
Abstract
The protein complex crystallographic beamline BL19U1 at the Shanghai Synchrotron Radiation Facility is one of the five beamlines dedicated to protein sciences operated by National Facility for Protein Science (Shanghai, China). The beamline, which features a small-gap in-vacuum undulator, has been officially open to users since March 2015. This beamline delivers X-ray in the energy range 7–15 keV. With its high flux, low divergence beam and a large active area detector, BL19U1 is designed for proteins with large molecular weight and large crystallographic unit cell dimensions. Good performance and stable operation of the beamline have allowed the number of Protein Data Bank (PDB) depositions and the number of articles published based on data collected at this beamline to increase steadily. To date, over 300 research groups have collected data at the beamline. More than 600 PDB entries have been deposited at the PDB (www.pdb.org). More than 300 papers have been published that include data collected at the beamline, including 21 research articles published in the top-level journals Cell, Nature, and Science.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Human health is one of the major concerns of modern society, and protein science has made a major contribution to it. To understand the detailed mechanisms by which macromolecules carry out their function in different biological processes, it is important to study the three-dimensional structure of these macromolecules [1]. These studies have greatly stimulated the study of protein structure in recent years in China and resulted in a huge expansion of the research community [2]. With more than 300 research groups in the protein crystallography field alone, such studies are producing high demand on the various advanced techniques and tools needed for the research on protein science. An advanced and comprehensive platform for protein science research, the National Facility for Protein Science (NFPS), was built at Shanghai a few years ago and officially opened to users in July 2015. A remarkable feature of this facility is the integration of versatile techniques, including nuclear magnetic resonance (NMR), electron microscopy (EM), atomic force microscopy (AFM), high-resolution optical microscopy, and the five beamlines at the Shanghai Synchrotron Radiation Facility (SSRF).
BL19U1 is one of the five beamlines operated by the NFPS at the 3.5 GeV storage ring at SSRF [3]. Along with BL18U1 and BL17B1, there are now three more beamlines serving macromolecular crystallography at the third-generation synchrotron X-ray source in mainland China. BL19U1 is designed to resolve the crystal structures of protein complexes and assemblies, especially those with large molecular weight and large unit cell dimensions. The large cell parameters in real space induce fine small reciprocal spacing, and the many diffraction points are difficult to distinguish. To improve the resolution between individual points, the divergence of the incident X-ray beam should be reduced as far as possible. Meanwhile, weak diffraction and radiation sensitivity are characteristics of large protein complex crystals. There is high demand for crystallography beamlines that can study these kinds of macromolecular structures [4]. The highly collimated X-ray will improve the diffraction data in the following ways: It will reduce the broadening of the diffraction spot, thereby improving the signal-to-noise ratio, and this will have the effect of increasing the resolution of the crystal structure that is being determined, especially for complex macromolecular crystals [5]. Reducing X-ray beam divergence is one of the major factors that should be considered for resolving a crystal structure with a large unit cell.
The pilot run of BL19U1 started in September 2014, and it was officially opened to the first users in March 2015. Access to BL19U1 is through the Sharing Service Platform of the Chinese Academy of Sciences (CAS) Large Research Infrastructure (http://lssf.cas.cn/). To date, BL19U1 has served more than 300 research groups in China and more than 600 Protein Data Bank (PDB) entries have been deposited from research using this beamline. As of July 2019, more than 300 papers had been published with data collected at this beamline, including 21 research articles published in the top-level journals Cell, Nature, and Science.
2 Beamline overview
The protein complex beamline is composed of the undulator source, the front end, the optical elements, and the experimental station. The photon source for BL19U1 is a small-gap in-vacuum undulator, which shares one straight section with the Bio-SAXS beamline BL19U2 [6] due to the limited number of straight sections in the storage ring. The two undulators of BL19U1 and BL19U2 are canted by 6 mrad to give enough horizontal beam separation between the two beamlines to provide high brilliance and tunable energy from 7 to 15 keV for both end stations [7]. The SSRF-made in-vacuum undulator has 80 periods and 2.5 cm period length. The RMS source size (FWHM) is 380 µm [horizontal (H)] × 25 µm [vertical (V)], and the divergence of central cone (FWHM@12 keV) is 80 µrad (H) × 22 µrad (V). The energy change is independent with BL19U1 and BL19U2. The optics elements are designed to provide state-of-the-art X-ray, to accommodate a wide range of measurement needs, and to maximize the availability of beamtime for the two beamline users. The key accelerator parameter that influences the design of the beamline is the canting angle between the two undulator sources, because this defines the separation between the two undulator beams along the sector length. The canted angle is enough to allow the placement of a first optical element for one of the beams without disturbing the other beam. For the two canted beamlines, substantial space is needed by the optical elements, such as monochromator and focusing mirror, underscoring the importance of providing adequate separation between the two undulator beams at their locations. The other undulator beam passes through this monochromator or mirror chamber undisturbed and continues farther downstream.
2.1 Characteristics of the beamline source
An intense and well-collimated source of X-ray is ideal for X-ray crystallography applications. An undulator is highly preferred due to its advantages of high brightness and low divergence. These turn out to be among the greatest merits for synchrotron-based protein crystallography, especially to determine atomic resolution structures of large macromolecular complexes. The electron energy of the SSRF storage ring is 3.5 GeV, so the higher harmonics in the spectrum of a short period in-vacuum undulator can provide high-brightness X-ray in the 7–15 keV range. The intensity of the beam against energy (scanned) was between 9 and 15 keV with a gap at 9.5 mm and the current of the storage ring at 180 mA (Fig. 1). Considering that undulators of shorter period length and larger period number can produce radiation of greater brightness, the U20 was used as a compromise between the desired brightness and wavelength tunability. The undulator parameters were optimized to cover the required energy range (7–15 keV) with easy tuning and with a very brilliant beam of around 1019 photons/(mm2 mrad2 s).

Intensity of the beam against energy was scanned between 9 and 15 keV with the gap at 9.5 mm and the current of storage ring at 180 mA
2.2 Optical elements
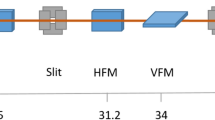
The schematic beamline optical layout of the BL19U1 is shown in Fig. 2. The beamline is 40.8 m long from the source to the sample position. The beamline front end finishes at the shielding wall, 17.5 m from the source. A collimator is installed at the exit of the front end to absorb the bremsstrahlung radiation. Water-cooled white-beam slits are used to limit the acceptance cone to 0.10 mrad (H) × 0.05 mrad (V).

Schematic optical layout of the BL19U1 beamline with the distances of major components from the source
2.2.1 Double-crystal monochromator (DCM)
The liquid-nitrogen-cooled double-crystal monochromator (DCM), 22.5 m downstream from the photon source, provides tunable energy in the range 7–15 keV with a fixed offset of 25 mm. Right after the DCM, another collimator is installed to further narrow the dimensions of the bremsstrahlung radiation. A flat reflection mirror is placed 25.5 m from the source to separate the two beams from the same straight section. With the incident gazing angle of 3.5 mrad, this reflection mirror increases the horizontal distance between the two beams.
The two crystals are in the (n, − n) achromatic arrangement. The exit beam height is fixed with respect to the incoming beam during energy scans. The beam height offset is set at 25 mm. Second crystal translation in the direction normal to the crystal surface is necessary to keep the beam-offset fixed. Water-cooled slits are set before the DCM, which absorbs a large part of the heat power and reduces the total power on the first crystal to about 170 W. However, the density of heat power absorbed by the first crystal is still very high and the maximum power density is about 18.4 W/mm2, which would cause a serious distortion of the crystal if it were not properly cooled. The crystal surface slope error caused by heat distortion can be constrained by liquid-nitrogen side cooling, which has little effect on the energy resolution.
The primary axis of Bragg rotation passes through the center of the first crystal reflecting face, perpendicular to the beam axis, so that the incident white beam is always centered on the axis of rotation. Due to the high-power loading involved, the first crystal is cryogenically cooled to dissipate the photon energy that is absorbed. The second crystal is indirectly cryogenically cooled to ensure perfect matching of the crystal plane dimensions. The double-crystal monochromator is located 22.5 m from the source, where the horizontal separation between the two undulator beams is 97.4 mm. This allows the beam from BL19U2 to pass undisturbed through the monochromator assembly.
2.2.2 Focusing mirror
The rhodium (Rh)-coated focusing mirror at 33.1 m from the source focuses the monochromatic beam from the DCM onto the sample position horizontally and vertically. The focusing mirror has a toroidal surface obtained from bending a cylinder about a horizontal axis and is oriented to reflect the beam upward on the vertical plane. The mirror focuses the beam horizontally using the sagittal radius and focuses the beam vertically using the dynamically bending radius, which is adjustable to optimize the focus. The grazing incident angle is 3.5 mrad, and the cutoff energy is 22 keV. Typically, the beam can be focused to 106 μm (H) × 41 μm (V) (FWHM and at 12.4 keV) at the sample position (Fig. 3). The focusing mirror has a toroidal surface obtained from bending a cylinder about a horizontal axis. The mirror is placed horizontally and reflects the beam upward in the vertical plane.

Focused beam size of BL19U1 beamline. a Horizontal beam size with blade scan at 12.4 keV; b vertical beam size with blade scan at 12.4 keV; c full view of the focused beam at 12.4 keV; d focused beam defined by a fixed aperture with 50 µm diameter
For this beamline, the distance from the focusing mirror to the focus point is relatively long. The surface figure errors contribute to a focused spot size roughly proportional to this distance. A critical physical parameter determining the focused beam size is the residual figure errors of the toroidal mirror surfaces. The RMS slope error of the focusing mirror is better than 1.5 μrad along the meridional direction.
2.2.3 Beamline properties
The properties of the beamline BL19U1 are shown in Table 1. The energy of the beamline is tunable over the range 7–15 keV. The energy resolution is less than 2 × 10−4 at 12.4 keV. The size for the focused beam at the sample position is about 106 µm (H) × 41 µm (V) (FWHM and at 12.4 keV). Although the normally used beam size is about 50 µm × 50 µm defined with a fixed aperture (Fig. 3), the flux of a focused beam is about 1.5 × 1012 ph/s at 12.4 keV with a 300 mA synchrotron beam current. The value of the divergence is about 77 μrad (H) × 54 μrad (V) at 12.4 keV, and it does not change significantly across the entire energy range. The divergence can be further reduced by adjusting the mono-beam slits before the focusing mirror.
2.3 Experimental station
The experimental station stands at the end of the protein complex crystallography beamline. This is because this beamline is designed to interpret the structure of complex biological samples, such as large proteins, protein complexes, and assemblies in particular, and membrane proteins with high mass weight (MW) and large unit cells (cell parameter > 500). The large cell parameters in real space produce small reciprocal spacing, resulting in many closely spaced diffraction points that cannot always be distinguished. In the experimental station, long sample-to-detector distance up to 900 mm can be achieved and a large area detector with 423.6 mm × 434.6 mm active area is used to improve this spatial resolution. Besides the large active area detector, the experimental station of this beamline also has devices similar to those found at other macromolecular crystallography beamlines, as shown in Figs. 2 and 4, for example a beryllium window, beam-position monitors, X-ray attenuator, slits, and ion chambers. They also include data acquisition and accessory devices such as detectors, fast shutters, detector support systems, sample alignment microscope, goniometer, sample cryocooler, and fluorescence detector. The layout of the experimental station is shown in Fig. 4. It has been designed to be suitable for multiple and single-wavelength anomalous diffraction (MAD and SAD) experiments in the energy range 7–15 keV. The monochromatic beam, which exits from a beryllium window upstream of the experimental station, can be attenuated using two XIA PF4 filter boxes with 6 aluminum foils of various thicknesses. The experimental station of BL19U1 is equipped with a Maatel/EMBL MD2 microdiffractometer [8], a Pilatus3 × 6 M detector [9], a fully automated sample changer (ACTOR, Rigaku), a cryostream system (Cryostream700, Oxford), and an X-ray fluorescence detector (Vortex-EX, Hitachi).

View of BL19U1 end station and sample environment. a Photographs of the experimental station of BL19U1 showing the Pilatus detector, MD2 microdiffractometer and ACTOR robot; b BL19U1 sample environment, showing the cryocooler, on-axis camera, beamstop, fluorescence detector and goniometer
The MD2 microdiffractometer is placed on a five-axis table (Bruker ASC) for beam alignment. This is a highly integrated piece of equipment designed for X-ray macromolecular crystallography experiments. The MD2 microdiffractometer includes a high-precision goniometer axis with omega rotation, a smart magnet nozzle, an on-axis viewing system, an XYZ alignment table, beam defining aperture, capillary–beamstop, scintillator–photodiode, and lighting system. The maximum rotation speed of the air-bearing goniometer axis is 120°/s. The magnetic force of the smart magnet nozzle can be turned off, which is quite useful for mounting by hand. The on-axis viewing camera with zoom lens and a transmission backlight are integrated in the MD2, which makes it possible to visualize and center small crystals of only a few microns size. A capillary–beamstop and scintillator–photodiode are moved down under the cover to protect them from being touched by users or a robot gripper. Beam-shaping apertures can be chosen to define the beam (20 µm, 50 µm, or 100 µm). The MD2 control rack with a Delta Tau UMAC control system is used to control the movement of the motors.
The sample environment is cooled by an Oxford Cryostream700, and the temperature of 100 K is typically used for macromolecular crystal studies. The sample environment temperature is also adjustable within the temperature range 80–350 K, which could also be used for studying the temperature effects on some small chemical molecular crystal structures.
The diffraction data are collected using a Pilatus3 × 6 M detector mounted on a three-axis support stage (Bruker ASC), which allows the sample-to-detector distance to be varied from 115 to 900 mm (resolution 0.95–4.1 Å at 12.4 keV) and a maximum 2θ angle of 30°. The Pilatus 3 × 6 M detector consists of 60 small modules with gaps between them. Although the inter-module gap takes about 8.5% of total area, the data processing is not affected significantly. The thickness of the Si sensor is 450 µm, providing high quantum efficiency of about 47.5% at 17.5 keV and 97.9% at 8 keV. The dynamic range is as high as 20 bits, which is quite beneficial for improving the resolution of diffraction data. The readout time is 0.95 ms. With the quick readout time, the shutter-free data collection method has normally been used. The frame rate of the detector can be as high as 100 Hz, which improves the efficiency of data collection. The detector can trigger the shutter with a 5 V TTL external trigger signal. The detector can also be triggered by the 5 V TTL external signal.
The sample can be mounted manually by the user, but usually the samples are mounted with the ACTOR automated sample changer, which is much more efficient. It takes about 1 min to remove a crystal sample from the goniometer and replace it with a new one. The ACTOR automated sample changer includes a Mitsubishi robot arm with a pin gripper and liquid-nitrogen Dewar with autofill and temperature control system. A barcode reader is installed for scanning the crystal information, which is useful for industrial users and high-throughput crystal screening. The ACTOR automated sample changer allows the use of universal pucks, and the capacity of the storage Dewar is 80 samples (5 pucks) at a time. Considering the moving range of the support table, a standard pin with the length of 18 mm is recommended.
To improve the performance of the beamline and protect the devices in the experimental hutch, an air-conditioning system has been installed. The temperature in the experimental hutch is 25 ± 1 °C, which is controlled by the air-conditioning system. The humidity of the experimental hutch is roughly controlled by an industrial dehumidifier, and an upgrade of the humidity control is being considered.
In addition, a Vortex-EX multi-element fluorescence detector is available for MAD scans and X-ray excitation, which can be used for setting the optimal energy before MAD/SAD data collection.
2.4 Experiment control and data analysis
Users are quite familiar with the SSRF’s revised version of Blu-Ice [10, 11], which has also been implemented on BL19U1 so that users will not feel any operational differences among macromolecular crystallographic beamlines (BL17U1 [12], BL17B1, BL18U1, and BL19U1) at the SSRF. Most of the important equipment is controlled by the software supplied by individual manufacturers. Movements of all the goniometer motors are controlled by MD2 control software. The on-site viewing camera and sample centering system are implemented using MD2 control software. Changing the beam size by choice of the beam aperture is also implemented using the MD2 control software, which also provides the functions of controlling the shutter, sample loop autocentering, raster scan, and beam adjustment. The MD2 control software communicates with the motors through the PMAC control system. The data collection from the Pilatus 3 × 6 M detector is controlled by Camserver and TVX. TVX is data acquisition and control software that contains an analysis tool and user interface. It communicates with Camserver, which is used for controlling the hardware of the Pilatus detector system, through a TCP/IP connection. The ACTOR autosample changer is controlled by CameraMan, which controls movement of the robot arm and the liquid-nitrogen level of the sample Dewar. All the hardware control software is implemented through Blu-Ice. The communication between Blu-Ice and hardware control software is through a DHS/DCS system. The Experimental Physics and Industrial Control System (EPICS) is used to control the movement of all the motors used for the optical and support systems. It is also used for signal readout of the beamline equipment following the protocols developed by the SSRF [13]. In addition, commonly used protein crystallographic software is available, including HKL3000 [14], XDS [15], CCP4 suite [16], PHENIX [17], and SHELX [18]. There is a storage server with 20 TB capacity located on site. Owing to storage limits, diffraction images are currently stored on the server for 1 month.
3 Facility access
BL19U1 provides about 4000 h of beamtime per year to general users following the SSRF operational schedule. There are two calls for proposals per year (March and September). Users can apply for proposals on the Web site of the sharing service platform of the CAS large research infrastructure (http://lssf.cas.cn/). The BL19U1 beamline can be found in the facility catalogue of the National Facility for Protein Science. The proposals are first reviewed by beamline staff to determine the beamline suitability and technical feasibility of the research plan. Then, the projects will be sent for peer review of scientific merit by members of a committee. Approved proposals are valid for 2 years. Beamtime is scheduled upon receipt of a beamtime request of a user against an active proposal, about one or 2 months in advance. Each group of users will receive on-site operational and safety training before their experiment. For safety issues, a training course must be taken and the training examination must be passed on the Web site (http://ssrf.sinap.ac.cn/proposals/) before on-site experiments. Users can get the beamtime arrangement information on the same Web site. For high credit users with high-level paper publication and high impacts, another mode called awarded beamtime could be applied. An awarded project is set based on user output after using this beamline. The total project beamtime awarded is about 10–15% of the total user time; the criteria of a reward project depend on the application situation, and they are applied in one round. Users must acknowledge the beamline or published PDBs researched at BL19U1 to get an award project. Emergency beamtime can be applied for by users with high-level papers under review, when higher quality data are needed for paper publication. In addition to academic users, industrial users are also welcome at the BL19U1 beamline. About 10% of the beamtime is reserved for industrial users. By now, about 10 industrial users have collected data at BL19U1. For higher efficiency, one beamtime shift is 6 h.
4 Productivity
BL19U1 has been officially opened to users since March 2015 and has been productive during the past 4 years. With the support of this beamline, many cutting-edge scientific research papers in the field of structural biology have been published. As of July 2019, 21 research articles had been published in the top-level journals Cell, Nature, and Science with data collected at this beamline [19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39]. Figure 5 shows the statistical data on PDB depositions and article publications up to May 22, 2019. The PDB statistic data are from the BioSync Web site (http://biosync.sbkb.org).

Structures and articles published by users since opening and up to July 20, 2019
4.1 Large cell constants: influenza A virus hemagglutinin H4
Influenza A viruses (IAVs) are important pathogens of humans and many other species. The classification of IAVs is based on two surface glycoproteins/hemagglutinin (HA) and neuraminidase (NA). Since the first novel low-pathogenicity avian influenza virus (LPAIV) H7N9 outbreak occurred in March 2013 in China, LPAIVs have caused global concern for public health. It has been determined that H4 LPAIV is widespread in wild and domesticated avian species in Asian, European, and North American countries. In addition to avian species, the H4 virus has been naturally isolated from mammalian animals including seals and pigs. Studies show that the H4 viruses have the potential to cause human pandemics. The research group of George F. Gao have resolved the structures of H4 hemagglutinin and its complex with avian or human receptor analogs, which crystallized in H32 space group with large cell dimensions of a = 100 Å, b = 100 Å, and c = 685 Å (the data collection statistics are in Table 2) [40]. The molecular basis for the receptor binding properties of H4 viruses has been evaluated using two representative virus isolates: avian H4N6 and swine H4N6. The structural basis for the receptor binding preference changes has been demonstrated by the crystal structures of the avian H4 and different mutants in complex with receptor analogs.
4.2 Several high-resolution cases
Hepatoma-derived growth factor (HDGF)-related proteins (HRPs) include HDGF, HRP1-3, and lens epithelium-derived growth factor (LEDGF), all of which are characterized by a conserved N-terminal PWWP domain and a variable C-terminal region. The PWWP domain, also known as the HATH domain, can bind both histone and DNA substrates. Zhanxin Wang Group have solved a series of crystal structures of the HRP3 PWWP domain in complex with various double-stranded DNAs with or without bound histone peptides. Among them, the apo form of the HRP3 PWWP domain crystallized in P212121 space group with the highest resolution of 0.951 Å (PDB entry: 6IIP) [41], which is also the highest resolution among structures from BL19U1. This study shows that HRP3 PWWP uses two adjacent surfaces to bind both DNA and histone substrates simultaneously and indicates that HRP3 PWWP is a new family of minor groove-specific DNA-binding proteins, which might help to elucidate further the biological impact of HRP3 recruitment on chromatin and its relationship with human pathogenesis.
DNA can form many diverse structures, including duplex, triplex, quadruplex, hairpin, and cruciform. In addition to the traditional types of base pairing, DNA is also capable of forming metallo-base pairs in the presence of metal ions by both artificial and natural nucleobases. Metal-mediated base pairs have great application potential in many research fields, including novel therapeutics development, genetic-code extension, and nanodevice design. A novel non-helical DNA crystal structure containing Ag1-mediated G:G and C:C base pairs has been reported [42], which was crystallized in P21212 space group with a high resolution of 0.98 Å (PDB entry: 5XJZ). These studies could not only help people understand the interactions between metal ions and nucleobases, but also provide a unique structural component for the rational design of new DNA nanodevices.
4.3 Different phasing methods
After inspecting all structures deposited with data collected on BL19U1 (related statistical data are from BioSync), there was one structure resolved by ab initio phasing, one structure by Fourier Synthesis, four structures resolved by multi-wavelength anomalous dispersion (MAD), 81 structures resolved by single-wavelength anomalous diffraction (SAD), and 452 structures by molecular replacement (MR). One using the ab initio method and one the SAD method are described below.
Collagen has a unique triple-helix conformation formed by three polypeptide chains and can be classified into two groups: the fibril-forming collagens and non-fibrillar collagens. A large number of naturally occurring interruptions in collagens have been observed, and they play key roles in molecular flexibility, collagen degradation, and ligand binding. The crystal structure of human Type XIX collagen shows a unique conformation [43], the structure of which was determined by an ab initio method ACORN [44] in the P21 space group at the resolution of 1.03 Å. The structural information could partly explain the different thermal stability between structures with different interruptions and could provide a structural basis for further functional studies.
The substance N6-methyladenosine (m6A) is an internal modification in messenger RNA and long noncoding RNA, which is commonly found in many species such as viruses, bacteria, yeast, plants, and mammalian animals. It affects RNA metabolism such as pre-mRNA processing, translation efficiency, transcript stability, and miRNA biogenesis, and could function in cell cycles, regulation of development, fate determination, and heat shock stress response. There are three different kinds of protein factors involved in the m6A modification functioning: the “writers”, the “erasers”, and the “readers”. The human METTL3 and METTL14 participate as “writer” in this modification. The METTL3-METTL14 complex crystal structure has been resolved by Ping Yin Group via SAD of bromide, in the P41212 space group with the resolution of 1.6 Å [19]. This structural information could help further investigation of the function and regulation of m6A.
4.4 Cutting-edge research
A series of protein and protein complex structures related to CRISPR–Cas systems have been resolved from data collected at BL19U1. The “clustered regularly interspaced short palindromic repeats” (CRISPR), together with CRISPR-associated (Cas) proteins, could protect bacteria and archaea against invading nucleic acids from phages and plasmids, which form the microbial adaptive immune system and which have potential applications in RNA technology. The structure of Cas1–Cas2-dual-forked DNA complexes has been reported, revealing how the protospacer DNA is sampled prior to insertion into the CRISPR loci in 2015 by the Yanli Wang Group [20]. The CRISPR–Cas systems are classified into two main groups: Those in Class 1 function as an effect complex composed of multiple subunits, and those in Class 2 function by single component effectors, comprising type II, type V, and type VI systems, among which Cas9, Cpf1, and C2c1 all have RNA-mediated DNA endonuclease activity. The crystal structure of Leptotrichia shahii C2c2-crRNA binary complex and apo form was reported in 2017, revealing that LshC2c2 exerts two different RNase activities through two independent active domains and provides a structural biological basis of great importance for studying the molecular mechanism of C2c2 to exert RNase activity [26]. The crystal structure of Leptotrichia buccalis (Lbu) Cas13a bound to crRNA, and its target RNA, was resolved in 2017 by the same group, as well as the cryo-EM structure of LbuCas13a-crRNA [28]. Cas13a is a type VI-A CRISPR–Cas RNA-guided RNA ribonuclease that can degrade invasive RNAs targeted by CRISPR RNA (crRNA). In late 2018, the structure of type III-A CRISPR-Csm complex in the presence of target RNAs was reported, which revealed a molecular basis for a unique mode of self/non-self discrimination [37]. Csm is a type III-A CRISPR–Cas interference complex, which is a crRNA-guided RNase that also has target RNA-dependent DNase and cyclic oligoadenylate (cOA) synthetase activities. These studies provide crucial insights into the mechanism of crRNA-meditated sequence-specific RNA cleavage, RNA target-dependent non-specific DNA cleavage, and cOA generation.
Several virus-related proteins and protein complexes have been reported, which shows how BL19U1 supports research related to critical public health events.
-
(1)
In 2016, the crystal structure of the primed Ebola virus glycoprotein in complex with domain C of the endosomal entry receptor Niemann-Pick C1 was published, which provided a structural basis for Ebola virus endosomal-receptor binding and revealed structural insights into filovirus fusion to the late endosome and a molecular basis for designing therapeutic inhibitors of viral entry [22].
-
(2)
In 2019, the same group reported the crystal structures of the mouse MXRA8 (mMXRA8) and the complex between human MXRA8 (hMXRA8) and the CHIKV E protein, which provides structural insights into how the Chikungunya virus binds to its receptor on human cells [39]. Chikungunya virus (CHIKV) is a type of arbovirus that causes fever, rash, and joint pain. It used to be concentrated in tropical and subtropical regions of Africa and Asia. With the factors of global warming and modern transportation, an epidemic involving this virus has been continuously expanding and spreading in recent years. CHIKV virus infection could cause acute and chronic peripheral joint pain and arthritis and may cause death in severe cases. Because of the long-lasting joint pain and large-scale epidemic outbreaks, spread of the CHIKV virus has brought widespread concern. Based on its structures, it is proposed that MXRA8 is a novel Ig-like receptor with a unique topology and inter-domain assembly, which not only helps in investigating the interactive mechanism between CHIKV E and MXR8, but also provides theoretical guidance for the development of novel vaccines and broad-spectrum neutralizing antibodies, as well as new targets for antiviral drug design.
-
(3)
In 2019, the crystal structure of Mycobacterium smegmatis MmpL3 was determined, alone and with MmpL3 in complex with four tuberculosis drug candidates [38]. MmpL3 is an important drug discovery target in anti-tuberculosis research. This research reveals the new molecular mechanism of innovative drugs to kill bacteria, and opens a new path for future development of new antibiotics.
5 Discussion and conclusion
Before the NFPS beamlines began operation, there was only one macromolecular crystallographic beamline on the third-generation synchrotron open to users in mainland China. Beamtime was far from meeting the demand of the developing macromolecular crystallography study groups. The new operation of the NFPS beamlines eased the shortage of crystallographic beamtime in China and made it possible for more research groups to collect data close to their laboratories.
With its parameters of high flux and low divergence, the protein complex crystallographic beamline BL19U1 is mainly designed for proteins with large molecular weight and cell dimensions. Highly integrated and high-quality devices such as the MD2 microdiffractometer, Pilatus3 × 6 M, and ACTOR sample changer were used to equip the beamline, which improved its performance and efficiency.
Based on the high-quality and high performance of the beamline, many high-level research papers have been published with data collected at the BL19U1 beamline. Although the beamline is a modern macromolecular crystallography beamline that has most of the equipment and automation a scientist would expect, many new techniques still need to be developed. To improve the performance of the beamline, a temperature and humidity control system will be installed. A method for in situ data collection at room temperature is also under testing. It is important for drug discovery projects to design candidate drugs based on protein structure. To screen drug candidates automatically, the automaticity performance needs to be improved. We plan to develop automatic processing of macromolecular crystallography X-ray diffraction data, and a raster scan method will also be developed to automatically locate the crystal position in the loop. To promote continuously, the development of structural biology in China, the BL19U1 staff, will cooperate with crystallographers to improve the performance of BL19U1.
References
S.E. Thomas, V. Mendes, S.Y. Kim et al., Structural biology and the design of new therapeutics: from HIV and cancer to mycobacterial infections—a paper dedicated to John Kendrew. J. Mol. Biol. 429(17), 2677–2693 (2017). https://doi.org/10.1016/j.jmb.2017.06.014
D.C. Wang, Structural biology in China. Prog. Biochem. Biophys. 41(10), 944–971 (2014). https://doi.org/10.3724/Sp.J.1206.2014.00240
M. Jiang, X. Yang, H. Xu et al., Shanghai Synchrotron Radiation Facility. Chin. Sci. Bull. 54(22), 4171 (2009). https://doi.org/10.1007/s11434-009-0689-y
R.L. Owen, J. Juanhuix, M. Fuchs, Current advances in synchrotron radiation instrumentation for MX experiments. Arch. Biochem. Biophys. 602, 21–31 (2016). https://doi.org/10.1016/j.abb.2016.03.021
W.R. Wikoff, W. Schildkamp, J.E. Johnson, Increased resolution data from a large unit cell crystal collected at a third-generation synchrotron X-ray source. Acta Crystallogr. Sect. D 56(7), 890–893 (2000). https://doi.org/10.1107/S0907444900005941
N. Li, X. Li, Y. Wang et al., The new NCPSS BL19U2 beamline at the SSRF for small-angle X-ray scattering from biological macromolecules in solution. J. Appl. Crystallogr. 49(Pt 5), 1428–1432 (2016). https://doi.org/10.1107/S160057671601195X
H. Qin, Y. Zhao, N. Wang et al., Layout and operation of the undulator canted beamlines on BL19U at SSRF. Nucl. Tech. 39(11), 110101 (2016). https://doi.org/10.11889/j.0253-3219.2016.hjs.39.110101. (in Chinese)
A. Perrakis, F. Cipriani, J.C. Castagna et al., Protein microcrystals and the design of a microdiffractometer: current experience and plans at EMBL and ESRF/ID13. Acta Crystallogr. D Biol. Crystallogr. 55(Pt 10), 1765–1770 (1999). https://doi.org/10.1107/s0907444999009348
T. Loeliger, C. Bronnimann, T. Donath, et al., The new PILATUS3 ASIC with instant retrigger capability, in: 2012 IEEE Nuclear Science Symposium and Medical Imaging Conference Record (Nss/Mic) (2012), pp. 610–615. https://doi.org/10.1109/nssmic.2012.6551180
T.M. McPhillips, S.E. McPhillips, H.-J. Chiu et al., Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9(6), 401–406 (2002). https://doi.org/10.1107/S0909049502015170
Q. Wang, S. Huang, B. Sun et al., Control and data acquisition system for the macromolecular crystallography beamline of SSRF. Nucl. Tech. 35, 5–11 (2012). (in Chinese)
Q.S. Wang, K.H. Zhang, Y. Cui et al., Upgrade of macromolecular crystallography beamline BL17U1at SSRF. Nucl. Sci. Tech. 29, 68 (2018). https://doi.org/10.1007/s41365-018-0398-9
P. Liu, Y.N. Zhou, Q.R. Mi et al., EPICS-based data acquisition system on beamlines at SSRF. Nucl. Tech. 33, 415–419 (2010). (in Chinese)
W. Minor, M. Cymborowski, Z. Otwinowski et al., HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. D 62(8), 859–866 (2006). https://doi.org/10.1107/S0907444906019949
W. Kabsch, Automatic indexing of rotation diffraction patterns. J. Appl. Crystallogr. 21(1), 67–72 (1988). https://doi.org/10.1107/S0021889887009737
M.D. Winn, C.C. Ballard, K.D. Cowtan et al., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67(Pt 4), 235–242 (2011). https://doi.org/10.1107/S0907444910045749
P.D. Adams, P.V. Afonine, G. Bunkoczi et al., PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66(Pt 2), 213–221 (2010). https://doi.org/10.1107/S0907444909052925
G.M. Sheldrick, A short history of SHELX. Acta Crystallogr. A 64(1), 112–122 (2008)
X. Wang, J. Feng, Y. Xue et al., Structural basis of N(6)-adenosine methylation by the METTL3–METTL14 complex. Nature 534(7608), 575–578 (2016). https://doi.org/10.1038/nature18298
J. Wang, J. Li, H. Zhao et al., Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR–Cas systems. Cell 163(4), 840–853 (2015). https://doi.org/10.1016/j.cell.2015.10.008
L. Hu, J. Lu, J. Cheng et al., Structural insight into substrate preference for TET-mediated oxidation. Nature 527, 118 (2015). https://doi.org/10.1038/nature15713
H. Wang, Y. Shi, J. Song et al., ebola viral glycoprotein bound to its endosomal receptor Niemann-Pick C1. Cell 164(1–2), 258–268 (2016). https://doi.org/10.1016/j.cell.2015.12.044
Y. Li, J. Han, Y. Zhang et al., Structural basis for activity regulation of MLL family methyltransferases. Nature 530(7591), 447–452 (2016). https://doi.org/10.1038/nature16952
J. Ding, K. Wang, W. Liu et al., Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535(7610), 111–116 (2016). https://doi.org/10.1038/nature18590
M. Zeng, Y. Shang, Y. Araki et al., Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell 166(5), 1163–1175e12 (2016). https://doi.org/10.1016/j.cell.2016.07.008
L. Liu, X. Li, J. Wang et al., Two distant catalytic sites are responsible for C2c2 RNase activities. Cell 168(1–2), 121–134e12 (2017). https://doi.org/10.1016/j.cell.2016.12.031
D. Dong, M. Guo, S. Wang et al., Structural basis of CRISPR–SpyCas9 inhibition by an anti-CRISPR protein. Nature 546(7658), 436–439 (2017). https://doi.org/10.1038/nature22377
L. Liu, X. Li, J. Ma et al., The molecular architecture for RNA-guided RNA cleavage by Cas13a. Cell 170(4), 714–726e10 (2017). https://doi.org/10.1016/j.cell.2017.06.050
H. Li, R. Liefke, J. Jiang et al., Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 549(7671), 287–291 (2017). https://doi.org/10.1038/nature23881
H. Chen, J. Xue, D. Churikov et al., Structural insights into yeast telomerase recruitment to telomeres. Cell 172(1–2), 331–343e13 (2018). https://doi.org/10.1016/j.cell.2017.12.008
Y. Wang, M. Shi, H. Feng et al., Structural insights into non-canonical ubiquitination catalyzed by SidE. Cell 173(5), 1231–1243e16 (2018). https://doi.org/10.1016/j.cell.2018.04.023
M. Mompean, W. Li, J. Li et al., The structure of the necrosome RIPK1–RIPK3 core, a human hetero-amyloid signaling complex. Cell 173(5), 1244–1253e10 (2018). https://doi.org/10.1016/j.cell.2018.03.032
Y. Dong, Y. Mu, Y. Xie et al., Structural basis of ubiquitin modification by the Legionella effector SdeA. Nature 557(7707), 674–678 (2018). https://doi.org/10.1038/s41586-018-0146-7
Y. Yan, Q. Liu, X. Zang et al., Resistance-gene-directed discovery of a natural-product herbicide with a new mode of action. Nature 559(7714), 415–418 (2018). https://doi.org/10.1038/s41586-018-0319-4
L. Zhang, A. Serra-Cardona, H. Zhou et al., Multisite substrate recognition in Asf1-dependent acetylation of histone H3 K56 by Rtt109. Cell 174(4), 818–830e11 (2018). https://doi.org/10.1016/j.cell.2018.07.005
P. Zhou, Y. She, N. Dong et al., Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature 561(7721), 122–126 (2018). https://doi.org/10.1038/s41586-018-0433-3
L. You, J. Ma, J. Wang et al., Structure studies of the CRISPR–Csm complex reveal mechanism of co-transcriptional interference. Cell 176(1–2), 239–253e16 (2019). https://doi.org/10.1016/j.cell.2018.10.052
B. Zhang, J. Li, X. Yang et al., Crystal structures of membrane transporter MmpL3, an anti-TB drug target. Cell 176(3), 636–648e13 (2019). https://doi.org/10.1016/j.cell.2019.01.003
H. Song, Z. Zhao, Y. Chai et al., Molecular basis of arthritogenic alphavirus receptor MXRA8 binding to Chikungunya virus envelope protein. Cell 177(7), 1714–1724e13 (2019). https://doi.org/10.1016/j.cell.2019.04.008
H. Song, J. Qi, H. Xiao et al., Avian-to-human receptor-binding adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep. 20(5), 1201–1214 (2017). https://doi.org/10.1016/j.celrep.2017.07.028
W. Tian, P. Yan, N. Xu et al., The HRP3 PWWP domain recognizes the minor groove of double-stranded DNA and recruits HRP3 to chromatin. Nucleic Acids Res. 47(10), 5436–5448 (2019). https://doi.org/10.1093/nar/gkz294
H. Liu, F. Shen, P. Haruehanroengra et al., A DNA structure containing Ag(I)-mediated G:G and C:C base pairs. Angew. Chem. Int. Ed. Engl. 56(32), 9430–9434 (2017). https://doi.org/10.1002/anie.201704891
T. Xu, C.-Z. Zhou, J. Xiao et al., Unique conformation in a natural interruption sequence of type XIX collagen revealed by its high-resolution crystal structure. Biochemistry 57(7), 1087–1095 (2018). https://doi.org/10.1021/acs.biochem.7b01010
J.X. Yao, ACORN in CCP4 and its applications. Acta Crystallogr. A 58(11), 1941–1947 (2002). https://doi.org/10.1107/S0907444902016621
Acknowledgements
We thank the staff of the SSRF MX team for design, installation, and continuing collaboration, along with the assistance of the SSRF research support groups.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhang, WZ., Tang, JC., Wang, SS. et al. The protein complex crystallography beamline (BL19U1) at the Shanghai Synchrotron Radiation Facility. NUCL SCI TECH 30, 170 (2019). https://doi.org/10.1007/s41365-019-0683-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41365-019-0683-2