
Abstract
Malaria is a fever sickness caused by Plasmodium parasites that are transmitted to humans by mosquito bites from infected female Anopheles mosquitos. Intracellular malaria parasites require lipids for the growth and replication. They possess a prokaryotic type II fatty acid synthesis (FAS II) pathway that localizes to the apicoplast plastid organelle and is assumed to be necessary for pathogenic blood stage replication. Considering widening resistance of resistant Plasmodium parasites and thus, failing conventional antimalarial agents, we herein analyzed a set of 109 flavonoids in four protein structures including three homology models and one experimentally obtained crystal structure were conducted to obtain the probable conformations of ligands and their binding affinities. Our results suggested Volkensiflavone, Bilobetin and Sciadopitysin as lead candidates for further detailed analysis and testing their synthetic analogues for their in-vitro anti-malarial potentials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Malaria is a fever sickness caused by Plasmodium parasites that are transmitted to humans by mosquito bites from infected female Anopheles mosquitos [1]. It is caused by five parasitic species, two of which—P. falciparum and P. vivax—are the most dangerous [2,3,4,5,6]. The P. falciparum is the most dangerous and a common malaria parasite. It causes symptoms like recurring fever, chills, and headaches. After the commencement of a fever, it settles for a while and then recurs. It can lead to unconsciousness or even death in extreme circumstances [7,8,9,10]. In 2020, there were an estimated 241 million cases of malaria with 627,000 people dying as a result. According to W.H.O, African continent bears a disproportionately large amount of the worldwide malaria burden. The 96% of malaria fatalities and 95% of malaria cases occurred in the same region. Around 80% of all malaria deaths in the region were in children under the age of 5 [11]. The WHO (The World Health Organization) now recommends artemisinin-based combination treatments (ACTs) for the treatment of multidrug-resistant P. falciparum malaria. In Southeast Asia, resistance to ACTs against P. falciparum has started to appear with other issues such as high treatment costs, toxicities, unsatisfactory physicochemical/pharmacokinetic properties and low abundance [11,12,13]. As a result, treating multidrug-resistant malaria has become more difficult in most malaria-endemic regions of the world, and requires the urgent development of newer and more effective antimalarial drugs or medicines [13,14,15,16]. Plants and/or plant-based traditional medicines [17, 18] are thought to be the most trustworthy and alternative means of discovering novel antimalarial compounds to address the above-enlisted issues. chemical compounds derived from nature are essentially secondary metabolites of plant or another natural origin that included several important natural product classes that have a wide range of biological properties, features and health advantages.
1.1 General Considerations and Chemistry of Flavonoids
Flavonoids (Fig. 1) are one of the natural compounds or phytochemical classes that have lately attracted significant attention because of their prospective chemo preventive and chemo-protective potentials in inflammatory disorders, cardiovascular illnesses, diabetic complications, neurodegenerative disorders, malignant sickness, malaria, and microbial infectious diseases for these reason medicinal chemists are interested in these structures [18,19,20,21]. Flavonoids have a chemical structure based on the flavonoid molecular framework (C6–C3–C6), which is a fifteen-carbon skeleton made up of two benzene rings (A ring and B ring) linked by a three-carbon heterocyclic pyran ring (C ring). At the C-2 (flavone), C-3 (e.g., iso-flavone), or C-4 (neo flavone) locations, the chroman ring (C ring) is connected to the second aromatic ring (ring B, benzenoid substituent). An acyclic moiety (chalcone) or a five-membered heterocyclic furan ring (aurone) can sometimes be found in place of a six-membered heterocyclic pyran ring (ring C). A-pyrone (flavones, flavanols, and isoflavones) or its dihydroderivatives are six-membered rings condensed with the benzene ring (flavanones and flavanols). Plant polyphenols are hydroxylated phenolic compounds, whereas flavonoids are hydroxylated phenolic substances. They are often hydroxylated in positions 3, 5, 6, 7, 3′, 4′, and 5′. Flavonoids are thought to block the fatty acid synthesis in parasite biochemistry in the parasite. They could inhibit the influx of l-glutamine and myoinositol in the infected intraerythrocytic phase and also inhibit the heme detoxification and degradation in the food vacuoles of the parasite [6, 18,19,20,21].

Basic flavonoid structure showing rings A, B and C and the numbering, flavonoids and chalcone chemical structures [Adapted from Open Access Article available at Int. J. Mol. Sci. 2021, 22(2), 646; https://doi.org/10.3390/ijms22020646]
1.2 Targeting Flavonoids for FAS-II (Fatty Acid Synthesis: FAS-II) Pathway
Genome sequencing of P. falciparum has opened many avenues for the drug discovery [22]. Fatty acids are required for various biological processes such as membrane lipid synthesis and lipid metabolism in the parasite. FAS-II (fatty acid synthesis: FAS-II) pathway appears to be a perfect target as they are non-homologous to humans. Many FAS-II inhibitors were found to be effective against blood stage parasites at nanomolar doses and also, and they could block three enzymes in the same pathway [23].
The FAS-II pathway in Apicoplast commences with the importation of substrates from the cytoplasm, leading to the creation of eight or more carbon as saturated fatty acid chains through a sequence of biochemical reactions using acyl-carrier proteins (ACPs) and nine enzymes [22,23,24,25,26]. Depending on the phases FAS-II can be divided into three steps called preparation, initiation and elongation. In the preparation phase importation of the glycolytic intermediate phosphoenolpyruvate (PEP) from the cytoplasm takes place and it is converted to acetyl-CoA and ATP [22,23,24,25,26]. The initiation phase is involved in the synthesis of malonyl-ACP from acetyl-CoA, this serves as the first substrate for the fatty acid elongation cycle. Finally, during the elongation phase, which uses four enzymes, the growing fatty acid chain is lengthened by two carbon units per turn, resulting in the generation of mature-length acyl-ACPs [22,23,24,25,26]. After the synthesis of malonyl-CoA from acetyl-CoA via acetyl-CoA carboxylase (ACC) the next stage in the initiation phase is catalyzed by Malonyl-CoA: ACP transacylase (FabD). In this phase the transfer of a malonyl group from the malonyl-CoA to ACP takes place which yields malonyl-ACP. The sequence characteristics of the Plasmodium and Toxoplasma gondii FabD are compatible with apicoplast targeting, and the activity of PfFabD has been established in-vitro [22,23,24,25,26]. In the final stage of the initiation phase, the condensation of malonyl-ACP with acetyl-CoA takes place along with the generation of CoA and Carbon dioxide which is catalyzed by FabH (beta-ketoacyl-ACP synthase III) [22,23,24,25,26]. During the first cycle of chain elongation, the four-carbon acetoacetyl-ACP generated in this phase is used. In-vitro, a variety of sulfides, sulfonyls and, sulfonamides, were found as potential FabH inhibitors, and they all showed efficacy against P. falciparum and PfFabH [22,23,24,25,26]. As the initial step in FAS-II elongation phase, FabB/F catalyzes (beta-ketoacyl-ACP synthase I/II) the condensation of malonyl-ACP with the acyl-ACP, resulting in carbon dioxide and a beta-ketoacyl-ACP product that has been extended by two carbon units. However, this step is bypassed by FabH enzyme in the first elongation cycle which also executes the same condensation reaction. Genetic studies on FabB/F were achieved in P. falciparum such that deletion of FabB/F blocked sporozoite development. Enoyl-ACP reductase, also known as FabI (enoyl-ACP reductase (FABI)), catalyzes the last step of FAS-II elongation phase, which entails converting enoyl-ACP to acyl-ACP using NADH as an electron donor. The crystal structure of P. falciparum, P. berghei and T. gondii has been elucidated, revealing essential insight into the structural biology of the enzyme. Most inhibition studies have been performed on P. falciparum FabI, more than on any other enzyme of the entire FAS-II pathway enzymes [27, 28]. The glycosylated flavonoid lutein-7O-glucoside has been reported to be the first malarial natural product to inhibit P. falciparum enoyl acyl carrier protein (ACP) reductase (FabI) [28].
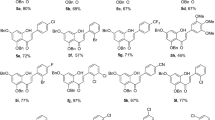
In light of this foregoing, and given the importance of flavonoids and analogues as a potent antimalarial class, we performed a molecular docking simulation on the FAS-II enzymes, two of which are involved in the initiation phase and the other two in the elongation phase, in the hopes of finding a potent antimalarial flavonoid capable of inhibiting multiple stages and eventually shutting down the entire FAS-II pathway (Fig. 2). Homology modeling techniques are employed when the experimental crystal structure of a protein is not reported on the PDB database but the three dimensional (3D)-crystal structure is required for example to aid in Computer-Aided Drug Design (CADD), to deepen the structural insights of the active site [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. In this current study, we constructed the homology models of FabD, FabH and FabB using the already reported experimental 3D-crystallographic structures of homologous proteins based on sequence alignment. Many studies have been conducted to analyse the flavonoids present in medicinal plants for antimalarial activity utilizing hypoxanthine assays, in-vitro assays, parasite growth assays etc. [45,46,47]. A thorough literature search was performed to identify the flavonoid structures which exhibited in-vitro or in-vivo antimalarial activity (see supporting information). Thereafter, a molecular docking simulation of 109 flavonoids in four protein structures including three homology models and one experimental crystal structure was conducted to obtain the conformations of ligands and their binding affinities. Docking interactions were visualized with the help of Discovery studio visualizer [48] and high binding affinity flavonoids (top 3 hits) were analyzed for ADMET (absorption, distribution, metabolism, and excretion and toxicity) profile (flow chart of docking methodology in Fig. 3).

Type II fatty acid biosynthesis and its enzymes; Numbers 1–4 indicates the targets selected along with the core structure of flavonoids

Flowchart of docking methodology
2 Methodology
2.1 Template Sequence Alignment
The Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [49] tool was used to align the query and best template sequences based on the identity parameter. The input format was FASTA Sequence of query and template, and the output format was ClustalW with the character counts using the default settings.
2.2 Homology Modeling
The protein sequences for FabD, FabH and FabB were retrieved from NCBI (The National Center for Biotechnology Information) and UniProtKB databases, with the following accession codes AAK83684.1, XP_001349620.1, respectively and UniProtKB—Q965D4 (Q965D4_PLAFA). Target sequences were retrieved in FASTA format and exported/submitted to the SWISS model [50] online workspace as input to build the homology model. QMEAN is a composite scoring function which is able to derive both global (i.e., for the entire structure) and local (i.e., per residue) absolute quality estimates on the basis of one single model [51, 52]. The Ramachandran plot is a plot of the torsional angles—phi (φ)and psi (ψ)—of the residues (amino acids) contained in a peptide [53]. The QSQE score, which ranges from 0 to 1, indicates how well interchain connections should be predicted for a model created using a specific alignment and template [51, 52]. A score above 0.7 can be deemed dependable to follow the projected quaternary structure in the modelling process, and a higher QSQE is often thought to be "better." Based on QSQE scores, a template for 3D prediction of protein was selected. A different strategy is to take into account the alignment and template search method's quality, which is reflected in the GMQE (Global Model Quality Estimation) score. The GMQE score, which is given as a number between 0 and 1 where larger numbers indicate more reliability, represents the expected correctness of that alignment [51, 52]. To refine the predicted structures and remove structural artefacts, forcefield based energy minimization was performed utilizing YASARA online server [53]. Furthermore, the dependability of all the modelled 3D protein structures was evaluated by using QMEAN (quality model energy analysis) server, ProSA (Protein structure analysis) web service, SAVES (https://saves.mbi.ucla.edu/) [54,55,56] and Molprobity servers [57].
2.3 Protein and Ligand Preparation
In protein preparation, water molecules and heteroatoms were removed along with the subsequent addition of polar hydrogens and Kollman charges using AUTODOCK tools4 [58]. Flavonoids with antimalarial activity either tested in in-vitro or in-vivo models were drawn manually using ‘Chemsketch’ software [59] and further optimized for the energy minimization using ‘AVOGADRO software’ [60] with MMFF94 forcefield using the Steepest Descent algorithm (number of steps = 1000). The conversion of ligand files from ‘.mol2’ to ‘.pdbqt’ (suitable for Autodock vina) were done using the ‘Openbabel’ interface [61].
2.4 Active Site Identification
In the case of FabD and FabH, the ligand bound in the template structure was aligned with the modelled protein to identify the active site. As there were no ligands present in FabB template protein an online server called Computed Atlas of Surface Topography of proteins (CASTp) [62] was utilized to predict the active site. Again, the active site for FabI (3LTO) was identified using already present ligands in the active site Table 1 summarizes the active site grid coordinates and grid box size.
2.5 Network Generation
Protein–protein interactions are crucial for predicting target protein function and drug-like properties of compounds. The bulk of genes and proteins use a variety of interactions to materialize the phenotype that results from their activity. The protein–protein interaction network (PIN) is a helpful tool for doing a systematic analysis of the intricate biological processes occurring within cells. PINs for numerous species, including viruses, bacteria, plants, animals, and humans, have been reconstructed due to the growing interest in proteome-wide interaction networks. STRING (https://string-db.org/) module was used to identify the protein–protein interaction partners of Plasmodium falciparum enoyl-ACP reductase. STRING is a biological database that is employed to create a Protein–Protein Interaction Network (PPIN) for various known and predicted protein interactions. Currently, the string database has 67,592,464 proteins from 14,094 different organisms [63].
2.6 Molecular Docking Simulation
A series of optimized 109 flavonoids’ structures were virtually screened in four protein structures among which three were predicted homology models (FabD, FabH & FabB) and one crystal structure of FabI with pdb Id-3LT0 [64] using Autodock vina. The virtual screening was performed on a personal computer utilizing AMD Ryzen 5 processor (12 cores) with parameters Energy range, num modes and exhaustiveness set to 4, 10 and 16 respectively. In order to visualize the various interaction made by the ligand in the active site of protein Discovery Visualizer Studio was used to generate 2D and 3D interaction profiles.
2.7 In-Silico ADMET Profiling
By using smiles descriptors as an input format for the search, ADMET (absorption, distribution, metabolism, excretion and toxicity) analysis of the top three binding affinity flavonoids was evaluated through SWISS ADME (http://www.swissadme.ch/) [65] and admetSAR (http://lmmd.ecust.edu.cn/admetsar1/) (for toxicity analysis) [66] respectively.
2.8 Molecular Dynamics
Using molecular dynamics simulations, the stability and binding flexibility of the chosen protein–ligand docking complexes were examined in real time. We conducted MD simulations to investigate the structural stability of complexes, residue behaviors, and atom behaviors using the Groningen Machine for Chemical Simulations (GROMACS v5.1.5) and GROMACS 96-53a6 force fields [67]. All ligand topology files were generated via the Dundee PRODRG3.0 server. To overcome these MDS models, the triclinic box type was built using an explicit simple point charge (SPC) water model (box size: 80 Å). Additionally, the simulation box was neutralized using the counter ions. As a result, equilibration with energy minimization utilizing NVT and NPT was carried out (parameters: Temperature (K) = 300, Pressure = 1 bar, and Simulation Time = 100 ns). Depending on the docking scores with best docked molecules and associated protein structure, we established the molecular dynamics stability analysis for FabI with Bilobetin (fg86); Volkensiflavone (fg36) and Sciadopitysin (fg89) for a period of 100 ns each. The root means square deviation (RMSD), a key statistic for assessing protein transformation changes during simulations, is generally recognized. Protein stability can also be investigated using the RMSD technique. The RMSF is a measure of the displacement of a particular atom, or group of atoms, relative to the reference structure, averaged over the number of atoms. The RMSD is useful for the analysis of time-dependent motions of the structure.
2.9 Calculations of Binding Free Energy Prime MMGBSA (Molecular Mechanics/Generalized Born Surface Area)
The most often used technique for determining the strength of interactions between a drug and its receptor is molecular mechanics with generalized born surface area (MM/GBSA) [68]. As a result, the Prime module from the Schrodinger suite was used to estimate binding free energies. OPLS molecular mechanics energies (EMM), a VSGB solvation model for polar solvation (GSGB), and a non-polar solvation term (GNP) made up of van der Waals interactions and the non-polar solvent-accessible surface area (SASA) make up Prime/MM-GBSA in its basic form. In MM/GBSA computations, the VSGB 2.0 model was employed, which uses an improved implicit solvent model to approximation the solvation free energy.
3 Results and Discussion
3.1 Sequence Alignment
Clustal Omega was used to determine alignment between the target and chosen sequences. (Figs. 4, 5 and 6). The Clustal Omega algorithm aligns sequences more quickly and precisely. To predict a higher-quality model of the query protein utilizing homology modelling, a strong alignment of template sequences and closely related template models is required.

Sequence alignment of the template protein 6U0J and the query protein sequences of FabD

Sequence alignment of the template protein 5BQS and the query protein sequences of FabH

Sequence alignment of the template protein 4LS6 and the query protein sequences of FabB
3.2 Homology Modeling
As the experimental crystal structures for FabD, FabH and FabB are not available in the PDB, such that a template library in the SWISS-MODEL was searched using BLAST to find an optimal template for building homology models. Sequence identity with the template structures is summarized in Table 2. If the sequence identity of two proteins is greater than 25%, investigations have shown that their 3D structures are comparable. The homology model of Plasmodium falciparum Malonyl-CoA:ACP transacylase or FabD was built by comparative modelling using the crystal structure of malonyl-CoA Acyl Carrier Protein Transacylase Escherichia coli K-12 (PDB ID: 6U0J) with a sequence identity of 29.69%. The template was chosen based on the GMQE score and the fact that it contained a ligand (9EF) in the crystal structure that could be exploited for the identification of the active site. Again, for the 3D structural prediction of beta-Ketoacyl-ACP synthase III or FabH crystal structure of Streptococcus pneumoniae FabH (5BQS) was selected which is a dimer with a sequence length of 323 amino acids. For the PfFabH sequence, the QSQE score was 0.72, sequence coverage from THR50 to TYR371 and sequence identity of 35.96 with respect to the template structure. The chosen template also had a small inhibitor molecule called 4VN; which could be used as a binding site for PfFabH through structural alignment. The selection of active site was decided based on in-bound ligand retained, for the protein which had higher sequence similarity while developing homology models for FabH and FabD. Usually, the in-bound or co-crystallized ligand’s key amino residues are considered as preferable binding site, when active binding site residues are unknown or fully explored or when co-crystal for that protein target is missing. for finally, the crystal structure of beta-ketoacyl-ACP synthase II (FabF) I108F mutant from Bacillus subtilis (PDB ID: 4lS6) was chosen for three-dimensional prediction of beta-ketoacyl-ACP synthase I or FabB owing to its high sequence identity (51.79%) and QSQE (0.84) score among the generated template structures. The sequence coverage of the model ranged from SER2 to VAL256. Even though the homology modeling process is one of the most robust modelling tools in bioinformatics, it commonly contains significant local distortions such as unphysical phi/psi angles, irregular H bonding networks, and steric clashes generating the structure models less practical for high-resolution functional analysis. Such that to encounter this problem YASARA forcefield [69] was used to minimize the homology models to improve the overall structure.
3.3 Validation and Verification of Homology Models
To validate the accuracy and reliability of the generated homology model’s various online validation servers were utilized. QMEAN is a composite scoring function that evaluates the protein structure's key geometrical characteristics. The models with a score of < 1 falls in the dark zone as shown in Fig. 7. Models are regarded as good if they fall in this dark zone. Here the red star marker indicates the generated target homology model, which in these cases is considered good based on its proximity to or within the dark zone. The Ramachandran plot is a visual depiction of the favoured areas for backbone dihedral angles (Psi) against (Phi) of amino acid residues. It also provides statistical information such as amino acids in residues residing within the favourable allowed and disallowed regions. Ramachandran plot statistics were obtained from the Molprobity server and the results are shown in Fig. 8 and tabulated in Table 3. The majority of amino acid residues were found to be residing within the energetically favoured regions (> 94%). Ramachandran distribution z scores values were all < 2 along with Bad bonds and bad angles within the optimal values thus indicating a good quality model for further use. ERRAT [70] overall quality factor was assessed using SAVES server. The overall quality factor values of all predicted structures were greater than 80% (Fig. 9) represented as the proportion of the protein for which the calculated values fall below the 95% rejection limit. In general, a high-quality model yields a value > 50. ProSA-web service is a tool to analyze the errors in the 3D structure of proteins. The analysis of the models showed a Z-score between − 8.86, − 8.38 & − 5.83 for FabD, FabH & FabB respectively (acceptable values are below 0.5) (Fig. 10). By comparing the 3D profile of a protein structure to its amino acid sequence, VERIFY 3D determines its accuracy. A structure's three-dimensional profile and its sequence are expected to have a high-score match. All modelled proteins passed the Verify-3D [71, 72] tests and showed a good 3D environment profile. Results of validation and verification of models using various tools are summarized in Table 4.

Estimation of absolute quality of homology model as a graph

General Ramachandran plot of homology model of A FabD, B FabH and C FabB using Molprobity server

Overall quality factor of homology model of A FabD, B FabH and C FabB using PROCHECK server

ProSA Z-Score values using homology models of A FabD, B FabH and C FabB using ProSA Server
3.4 String Interactions
The protein–protein interaction of PfENR or FabI was determined by the STRING and the interaction as given in Fig. 11. PPI networks provide an understanding of complicated molecular mechanisms and enable the identification of novel modulators of disease progression. PfENR was found to interact with 10 other proteins in which the FabD, FabH and FabB were also found to be present in the network with known and predicted interactions. PfENR was predicted to have a high score as functional partners (0.998) with MCAT or FabD which catalyzes the transfer of the malonyl group from the malonyl-CoA to ACP. Along with the FAS-II pathway proteins, a key enzyme in folate metabolism called dihydrofolate reductase-thymidylate synthase (DHFR-TS) was also present in the generated PPI network. DHFR-TS is already a target of anti-malarial drugs (pyrimethamine and cycloguanil).

Protein–Protein interaction (PPI) network of PfENR or FabI (present in the centre) generated by STRING server
3.5 In-silico Molecular Docking
Molecular docking simulations are sophisticated bioinformatic tools which are utilized to predict and find the best binding conformation of the ligand within the active site cavity of the target protein. A high negative magnitude of binding affinity depicts the best configuration of the ligand in the active site of a protein. The therapeutic efficacy of FAS-II inhibitors has been verified by studies using pathogenic microorganisms. One of them is triclosan, a microbicide that is frequently found in consumer goods. There is a lot of interest in this drug and its expected target, FabI, as a result of a widely referenced study that demonstrated triclosan's antimalarial effectiveness in-vitro against P. falciparum and in vivo against the rodent parasite P. berghei, aimed towards the pathogenic asexual blood stages. As a result, we used FabI to examine the top 3 phytoconstituents (based on docking scores). Tables 6 and 7 are included with information on the docking scores and the implicated amino acid residues for the other three targets. The Binding affinities of the 109 flavonoids against the selected target proteins in FAS-II pathway are tabulated in Table 5 (Figs. 12, 13, 14, 15). Virtual screening of flavonoids yielded top three hits Bilobetin (fg86), Volkensiflavone (fg36) and Sciadopitysin (fg89), they also made high binding affinities with all target enzymes. The binding affinities of these three flavonoids were greater than the standard drug artemisinin and are given in Table 6 along with their interacting residues in Table 7. Ligand fg86 or Bilobetin was found to have the highest affinity (−12.6 KCal/mol) towards FabI protein among the 109 flavonoids which were analyzed. The docking interactions shown by Bilobetin is depicted in Fig. 12C, D, it had interactions with ALA169, SER215, GLY104, ALA319 and ALA219 residues through conventional hydrogen bonding while the π-electron in benzene of chromen ring established a π–π stacking, two π-alkyl & two π-sigma interactions through TRP131, MET281, VAL222, LEU216 & ALA217 respectively. Bilobetin also had high binding affinities against the FabB, FabD and FabH receptors (Table 6) and it formed hydrogen bonds in every active site pocket. Volkensiflavone scored second best affinity against FabI with a binding affinity of −12.4 kcal/mol. Inside the active cavity Volkensiflavone made three hydrogens bonds with ARG318, ASP168 and SER170, pi alkyl bonding with LEU216, ALA169 and ALA319. Again, TRP131 was found to be making Pi-Pi stacking with the chromen ring (Fig. 12A, B). It was also revealed that Volkensiflavone had the best binding affinity in FabH active site cavity. Sciadopitysin had a binding affinity of −12.0 KCal/mol in FabI protein, it formed hydrogen bonding interactions with ALA217, ALA319, GLY104 and PHE167, unfavourable donor–donor interaction with ALA169 and Pi–Pi stacking with TRP131(Fig. 12E, F). The binding cavity residues for inbound ligand (FabI) were found to be TYR111, LEU315, SER317, LYS285, ALA319, ASP168, ALA169 and SER170.

2-D and 3-D interactions of the top hits fg38 (A, B) fg86 (C, D) and fg89 (E, F) with FabI

2-D and 3-D interactions of the top hits fg38 (A, B) fg86 (C, D) and fg89 (E, F) with FabD

2-D and 3-D interactions of the top hits fg38 (A, B) fg86 (C, D) and fg89 (E, F) with FabH

2-D and 3-D interactions of the top hits fg38 (A, B) fg86 (C, D) and fg89 (E, F) with FaB
3.6 Molecular Dynamics (MD) Simulation Analysis and MMGBSA Calculations
In simulating conditions that are similar to actual physiological environmental conditions, MD simulation analyses the protein–ligand stability (of the complex). Here, we have simulated three complexes for 100 ns each: FabI with Bilobetin (fg86); Volkensiflavone (fg36); and Sciadopitysin (fg89) (Fig. 16). FabI with Bilobetin (fg86), Volkensiflavone (fg36), and Sciadopitysin (fg89) complex RMSD values were maintained below 0.4, 0.5, and 0.4 nm, respectively. Furthermore, we found no deterioration in these three complexes compactness characteristics during simulations. Root mean square fluctuations (RMSF) measurements were also recollected below acceptable ranges (0.6, respectively for complexes, FabI with Bilobetin (fg86); Volkensiflavone (fg36) and Sciadopitysin (fg89)) [72]. Moreover, RMSD and RMSF analysis for FabI with Artemisinin was also found to be stable. A value of ΔRMSF > 0.6 Å was used as the threshold value of RMSF that indicates a significant change in structural movements [72]. All protein frames are first aligned on the reference frame backbone, and then the RMSD is calculated based on the atom selection. Monitoring the RMSD of the protein can give insights into its structural conformation throughout the simulation. RMSD analysis can indicate if the simulation has equilibrated—its fluctuations towards the end of the simulation are around some thermal average structure. Changes of the order of 1–3 Å are perfectly acceptable for small, globular proteins. Changes much larger than that, however, indicate that the protein is undergoing a large conformational change during the simulation. It is also important that your simulation converges—the RMSD values stabilize around a fixed value. If the RMSD of the protein is still increasing or decreasing on average at the end of the simulation, then your system has not equilibrated, and your simulation may not be long enough for rigorous analysis. Similarly, for RMSD results, we found that RMSD is within acceptable ranges [73]. We also noticed that increments in the number of hydrogen bonds over 100 ns simulation time for these three complexes might explain their stabilities. Thus, based on RMSD, RMSF, and Rg values, we concluded that these three complexes were stable. Furthermore, Prime MMGBSA energies (MMGBSA dG Bind) for complexes, FabI with Bilobetin (fg86); Volkensiflavone (fg36) and Sciadopitysin (fg89)) were observed as −52.53 kcal/mol; −47.93 kcal/mol and −51.34 kcal/mol, respectively. The FabI with artemisinin MMGBSA dG Bind energy was found to be −50.72 kcal/mol. This analysis showed us that phytochemicals Bilobetin (fg86); Volkensiflavone (fg36) and Sciadopitysin (fg89)) had strong binding potentials for target FabI (Fig. 17).

RMSD and RMSF analysis profiles for FabI with Bilobetin (fg86); Volkensiflavone (fg36); and Sciadopitysin (fg89) over 100 ns simulation period

RMSD and RMSF analysis profiles for FabI with over 100 ns simulation period
3.7 In-Silico ADMET Analysis
To examine ADMET characteristics (summarized in Table 8) of the top three hits, absorption, distribution, metabolism, excretion and toxicity as well as some physicochemical aspects, the SWISS ADME server and the admetSAR online database were utilized. The three flavonoids Bilobetin, Volkensiflavone and Sciadopitysin expressed positive human intestinal absorption, a non-AMES toxic, non-carcinogens, weak inhibitor of human ether a-go-go related genes and non-substrates CYP450 2C9 and 2D6. Bilobetin and Sciadopitysin were identified to be inhibitors of p-glycoproteins as well as a substrate for CYP450 3A4 isoenzyme. Many medications that induce or inhibit P-glycoprotein have a comparable effect on the drug metabolizing isoenzyme CYP450 3A4, implying a synergistic involvement in xenobiotic detoxification [43]. Furthermore, Volkensiflavone was classified as class II in acute oral toxicity (50 < LD50 ≤ 500 mg/kg) and it was also found to be BBB permeating. Bilobetin and Sciadopitysin were found to be slightly toxic as they were both classified as class III in toxicity profile (500 < LD50 ≤ 5000 mg/kg). The ADMET parameters and physicochemical properties of the top hits Bilobetin (fg86) and Sciadopitysin (fg89) were qualified for Lipinski rule of five with one violation each (MW > 500) whereas fg36 had two violations (MW > 500, NH or OH > 5) thus failing Lipinski's rule of five. Natural products, in most situations, do not necessarily follow Lipinski's rule since they are anticipated to enter the human body by more complex methods such as active transportation rather than passive diffusion, and hence are not expected to conform with bioavailability standards [44]. Bilobetin and Sciadopitysin had the same bioavailability score of 0.55 whereas Volkensiflavone scored a low bioavailability score (0.17). The top hits showed no structural alerts against the PAINS filter [72].
4 Conclusion
In this study, we have performed a three-dimensional construction of homology proteins of FabD, FabH and FabB using a SWISS-MODEL server. Subsequently, validation and verification of produced models were assessed via SAVES, QMEAN, ProSA-web, Verify3D and Molprobity servers. The predicted models resembled the template structure, suggesting their reliability. This study also gives insight on the binding modes and binding interaction of flavonoids with homology modelled protein targets. The flavonoid Bilobetin (fg86) showed strong interactions with FabI (3LT0) enzyme (binding affinity—12.6 K Cal/mol) with the following amino acid residues ALA219, ALA319, GLY104, SER215 and ALA169. Furthermore, the binding affinity of Volkensiflavone (fg36) and Sciadopitysin (fg89) ranged from − 7.5 to − 12.4 kcal/mol in all targets. The docking output of hits was found to be greater than the standard drug artemisinin (− 6.5 to − 9.5 kcal/mol) against four enzymes used in this study. This suggests that ligand forms better conformation within the active site(s) when compared with the standard drug artemisinin. In our ADMET analysis, it was observed that Bilobetin and Sciadopitysin had good pharmacokinetics profiles along with low oral toxicity (Class III) and inhibitors of P-glyco proteins. The three hits also showed the lowest binding affinity and also showed good antimalarial activity despite they violated the Lipinski's rule of five. Moreover, Bilobetin (fg86); Volkensiflavone (fg36) and Sciadopitysin (fg89)) had strong binding potentials for target FabI as depicted from MD simulation stability of 100 ns and MMGBSA energies. ∆G of Volkensiflavone is higher than the reference compound Artemisinin. ADMET results also showed that this substance has group II toxicity, penetrates into BBB, RMSD value is unstable. Thus, we believe that this hit can be more optimized for safer ADMET profiles, by forming their synthetic analogues or carrying out higher experimental in vitro, in vivo analysis. As a preliminary work, we are putting forward this lead and suggested for its better modifications. Considering previous literature reports on the flavonoids with the antimalarial activity we conducted the same via in-silico molecular docking analysis against key enzymes in FAS-II pathway. We identified Volkensiflavone (needs to be optimized further), Bilobetin and Sciadopitysin as lead candidates for further detailed in-silico analysis by molecular dynamics simulations, and testing their synthetic analogues for in-vitro anti-malarial potentials. To combat this deadly disease malaria, research should be carried out on this path to save mankind. Natural products such as flavonoids are expected to have high therapeutic efficacy against resistant malaria and deserve future research.
References
Wiesner J, Ortmann R, Jomaa H, Schlitzer M (2003) New antimalarial drugs. Angew Chem Int Ed 42(43):5274–5293
Cruz JN, Mali SN (2022) Antimalarial hemozoin inhibitors (β-hematin formation inhibition): latest updates. Combinat Chem High Throughput Screen. https://doi.org/10.2174/1386207325666220117145351
Mali SN, Pandey A (2022) Hemozoin (beta-hematin) formation inhibitors; a promising target for the development of new antimalarials: current update and a future prospect. Combinat Chem High Throughput Screen. https://doi.org/10.2174/1386207325666210924104036
Mali SN, Pandey A (2021) Molecular modeling studies on 2, 4-disubstituted imidazopyridines as anti-malarials: atom-based 3D-QSAR, molecular docking, virtual screening, in-silico ADMET and theoretical analysis. J Comput Biophys Chem 20(03):267–282
Mali SN, Tambe S, Pratap AP, Cruz JN (2022) Molecular modeling approaches to investigate essential oils (volatile compounds) interacting with molecular targets. In: Santana de Oliveira M (ed) Essential oils. Springer, Cham. https://doi.org/10.1007/978-3-030-99476-1_18
Mali SN, Pandey A (2022) Synthesis of new hydrazones using a biodegradable catalyst, their biological evaluations and molecular modeling studies (Part II). J Comput Biophys Chem. https://doi.org/10.1142/S2737416522500387
Rodrigues T, Moreira R, Lopes F (2011) New hope in the fight against malaria? Future Med Chem 3(1):1–3
Narender T, Tanvir K, Rao MS, Srivastava K, Puri SK (2005) Prenylated chalcones isolated from Crotalaria genus inhibits in vitro growth of the human malaria parasite Plasmodium falciparum. Bioorg Med Chem Lett 15(10):2453–2455
Ramalhete C, da Cruz FP, Mulhovo S, Sousa IJ, Fernandes MX, Prudêncio M, Ferreira MJU (2014) Dual-stage triterpenoids from an African medicinal plant targeting the malaria parasite. Bioorg Med Chem 22(15):3887–3890
Pérez B, Teixeira C, Gomes AS, Albuquerque IS, Gut J, Rosenthal PJ, Prudêncio M, Gomes P (2013) In vitro efficiency of 9-(N-cinnamoylbutyl) aminoacridines against blood-and liver-stage malaria parasites. Bioorg Med Chem Lett 23(3):610–613
World Health Organization (2020) WHO technical brief for countries preparing malaria funding requests for the Global Fund (2020–2022)
Wellems TE, Panton LJ, Gluzman IY, Do Rosario VE, Gwadz RW, Walker-Jonah A, Krogstad DJ (1990) Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature 345(6272):253–255
Ross LS, Fidock DA (2019) Elucidating mechanisms of drug-resistant Plasmodium falciparum. Cell Host Microbe 26(1):35–47
Tisnerat C, Dassonville-Klimpt A, Gosselet F, Sonnet P (2022) Antimalarial drug discovery: from quinine to the most recent promising clinical drug candidates. Curr Med Chem 29(19):3326–3365
Hodoameda P, Duah-Quashie NO, Quashie NB (2022) Assessing the roles of molecular markers of antimalarial drug resistance and the host pharmacogenetics in drug-resistant malaria. J Trop Med
Wright CW (2005) Traditional antimalarials and the development of novel antimalarial drugs. J Ethnopharmacol 100(1–2):67–71
Lopatriello A, Sore H, Habluetzel A, Parapini S, D’Alessandro S, Taramelli D, Taglialatela-Scafati O (2019) Identification of a potent and selective gametocytocidal antimalarial agent from the stem barks of Lophira lanceolata. Bioorg Chem 93:103321
Omar F, Tareq AM, Alqahtani AM, Dhama K, Sayeed MA, Emran TB, Simal-Gandara J (2021) Plant-based indole alkaloids: a comprehensive overview from a pharmacological perspective. Molecules 26(8):2297
Awad HM, Boersma MG, Boeren S, van Bladeren PJ, Vervoort J, Rietjens IM (2001) Structure−activity study on the quinone/quinone methide chemistry of flavonoids. Chem Res Toxicol 14(4):398–408
Yoshida K, Oyama KI, Kondo T (2012) Chemistry of flavonoids in color development. Recent Adv Polyphenol Res 3:99–129
Harborne JB (1967) Comparative biochemistry of the flavonoids-IV: correlations between chemistry, pollen morphology and systematics in the family plumbaginaceae. Phytochemistry 6(10):1415–1428
Vaughan AM, O’Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, Cowman AF, Kappe SH (2009) Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol 11(3):506–520
Schrader FC, Glinca S, Sattler JM, Dahse HM, Afanador GA, Prigge ST, Lanzer M, Mueller AK, Klebe G, Schlitzer M (2013) Novel type II fatty acid biosynthesis (FAS II) inhibitors as multistage antimalarial agents. ChemMedChem 8(3):442–461
Baschong W, Wittlin S, Inglis KA, Fairlamb AH, Croft SL, Kumar TR, Fidock DA, Brun R (2011) Triclosan is minimally effective in rodent malaria models. Nat Med 17(1):33–34
Tasdemir D (2006) Type II fatty acid biosynthesis, a new approach in antimalarial natural product discovery. Phytochem Rev 5(1):99–108
Van Ooij C (2009) The fatty liver stage of malaria parasites. Nat Rev Microbiol 7(2):95–95
Rudrapal M, Chetia D (2017) Plant flavonoids as potential source of future antimalarial leads. Syst Rev Pharm 8(1):13
Tasdemir D, Lack G, Brun R, Rüedi P, Scapozza L, Perozzo R (2006) Inhibition of Plasmodium falciparum fatty acid biosynthesis: evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. J Med Chem 49(11):3345–3353
Jadhav BS, Yamgar RS, Kenny RS, Mali SN, Chaudhari HK, Mandewale MC (2020) Synthesis, in silico and biological studies of thiazolyl-2h-chromen-2-one derivatives as potent antitubercular agents. Curr Comput Aided Drug Des 16(5):511–522
Jejurkar VP, Mali SN, Kshatriya R, Chaudhari HK, Saha S (2019) Synthesis, antimicrobial screening and in silico appraisal of iminocarbazole derivatives. ChemistrySelect 4(32):9470–9475
Anuse DG, Mali SN, Thorat BR, Yamgar RS, Chaudhari HK (2020) Synthesis, SAR, in silico appraisal and anti-microbial study of substituted 2-aminobenzothiazoles derivatives. Curr Comput Aided Drug Des 16(6):802–813
Desale VJ, Mali SN, Chaudhari HK, Mali MC, Thorat BR, Yamgar RS (2020) Synthesis and anti-mycobacterium study on halo-substituted 2-aryl oxyacetohydrazones. Curr Comput Aided Drug Des 16(5):618–628
Thorat BR, Mali SN, Rani D, Yamgar RS (2021) Synthesis, in silico and in vitro analysis of hydrazones as potential antituberculosis agents. Curr Comput Aided Drug Des 17(2):294–306
Kapale SS, Mali SN, Chaudhari HK (2019) Molecular modelling studies for 4-oxo-1, 4-dihydroquinoline-3-carboxamide derivatives as anticancer agents. Med Drug Discov 2:100008
Mali SN, Pandey A (2022) Balanced QSAR and molecular modeling to identify structural requirements of imidazopyridine analogues as anti-infective agents against trypanosomiases. J Comput Biophys Chem 21(01):83–114
Desale VJ, Mali SN, Thorat BR, Yamgar RS (2021) Synthesis, admetSAR predictions, DPPH radical scavenging activity, and potent anti-mycobacterial studies of hydrazones of substituted 4-(anilino methyl) benzohydrazides (Part 2). Curr Comput Aided Drug Des 17(4):493–503
Kshatriya R, Shelke P, Mali S, Yashwantrao G, Pratap A, Saha S (2021) Synthesis and evaluation of anticancer activity of pyrazolone appended triarylmethanes (TRAMs). Chem Sel 6(24):6230–6239
Mali SN, Pandey A (2021) Multiple QSAR and molecular modelling for identification of potent human adenovirus inhibitors. J Indian Chem Soc 98(6):100082
Mali SN, Pandey A, Thorat BR, Lai CH (2022) Multiple 3D-and 2D-quantitative structure–activity relationship models (QSAR), theoretical study and molecular modeling to identify structural requirements of imidazopyridine analogues as anti-infective agents against tuberculosis. Struct Chem 33(3):679–694
Nagre DT, Mali SN, Thorat BR, Thorat SA, Chopade AR, Farooqui M, Agrawal B (2021) Synthesis, in-silico potential enzymatic target predictions, pharmacokinetics, toxicity, anti-microbial and anti-inflammatory studies of bis-(2-methylindolyl) methane derivatives. Curr Enzym Inhib 17(2):127–143
Ghosh S, Mali SN, Bhowmick DN, Pratap AP (2021) Neem oil as natural pesticide: Pseudo ternary diagram and computational study. J Indian Chem Soc 98(7):100088
Bhosale D, Mali SN, Thorat BR, Wavhal SS, Bhagat DS, Borade RM (2022) Synthesis, molecular docking and in-vitro antimycobacterial studies on N’-arylidene-4-nitrobenzohydrazides. Recent Adv Anti-Infect Drug Discov 2022:17. https://doi.org/10.2174/1570193X19666220531154544
Nagre DT, Thorat BR, Mali SN, Farooqui M, Agrawal B (2021) Experimental and computational insights into bis-indolylmethane derivatives as potent antimicrobial agents inhibiting 2, 2-dialkylglycine decarboxylase. Curr Enzym Inhib 17(3):204–216
Thorat Bapu R, Mali Suraj N, Wagh Rahul R, Yamgar Ramesh S (2022) Synthesis, molecular docking, antioxidant, anti-TB, and potent MCF-7 anticancer studies of novel aryl-carbohydrazide analogues. Curr Comput Aided Drug Des. https://doi.org/10.2174/1573409918666220610162158
Lim SS, Kim HS, Lee DU (2007) In vitro antimalarial activity of flavonoids and chalcones. Bull Korean Chem Soc 28(12):2495–2497
Oliveira FQ, Andrade-Neto V, Krettli AU, Brandão MGL (2004) New evidences of antimalarial activity of Bidens pilosa roots extract correlated with polyacetylene and flavonoids. J Ethnopharmacol 93(1):39–42
de Monbrison F, Maitrejean M, Latour C, Bugnazet F, Peyron F, Barron D, Picot S (2006) In vitro antimalarial activity of flavonoid derivatives dehydrosilybin and 8-(1; 1)-DMA-kaempferide. Acta Trop 97(1):102–107
Studio D (2008) Discovery studio. Accelrys [2.1]
Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, Madhusoodanan N, Kolesnikov A, Lopez R (2022) Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. https://doi.org/10.1093/nar/gkac240
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350
Studer G, Rempfer C, Waterhouse AM, Gumienny G, Haas J, Schwede T (2020) QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 36:1765–1771
Gopalakrishnan K, Sowmiya G, Sheik SS, Sekar K (2007) Ramachandran plot on the web (2.0). Protein Peptide Lett 14(7):669–671
Land H, Humble MS (2018) YASARA: a tool to obtain structural guidance in biocatalytic investigations. Protein engineering. Springer, Berlin, pp 43–67
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(suppl 2):W407–W410
SAVESv6.0—structure validation server, UCLA-DOE LAB. https://saves.mbi.ucla.edu/
Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27(1):293–315
Morris GM et al (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
ACD/ChemSketch (2022) Version 2021.2.1. Advanced Chemistry Development, Inc., Toronto, ON, Canada, www.acdlabs.com
Hanwell MD et al (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminf 4(1):1–17
O’Boyle NM et al (2011) Open Babel: an open chemical toolbox. J Cheminf 3(1):1–14
Tian W, Chen C, Lei X, Zhao J, Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46(W1):W363–W367
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ (2021) The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49(D1):D605–D612
Trajtenberg F, Altabe S, Larrieux N, Ficarra F, de Mendoza D, Buschiazzo A, Schujman GE (2014) Structural insights into bacterial resistance to cerulenin. FEBS J 281(10):2324–2338
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):1–13
Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, Li W, Liu G, Tang Y (2019) admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35(6):1067–1069
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25
Du J, Sun H, Xi L, Li J, Yang Y, Liu H, Yao X (2011) Molecular modeling study of checkpoint kinase 1 inhibitors by multiple docking strategies and prime/MM–GBSA calculation. J Comput Chem 32(13):2800–2809
Krieger E, Vriend G (2015) New ways to boost molecular dynamics simulations. J Comput Chem 36(13):996–1007
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2(9):1511–1519
Bowie JU, Lüthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253(5016):164–170
Baell JB, Nissink JWM (2018) Seven year itch: pan-assay interference compounds (PAINS) in 2017 utility and limitations. ACS Chem Biol 13(1):36–44
Dong YW, Liao ML, Meng XL, Somero GN (2018) Structural flexibility and protein adaptation to temperature: molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc Natl Acad Sci 115(6):1274–1279
Desmond M (2022) Schrodinger. LLC, NY
Acknowledgements
Authors of this article are thankful to Dept. Of Pharmaceutical Sciences, Birla Institute of Technology, Mesra for their facility and academic support. SM also wish to thank the BIT, Mesra for the provision of IRF for year 2022, session SP.22.
Funding
None to report.
Author information
Authors and Affiliations
Contributions
All authors of this manuscript contributed equally.
Corresponding authors
Ethics declarations
Conflict of Interest
Not applicable.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pandey, A., Shyamal, S.S., Shrivastava, R. et al. Inhibition of Plasmodium falciparum Fatty Acid Biosynthesis (FAS-II Pathway) by Natural Flavonoids: A Computer-Aided Drug Designing Approach. Chemistry Africa 5, 1469–1491 (2022). https://doi.org/10.1007/s42250-022-00449-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-022-00449-7