serine as seen in other carbohydrate esterases. Despite the structural differences in right-handed β-helix, (α/α)3/7-barrel, and β-propeller class, the β-elimination mechanism utilizes metal (Ca2+/Mn2+)- assisted neutralization involving arginine or lysine as proton abstractor and water as a proton donor. Interestingly, RGLs of the PL4 and PL26 family, which displays β-sandwich + β-sheets and β-sandwich + (α/α)6-barrel topology, respectively, eliminates the requirement of ions during the β-elimination mechanism. Sequence and structural exploration reveal RGAE, which belong to SGNH-hydrolase family, adopts α/β/α-sandwich fold with the catalytic triad: serine-aspartate-histidine.
Although significant progress on the structures of pectinase has been done, still ample of scope available to explore structures from different sources which are not yet known. Current structural knowledge could be used to establish the three-dimensional structure of other pectinases using computational modeling approach and for an in-depth understanding of the structure–function association at the molecular level.
References
Abbott DW, Boraston AB (2007a) A family 2 pectate lyase displays a rare fold and transition metal-assisted β-elimination. J Biol Chem 282:35328–35336
Abbott DW, Boraston AB (2007b) The structural basis for exopolygalacturonase activity in a family 28 glycoside hydrolase. J Mol Biol 368:1215–1222
Abbott DW, Gilbert HJ, Boraston AB (2010) The active site of oligogalacturonate lyase provides unique insights into cytoplasmic oligogalacturonate β-elimination. J Biol Chem 285:39029–39038
Abbott DW, Thomas D, Pluvinage B, Boraston AB (2013) An ancestral member of the polysaccharide lyase family 2 displays endolytic activity and magnesium dependence. Appl Biochem Biotechnol 171:1911–1923
Ahlawat S, Mandhan R, Dhiman SS, Kumar R, Sharma J (2008) Potential application of alkaline pectinase from Bacillus subtilis SS in pulp and paper industry. Appl Biochem Biotechnol 149:287–293
Akita M, Suzuki A, Kobayashi T, Ito S, Yamane T (2001) The first structure of pectate lyase belonging to polysaccharide lyase family 3. Acta Crystallogr D Biol Crystallogr 57:1786–1792
Alahuhta M, Chandrayan P, Kataeva I, Adams MW, Himmel ME, Lunin VV (2011) A 1.5 Å resolution X-ray structure of the catalytic module of Caldicellulosiruptor bescii family 3 pectate lyase. Acta Crystallogr F Struct Biol Cryst Commun 67(12):1498–1500
Alahuhta M et al (2015) The catalytic mechanism and unique low pH optimum of Caldicellulosiruptor bescii family 3 pectate lyase. Acta Crystallogr D Biol Crystallogr 71:1946–1954
Amin F, Bhatti HN, Bilal M (2019) Recent advances in the production strategies of microbial pectinases—a review. Int J Biol Macromol 122:1017–1026
Armand S et al (2000) The active site topology of Aspergillus niger endopolygalacturonase II as studied by site-directed mutagenesis. J Biol Chem 275:691–696
Atmodjo MA, Hao Z, Mohnen D (2013) Evolving views of pectin biosynthesis. Annu Rev Plant Biol 64:747–779
Azadi P, O’Neill MA, Bergmann C, Darvill AG, Albersheim P (1995) The backbone of the pectic polysaccharide rhamnogalacturonan I is cleaved by an endohydrolase and an endolyase. Glycobiology 5:783–789
Bolvig PU, Pauly M, Orfila C, Scheller HV, Schnorr K (2003) Sequence analysis and characterisation of a novel pectin acetyl esterase from Bacillus subtilis. In: Voragen F, Schols H, Visser R (eds) Advances in pectin and pectinase research. Springer, Berlin, pp 315–330
Bonivento D et al (2008) Crystal structure of the endopolygalacturonase from the phytopathogenic fungus Colletotrichum lupini and its interaction with polygalacturonase-inhibiting proteins. Proteins Struct Funct Bioinform 70:294–299
Bonnin E, Garnier C, Ralet M-C (2014) Pectin-modifying enzymes and pectin-derived materials: applications and impacts. Appl Microbiol Biotechnol 98:519–532
Boraston AB, Abbott D (2012) Structure of a pectin methylesterase from Yersinia enterocolitica. Acta Crystallogr F Struct Biol Cryst Commun 68:129–133
Bosch M, Cheung AY, Hepler PK (2005) Pectin methylesterase, a regulator of pollen tube growth. Plant Physiol 138:1334–1346
Brummell DA, Harpster MH (2001) Cell wall metabolism in fruit softening and quality and its manipulation in transgenic plants. In: Carpita NC, Campbell M, Tierney M (eds) Plant cell walls. Springer, Berlin, pp 311–340
Caffall KH, Mohnen D (2009) The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydr Res 344:1879–1900
Charnock SJ, Brown IE, Turkenburg JP, Black GW, Davies GJ (2002) Convergent evolution sheds light on the anti-β-elimination mechanism common to family 1 and 10 polysaccharide lyases. Proc Natl Acad Sci 99:12067–12072
Cho SW, Lee S, Shin W (2001) The X-ray structure of Aspergillus aculeatus polygalacturonase and a modeled structure of the polygalacturonase-octagalacturonate complex. J Mol Biol 311:863–878
Choi D-H, Kim Y-D, Chung I-S, Lee S-H, Kang S-M, Kwon T-J, Han K-S (2000) Gene cloning and expression of cephalosporin-C deacetylase from Bacillus sp. KCCM10143. J Microbiol Biotechnol 10:221–226
Creze C, Castang S, Derivery E, Haser R, Hugouvieux-Cotte-Pattat N, Shevchik VE, Gouet P (2008) The crystal structure of pectate lyase peli from soft rot pathogen Erwinia chrysanthemi in complex with its substrate. J Biol Chem 283:18260–18268
Dehdashti SJ, Doan CN, Chao KL, Yoder MD (2003) Effect of mutations in the T1. 5 loop of pectate lyase A from Erwinia chrysanthemi EC16. Acta Crystallogr D Biol Crystallogr 59:1339–1342
Derbyshire P, McCann MC, Roberts K (2007) Restricted cell elongation in Arabidopsis hypocotyls is associated with a reduced average pectin esterification level. BMC Plant Biol 7:31
Di Matteo A et al (2005) Structural basis for the interaction between pectin methylesterase and a specific inhibitor protein. Plant Cell 17:849–858
Federici L et al (2001) Structural requirements of endopolygalacturonase for the interaction with PGIP (polygalacturonase-inhibiting protein). Proc Natl Acad Sci 98:13425–13430
Fries M, Ihrig J, Brocklehurst K, Shevchik VE, Pickersgill RW (2007) Molecular basis of the activity of the phytopathogen pectin methylesterase. EMBO J 26:3879–3887
Garg G, Singh A, Kaur A, Singh R, Kaur J, Mahajan R (2016) Microbial pectinases: an ecofriendly tool of nature for industries. 3 Biotech 6:47
Garron M-L, Cygler M (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20:1547–1573
Gou J-Y, Miller LM, Hou G, Yu X-H, Chen X-Y, Liu C-J (2012) Acetylesterase-mediated deacetylation of pectin impairs cell elongation, pollen germination, and plant reproduction. Plant Cell 24:50–65
Gough J, Karplus K, Hughey R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313:903–919
Harholt J, Suttangkakul A, Scheller HV (2010) Biosynthesis of pectin. Plant Physiol 153:384–395
Hatanaka C, Ozawa I (1971) Enzymic degradation of pectic acid XIII. A new exopolygalacturonase producing digalacturonic acid from pectic acid. Berichte des Ohara Instituts für landwirtschaftliche Biologie, Okayama Universität 15:47–60
Held MA, Jiang N, Basu D, Showalter AM, Faik A (2015) Plant cell wall polysaccharides: structure and biosynthesis. In: Ramawat KG, Mérillon JM (eds) Polysaccharides. Springer, Switzerland, pp 3–54
Herron SR, Benen JA, Scavetta RD, Visser J, Jurnak F (2000) Structure and function of pectic enzymes: virulence factors of plant pathogens. Proc Natl Acad Sci 97:8762–8769
Herron SR, Scavetta RD, Garrett M, Legner M, Jurnak F (2003) Characterization and implications of Ca2+ binding to pectate lyase C. J Biol Chem 278:12271–12277
Ho YS et al (1997) Brain acetylhydrolase that inactivates platelet-activating factor is a G-protein-like trimer. Nature 385:89–93
Jacob N (2009) Pectinolytic enzymes. In: Singh nee’ Nigam P, Pandey A (eds) Biotechnology for agro-industrial residues utilisation. Springer, Berlin, pp 383–396
Jenkins J, Mayans O, Smith D, Worboys K, Pickersgill RW (2001) Three-dimensional structure of Erwinia chrysanthemi pectin methylesterase reveals a novel esterase active site. J Mol Biol 305:951–960
Jenkins J, Shevchik VE, Hugouvieux-Cotte-Pattat N, Pickersgill RW (2004) The crystal structure of pectate lyase Pel9A from Erwinia chrysanthemi. J Biol Chem 279:9139–9145
Jensen MH, Otten H, Christensen U, Borchert TV, Christensen LL, Larsen S, Leggio LL (2010) Structural and biochemical studies elucidate the mechanism of rhamnogalacturonan lyase from Aspergillus aculeatus. J Mol Biol 404:100–111
Johansson K, El-Ahmad M, Friemann R, Jörnvall H, Markovič O, Eklund H (2002) Crystal structure of plant pectin methylesterase. FEBS Lett 514:243–249
Jolie RP, Duvetter T, Van Loey AM, Hendrickx ME (2010) Pectin methylesterase and its proteinaceous inhibitor: a review. Carbohydr Res 345:2583–2595
Kashyap D, Vohra P, Chopra S, Tewari R (2001) Applications of pectinases in the commercial sector: a review. Bioresour Technol 77:215–227
Kent LM, Loo TS, Melton LD, Mercadante D, Williams MA, Jameson GB (2016) Structure and properties of a non-processive, salt-requiring, and acidophilic pectin methylesterase from Aspergillus niger provide insights into the key determinants of processivity control. J Biol Chem 291:1289–1306
Kester H, Someren MA, Müller Y, Visser J (1996) Primary structure and characterization of an exopolygalacturonase from Aspergillus tubingensis. Eur J Biochem 240:738–746
Khan M, Nakkeeran E, Umesh-Kumar S (2013) Potential application of pectinase in developing functional foods. Annu Rev Food Sci Technol 4:21–34
Kunishige Y, Iwai M, Nakazawa M, Ueda M, Tada T, Nishimura S, Sakamoto T (2018) Crystal structure of exo-rhamnogalacturonan lyase from Penicillium chrysogenum as a member of polysaccharide lyase family 26. FEBS Lett 592:1378–1388
Langkilde A, Kristensen SM, Lo Leggio L, Mølgaard A, Jensen JH, Houk AR, Navarro Poulsen J-C, Kauppinen S, Larsen S (2008) Short strong hydrogenbonds in proteins: a case study of rhamnogalacturonan acetylesterase. Acta Crystallogr D Biol Crystallogr 64:851–863
Le Goff A, Renard C, Bonnin E, Thibault J-F (2001) Extraction, purification and chemical characterisation of xylogalacturonans from pea hulls. Carbohydr Polym 45:325–334
Lee W, Yusof S, Hamid NSA, Baharin BS (2006) Optimizing conditions for enzymatic clarification of banana juice using response surface methodology (RSM). J Food Eng 73:55–63
Leroux C et al (2015) Pectin methylesterase 48 is involved in Arabidopsis pollen grain germination. Plant Physiol 167:367–380
Lietzke SE, Yoder MD, Keen NT, Jurnak F (1994) The three-dimensional structure of pectate lyase E, a plant virulence factor from Erwinia chrysanthemi. Plant Physiol 106:849–862
Lietzke SE, Scavetta RD, Yoder MD, Jurnak F (1996) The refined three-dimensional structure of pectate lyase E from Erwinia chrysanthemi at 2.2 A resolution. Plant Physiol 111:73–92
Lionetti V, Cervone F, Bellincampi D (2012) Methyl esterification of pectin plays a role during plant–pathogen interactions and affects plant resistance to diseases. J Plant Physiol 169:1623–1630
Liu C-Q et al (2017) Polygalacturonase gene pgxB in Aspergillus niger is a virulence factor in apple fruit. PLoS ONE 12:e0173277
Lombard V, Bernard T, Rancurel C, Brumer H, Coutinho PM, Henrissat B (2010) A hierarchical classification of polysaccharide lyases for glycogenomics. Biochem J 432:437–444
Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495
Markovič O, Janeček Š (2004) Pectin methylesterases: sequence-structural features and phylogenetic relationships. Carbohydr Res 339:2281–2295
Martens-Uzunova ES et al (2006) A new group of exo-acting family 28 glycoside hydrolases of Aspergillus niger that are involved in pectin degradation. Biochem J 400:43–52
Martínez-Martínez I, Navarro-Fernández J, Daniel Lozada-Ramírez J, García-Carmona F, Sánchez-Ferrer Á (2008) YesT: a new rhamnogalacturonan acetyl esterase from Bacillus subtilis. Proteins Struct Funct Bioinform 71:379–388
Mayans O et al (1997) Two crystal structures of pectin lyase A from Aspergillus reveal a pH driven conformational change and striking divergence in the substrate-binding clefts of pectin and pectate lyases. Structure 5:677–689
McDonough MA, Kadirvelraj R, Harris P, Poulsen J-CN, Larsen S (2004) Rhamnogalacturonan lyase reveals a unique three-domain modular structure for polysaccharide lyase family 4. FEBS Lett 565:188–194
McLean R, Hobbs JK, Suits MD, Tuomivaara ST, Jones DR, Boraston AB, Abbott DW (2015) Functional analyses of resurrected and contemporary enzymes illuminate an evolutionary path for the emergence of exolysis in polysaccharide lyase family 2. J Biol Chem 290:21231–21243
Mohnen D (2008) Pectin structure and biosynthesis. Curr Opin Plant Biol 11:266–277
Mohnen D, Bar-Peled M, Somerville C (2008) Cell wall polysaccharide synthesis. In: Himmel M (ed) Biomass recalcitrance: deconstructing the plant cell wall bioenergy. Blackwell Publishing, Oxford, pp 94–187
Mølgaard A, Larsen S (2002) A branched N-linked glycan at atomic resolution in the 1.12 Å structure of rhamnogalacturonan acetylesterase. Acta Crystallogr D Biol Crystallogr 58:111–119
Mølgaard A, Kauppinen S, Larsen S (2000) Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure 8:373–383
Munoz-Munoz J, Cartmell A, Terrapon N, Baslé A, Henrissat B, Gilbert HJ (2017) An evolutionarily distinct family of polysaccharide lyases removes rhamnose capping of complex arabinogalactan proteins. J Biol Chem M117:794578
Murthy PS, Naidu MM (2011) Improvement of robusta coffee fermentation with microbial enzymes. Eur J Appl Sci 3:130–139
Mutter M, Beldman G, Schols HA, Voragen AGJ (1994) Rhamnogalacturonan α-l-rhamnopyranohydrolase (A novel enzyme specific for the terminal nonreducing rhamnosyl unit in rhamnogalacturonan regions of pectin). Plant Physiol 106:241–250
Mutter M, Beldman G, Pitson SM, Schols HA, Voragen AG (1998) Rhamnogalacturonan α-d-galactopyranosyluronohydrolase: an enzyme that specifically removes the terminal nonreducing galacturonosyl residue in rhamnogalacturonan regions of pectin. Plant Physiol 117:153–163
Najafian L, Ghodsvali A, Khodaparast MH, Diosady L (2009) Aqueous extraction of virgin olive oil using industrial enzymes. Food Res Int 42:171–175
Nakamura A, Furuta H, Maeda H, Takao T, Nagamatsu Y (2002) Analysis of the molecular construction of xylogalacturonan isolated from soluble soybean polysaccharides. Biosci Biotechnol Biochem 66:1155–1158
Navarro-Fernández J, Martínez-Martínez I, Montoro-García S, García-Carmona F, Takami H, Sánchez-Ferrer Á (2008) Characterization of a new rhamnogalacturonan acetyl esterase from Bacillus halodurans C-125 with a new putative carbohydrate binding domain. J Bacteriol 190:1375–1382
Normand J, Ralet M-C, Thibault J-F, Rogniaux H, Delavault P, Bonnin E (2010) Purification, characterization, and mode of action of a rhamnogalacturonan hydrolase from Irpex lacteus, tolerant to an acetylated substrate. Appl Microbiol Biotechnol 86:577–588
Novoa de Armas H, Verboven C, De Ranter C, Desair J, Vande Broek A, Vanderleyden J, Rabijns A (2004) Azospirillum irakense pectate lyase displays a toroidal fold. Acta Crystallogr D Biol Crystallogr 60:999–1007
O’Neill MA, York WS (2003) The composition and structure of plant primary cell walls. In: Rose JKC (ed) The plant cell wall. Blackwell Publishing, Oxford, pp 1–54
Ochiai A, Itoh T, Maruyama Y, Kawamata A, Mikami B, Hashimoto W, Murata K (2007) A novel structural fold in polysaccharide lyases Bacillus subtilis family 11 rhamnogalacturonan lyase YesW with an eight-bladed β-propeller. J Biol Chem 282:37134–37145
Ochiai A, Itoh T, Mikami B, Hashimoto W, Murata K (2009) Structural determinants responsible for substrate recognition and mode of action in family 11 polysaccharide lyases. J Biol Chem 284:10181–10189
Pagès S, Heijne WH, Kester HC, Visser J, Benen JA (2000) Subsite Mapping of Aspergillus niger endopolygalacturonase II by site-directed mutagenesis. J Biol Chem 275:29348–29353
Paniagua C et al (2017) Structural changes in cell wall pectins during strawberry fruit development. Plant Physio Biochem 118:55–63
Pelletier S et al (2010) A role for pectin de-methylesterification in a developmentally regulated growth acceleration in dark-grown Arabidopsis hypocotyls. New Phytol 188:726–739
Pelloux J, Rusterucci C, Mellerowicz EJ (2007) New insights into pectin methylesterase structure and function. Trends Plant Sci 12:267–277
Petersen TN, Kauppinen S, Larsen S (1997) The crystal structure of rhamnogalacturonase A from Aspergillus aculeatus: a right-handed parallel β helix. Structure 5:533–544
Pickersgill R, Jenkins J, Harris G, Nasser W, Robert-Baudouy J (1994) The structure of Bacillus subtilis pectate lyase in complex with calcium. Nat Struct Mol Biol 1:717–723
Pickersgill R, Smith D, Worboys K, Jenkins J (1998) Crystal structure of polygalacturonase from Erwinia carotovora ssp. carotovora. J Biol Chem 273:24660–24664
Pijning T, van Pouderoyen G, Kluskens L, van der Oost J, Dijkstra BW (2009) The crystal structure of a hyperthermoactive exopolygalacturonase from Thermotoga maritima reveals a unique tetramer. FEBS Lett 583:3665–3670
Rosenthal PB et al (1998) Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 396:92
Rozeboom HJ, Beldman G, Schols HA, Dijkstra BW (2013) Crystal structure of endo-xylogalacturonan hydrolase from Aspergillus tubingensis. FEBS J 280:6061–6069
Rytioja J, Hildén K, Yuzon J, Hatakka A, de Vries RP, Mäkelä MR (2014) Plant-polysaccharide-degrading enzymes from basidiomycetes. Microbiol Mol Biol Rev 78:614–649
Sandri IG, Fontana RC, Barfknecht DM, da Silveira MM (2011) Clarification of fruit juices by fungal pectinases. LWT Food Sci Technol 44:2217–2222
Scavetta RD et al (1999) Structure of a plant cell wall fragment complexed to pectate lyase C. Plant Cell 11:1081–1092
Seyedarabi A, To TT, Ali S, Hussain S, Fries M, Madsen R, Clausen MH, Teixteira S, Brocklehurst K, Pickersgill RW (2009) Structural insights intosubstrate specificity and the anti β-elimination mechanism of pectate lyase. Biochemistry 49:539–546
Shallom D et al (2005) Biochemical characterization and identification of the catalytic residues of a family 43 β-d-xylosidase from Geobacillus stearothermophilus T-6. Biochemistry 44:387–397
Sharma N, Rathore M, Sharma M (2013) Microbial pectinase: sources, characterization and applications. Rev Environ Sci Biotechnol 12:45–60
Shimizu T, Nakatsu T, Miyairi K, Okuno T, Kato H (2002) Active-site architecture of endopolygalacturonase I from Stereum purpureum revealed by crystal structures in native and ligand-bound forms at atomic resolution. Biochemistry 41:6651–6659
Silva IR et al (2014) Design of thermostable rhamnogalacturonan lyase mutants from Bacillus licheniformis by combination of targeted single point mutations. Appl Microbiol Biotechnol 98:4521–4531
Silva IR, Jers C, Meyer AS, Mikkelsen JD (2016) Rhamnogalacturonan I modifying enzymes: an update. New Biotechnol 33:41–54
Singh J, Kundu D, Das M, Banerjee R (2019) Enzymatic processing of juice from fruits/vegetables: an emerging trend and cutting edge research in food biotechnology. In: Kuddus M (ed) Enzymes in food biotechnology. Academic Press, Cambridge, pp 419–432
Tang Q, Liu YP, Ren ZG, Yan XX, Zhang LQ (2013) 1.37 Å crystal structure of pathogenic factor pectate lyase from Acidovorax citrulli. Proteins Struct Funct Bioinform 81:1485–1490
Teller DC, Behnke CA, Pappan K, Shen Z, Reese JC, Reeck GR, Stenkamp RE (2014) The structure of rice weevil pectin methylesterase. Acta Crystallogr F Struct Biol Cryst Commun 70:1480–1484
Thomas LM, Doan CN, Oliver RL, Yoder MD (2002) Structure of pectate lyase A: comparison to other isoforms. Acta Crystallogr D Biol Crystallogr 58:1008–1015
Tian G-W, Chen M-H, Zaltsman A, Citovsky V (2006) Pollen-specific pectin methylesterase involved in pollen tube growth. Dev Biol 294:83–91
van den Brink J, de Vries RP (2011) Fungal enzyme sets for plant polysaccharide degradation. Appl Microbiol Biotechnol 91:1477
van Pouderoyen G, Snijder HJ, Benen JA, Dijkstra BW (2003) Structural insights into the processivity of endopolygalacturonase I from Aspergillus niger. FEBS Lett 554:462–466
van Santen Y, Benen JA, Schröter K-H, Kalk KH, Armand S, Visser J, Dijkstra BW (1999) 1.68-Å crystal structure of endopolygalacturonase II from Aspergillus niger and identification of active site residues by site-directed mutagenesis. J Biol Chem 274:30474–30480
Vincken J-P, Schols HA, Oomen RJ, Beldman G, Visser RG, Voragen AG (2003) Pectin—the hairy thing. In: Voragen F, Schols H, Visser R (eds) Advances in pectin and pectinase research. Springer, Berlin, pp 47–59
Vitali J, Schick B, Kester HC, Visser J, Jurnak F (1998) The three-dimensional structure of Aspergillus niger pectin lyase B at 1.7-Å resolution. Plant Physiol 116:69–80
Walker SG, Ryan ME (2003) Cloning and expression of a pectate lyase from the oral spirochete Treponema pectinovorum ATCC 33768. FEMS Microbiol Lett 226:385–390
Wang Z-Y, MacRae EA, Wright MA, Bolitho KM, Ross GS, Atkinson RG (2000) Polygalacturonase gene expression in kiwifruit: relationship to fruit softening and ethylene production. Plant Mol Biol 42:317–328
Wang D, Yeats TH, Uluisik S, Rose JK, Seymour GB (2018) Fruit softening: revisiting the role of pectin. Trends Plant Sci 23:302–310
Wei Y, Schottel JL, Derewenda U, Swenson L, Patkar S, Derewenda ZS (1995) A novel variant of the catalytic triad in the Streptomyces scabies esterase. Nat Struct Mol Biol 2:218–223
Wong D (2008) Enzymatic deconstruction of backbone structures of the ramified regions in pectins. Protein J 27:30–42
Wu Y, Yin Z, Xu L, Feng H, Huang L (2018) VmPacC is required for acidification and virulence in Valsa mali. Front Microbiol 9:1981
Xiao Z, Bergeron H, Grosse S, Beauchemin M, Garron M-L, Shaya D, Sulea T, Cygler M, Lau PC (2008) Improvement of the thermostability and activityof a pectate lyase by single amino acid substitutions, using a strategy based on melting-temperature-guided sequence alignment. Appl Environ Microbiol 74:1183–1189
Yapo BM (2011) Pectic substances: from simple pectic polysaccharides to complex pectins—a new hypothetical model. Carbohydr Polym 86:373–385
Yip VL, Withers SG (2006) Breakdown of oligosaccharides by the process of elimination. Curr Opin Chem Biol 10:147–155
Yoder M, Jurnak F (1995a) Protein motifs. 3. The parallel beta helix and other coiled folds. FASEB J 9:335–342
Yoder MD, Jurnak F (1995b) The refined three-dimensional structure of pectate lyase C from Erwinia chrysanthemi at 2.2 angstrom resolution (implications for an enzymatic mechanism). Plant Physiol 107:349–364
Yoder MD, Keen NT, Jurnak F (1993a) New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science 260:1503–1507
Yoder MD, Lietzke SE, Jurnak F (1993b) Unusual structural features in the parallel β-helix in pectate lyases. Structure 1:241–251
Yue X, Lin S, Yu Y, Huang L, Cao J (2018) The putative pectin methylesterase gene, BcMF23a, is required for microspore development and pollen tube growth in Brassica campestris. Plant Cell Rep 37:1003–1009
Zandleven J, Beldman G, Bosveld M, Benen J, Voragen A (2005) Mode of action of xylogalacturonan hydrolase towards xylogalacturonan and xylogalacturonan oligosaccharides. Biochem J 387:719–725
Zandleven J, Beldman G, Bosveld M, Schols H, Voragen A (2006) Enzymatic degradation studies of xylogalacturonans from apple and potato, using xylogalacturonan hydrolase. Carbohydr Polym 65:495–503
Zega A, D’Ovidio R (2016) Genome-wide characterization of pectin methyl esterase genes reveals members differentially expressed in tolerant and susceptible wheats in response to Fusarium graminearum. Plant Physiol Biochem 08:1–11
Zheng Y et al (2012) Crystal structure and substrate-binding mode of a novel pectate lyase from alkaliphilic Bacillus sp. N16-5. Biochem Biophys Res Commun 420:269–274
Acknowledgements
The author would like to thank Dr. Bamaprasad Bag at Institute of Minerals and Materials Technology, Bhubaneswar and Dr. Hirak Chakraborty, Department of Chemistry, Sambalpur University for critical reading of the manuscript and fruitful suggestions.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
This review does not contain any studies with human participants or animals performed by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kanungo, A., Bag, B.P. Structural insights into the molecular mechanisms of pectinolytic enzymes. J Proteins Proteom 10, 325–344 (2019). https://doi.org/10.1007/s42485-019-00027-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42485-019-00027-5