
Opinion statement
Purpose of review Systemic sclerosis (SSc) has been difficult to treat due to disease heterogeneity and an incomplete understanding of the molecular mechanisms underlying pathogenesis. Some patients show improvement on existing or experimental therapies while others show no significant response. Due to these challenges, clinical trials often do not meet their primary clinical endpoints. Here we review the data-driven genomewideapproaches that have recently been employed to characterize the molecular changes observed in SSc patients.
Recent findings Understanding an individual patient’s gene expression phenotype or genotype, a cornerstone of personalized medicine, has been proposed to help determine which therapies are most likely to treat their disease. Network and systems biology methods have been applied to the compendium of publicly available SSc data and suggest that there are shared mechanisms driving the disease in different organs. Computational methods have also been applied to meta-analyses of SSc clinical trials. These approaches have led to the prediction of potential combination therapies that target the multiple pathways deregulated in SSc, which can now be tested in the clinic.
Summary Data driven methods to analyze compendia of data are providing additional insights into SSc and related conditions. Modern bioinformatics and systems biology are ecosystems of data and code that are growing exponentially. Data integration allows researchers to combine multiple underpowered studies, a concern in a rare disorder such as SSc, for greater gain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genome-scale gene expression data allow us to infer, from relative mRNA expression measurements, pathways that are active in a whole tissue biopsy or sample. Genome-wide gene expression has been studied in multiple affected tissues from patients with systemic sclerosis (SSc), including skin [1,2,3], blood [4,5,6], lung [7, 8], and esophagus [9•]. Perhaps, the most prominent feature of these data is the inherent heterogeneity of molecular processes perturbed in SSc patients. To that end, “intrinsic” molecular subsets have been identified by examining SSc skin gene expression data from three independent cohorts [1,2,3]. Furthermore, subsets have been observed in a small study of esophagus [9•]. A recent multi-tissue study also suggests that inflammatory subsets found in skin and esophagus share underlying gene expression patterns and pathways with fibrotic lung disease [10••].
Molecular heterogeneity is observed in rheumatic diseases beyond SSc, and characterizing this heterogeneity has proven critical to optimizing treatment for patients. Individual studies for connective tissue disease (CTD) have often been underpowered, which is a distinct limitation. Distinct molecular patterns in blood are found in patients with rheumatoid arthritis (RA) that have differential responses to TNF-α [11]. In a longitudinal analysis of pediatric systemic lupus erythematosus (SLE) blood gene expression, Banchereau, et al. identified molecular subsets of patients. In addition, the authors found that plasmablast signatures correlate with disease activity and that neutrophil transcripts increased during lupus nephritis progression [12••]. These results indicate that molecular phenotyping can simultaneously measure clinical heterogeneity, response to treatment, and help us understand disease mechanisms. Some early studies have identified patterns in cell types thought to drive disease that are correlated with response to treatment (e.g., methylation patterns in CD4 T cells in RA [13]). Perhaps the most important recent contribution of gene expression studies to SSc is the identification of specific pathways and cell types requiring further study [10••, 14]. These associations show significant progress towards precision medicine in SSc—a necessary prelude to optimizing clinical practice—that is rooted in an understanding of disease mechanism.
Herein, we review recent advances in the study of genome-scale gene expression data in SSc. We discuss the special considerations when it comes to high-throughput data analysis of this rare disease. In addition, we discuss the value of integrating SSc data with the comprehensive body of biological knowledge outside of SSc. In the authors’ view, this is necessary because human tissue biopsies are cell mixtures and because the reproducible and/or important signals within tissues are not necessarily single transcript levels, but the coexpression (correlation of expression) of sets of genes that represent biological processes [15••].
Genome-wide gene expression in SSc shows systemic molecular changes within a patient and multiple gene expression subsets across the patient population
Prior studies of whole genome gene expression in SSc have demonstrated both “global” molecular changes in patients with SSc as compared to healthy controls and molecular heterogeneity within the patient population. Whitfield, et al. found that lesional and non-lesional skin show highly similar, disease-specific patterns of expression despite one site showing fibrosis and the other showing little clinical involvement [16]. This result has been one of the most reproducible findings and has since been recapitulated in multiple studies across multiple independent cohorts in skin [1,2,3, 17•]. A similar result is observed in lower vs. upper esophagus [9•]. These results suggested the molecular phenotype that drives SSc disease is truly systemic and is therefore observed consistently even in tissues that are considered clinically unaffected. The reason why one of these tissues is clinically affected and one clinically unaffected despite the same deregulated molecular pathways remains a mystery.
A second observation that has been reproducible across cohorts is the existence of multiple gene expression subsets that can be found across the patient population. Gene expression subsets were first observed in a cross sectional study of SSc patients that included patients with diffuse and limited cutaneous disease (dcSSc and lcSSc, respectively), as well as patients with morphea and healthy controls [1]. Patient biopsies fell into one of four major subgroups, each with distinct patterns of gene expression with different deregulated pathways. These were the inflammatory subset, which included patients with dcSSc, lcSSc, and morphea, a fibroproliferative subset composed only of dcSSc patients, a limited subset composed only of patients with lcSSc, and normal-like subset is comprised of a small number of SSc patients that resemble healthy controls and may represent late stage disease (described in more detail below). Therefore, the subsets mirror the clinical subtypes to a certain extent, but add supplemental information. These are referred to as “intrinsic” gene expression subsets because they are “intrinsic” to a patient. The subsets were re-identified by Pendergrass et al. in a SSc cohort from Boston University that contained a small number of longitudinal biopsies [2]. In that study, it was shown that a patient’s gene expression subset was stable over periods of up to 1 year.
Study of the intrinsic subsets has since moved to studies of therapeutics, multi-cohort meta-analyses, and other organs. The subsets were again found in an SSc cohort from Northwestern University examining response to mycophenolate mofetil (MMF) in SSc patients [3]. This study showed that inflammatory patients were most likely to improve on MMF. A meta-analysis of these three datasets [1,2,3] using network-based methods identified a set genes consistently expressed across the cohorts that recapitulate the intrinsic gene expression subsets [15••]. A recent study by Assassi et al. [17•], a completely independent set of investigators from the first three studies, partially recapitulated the intrinsic gene expression subsets. An important result from the Assassi et al. paper is the finding that the normal-like subset is likely to be late stage disease resulting from patients whose disease has spontaneously improved. This result is consistent with the Mahoney et al. meta-analysis that found that normal-like subjects were characterized by a lack of inflammatory and proliferative gene expression, but no distinct abnormal gene expression.
In other tissues, a study by Taroni et al. analyzed gene expression data from esophageal biopsies from patients with SSc and found molecular subsets in this second tissue that appeared to be intrinsic to patients [9•]. Further work investigated whether the molecular patterns perturbed in skin were similar to those disrupted in other tissues, including esophagus and lung [10••] (discussed in detail in “Systems biology of SSc, data integration, and the importance of public data” below). These data suggest that intrinsic molecular subsets are a common feature of skin and esophagus in patients with SSc and may share expression patterns with one another and other tissues.
Systems biology of SSc, data integration, and the importance of public data
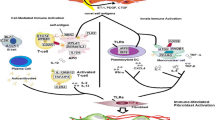
The wealth of high-throughput gene expression data collected on SSc can now be analyzed in aggregate to gain mechanistic insight into the disease. Systems biology approaches provide a powerful method by which to analyze these data (Fig. 1 ). By studying these data, we hope to understand the molecular abnormalities that distinguish patients with SSc from healthy controls (i.e., identify differentially expressed genes) and to quantify heterogeneity (i.e., subset detection). From these molecular abnormalities, we can discern higher-order biological information, such as pathways, biological processes, and/or cell type activation states that are aberrant in SSc, as these may drive disease and be rational targets for therapeutic intervention. Often, this higher-order information resides in curated ontologies, where human experts systematically review the literature to establish gene-pathway, gene-process, and gene-disease associations.

Schematic overview of systems-level approaches to SSc genome-scale data analysis. As a rare disease with no approved treatments, systemic sclerosis has special considerations when it comes to high-throughput data analysis. When genome-scale technologies are used to measure mRNA levels, there is considerable noise and typically small sample sizes, particularly in the case of pilot drug studies. The goal of molecular profiling is to take a steady state snapshot of the biological processes occurring in a whole tissue at the time of biopsy and infer what cellular states are perturbed in patients as compared to healthy subjects. Recent advances in systems biology and bioinformatics, notably the development of tissue and cell type-specific gene-gene functional networks [18] allow us to untangle cell lineages and compare disease-associated genes functions in different tissues, e.g., the major organs affected by fibrosis in SSc, skin, and lung. State-of-the-art machine learning algorithms can also be used to to “bolster” differential gene expression analyses in small studies.
Since all currently available SSc gene expression datasets are composed of whole tissue samples, they are mixtures of cell types. Recently, functional genomic networks have been built to study tissue- and cell type-specific genes’ interactions at the system-level [19•]. In addition, tools for assessing cell type-specific changes in expression data are now also available [18, 20,21,22]. These tools pave the way for understanding the critical similarities and differences between affected tissues in SSc—and thus, clinical manifestations—and ultimately how cellular state and tissue microenvironment give rise to SSc manifestations (e.g., skin fibrosis).
In addition to the biological variation within any cohort, one must also take into account the considerable technical noise. Moreover, because SSc is a rare disease, studies are often underpowered due to small sample sizes, making analysis and interpretation of these data challenging. It is here where functional genomic networks prove most useful, as they can be used to rigorously extract meaningful pathway and cell type signals from underpowered studies. This approach can result in biologically significant findings beyond what can be gleaned from an analysis of each dataset alone.
Mahoney et al. [15••] applied these methods to three independent SSc skin datasets to identify the genes that were consistently expressed across the patients and to determine how genetic polymorphisms found in genome-wide association studies (GWAS) were connected with the SSc intrinsic gene expression subsets. Mahoney et al. used the IMP functional genomic network (see Box 1 for definitions) [23] to understand the relationship between sets of genes that had similar coexpression patterns across three independent SSc skin cohorts. The IMP network was constructed using a large compendium of publicly available gene expression data (v2.0 lists over 3700 datasets from NCBI Gene Expression Omnibus http://imp.princeton.edu/networks/data/). Only two datasets contain the term “scleroderma” or “systemic sclerosis” in their titles. Thus, the overwhelming majority of experiments in these networks do not directly examine SSc. Nevertheless, when queried with the consensus genes from the meta-analysis of SSc skin, the resulting network captured information highly relevant to SSc pathobiology. For instance, the hubs of the functional modules in the network included FBN1, which had been implicated in SSc pathogenesis in prior work (Alterations in or duplications in this gene result in skin abnormalities in the Tsk1 and Stiff Skin Syndrome mouse models of SSc [24, 25].) Functional hubs identified in these data included a hub associated with alternatively activated, M2 macrophages, interferon signaling, cell proliferation, and TGFβ signaling and ECM deposition. An important result from this study was the finding that genetic risk polymorphisms identified in candidate gene studies and GWAS were almost exclusively connected in the immune system [15••]. These data also suggest that the gene expression subsets are mechanistically interconnected and strongly suggest the initiating events in the SSc reside in the immune system with aberrant immune responses.
This work was extended in Taroni et al. in which ten SSc datasets sampling four different affected tissues (skin, lung, esophagus, and peripheral blood mononuclear cells [PBMCs]) were examined and subsequently analyzed in the context of the GIANT [25] tissue-specific networks [10]. An important finding from this analysis is that the inflammatory subsets identified in skin and esophagus shared coexpression patterns with severe phenotypes in other tissues (e.g., pulmonary fibrosis and pulmonary arterial hypertension). These patterns included innate immune signatures. The use of tissue-specific GIANT networks allowed for more detailed comparison of tissues (e.g., lung and skin), which lead to further analyses that suggested alternatively activated macrophages play a role in both tissues, but may have slightly different phenotypes depending on the tissue context. A limitation of this study was that the biopsies in the multiple cohorts were largely from different patients (i.e., there was minimal overlap between cohorts). Further work might include the study of multiple tissues from patients to test whether patterns are conserved across tissues within a patient.
Lofgren et al. took a different approach to the integration of data in SSc and identified an SSc skin severity score (4S) that was significantly correlated with mRSS [32••]. This study analyzed seven different SSc datasets from six independent clinical centers. They used samples from two centers as discovery cohorts and validated these results in five independent cohorts. Lofgren et al. show that this 4S signature was significantly correlated with mRSS and could predict mRSS change at 24 months.
These studies demonstrate the value of analyzing gene expression data from multiple studies in the context of global and tissue-specific networks to examine disease and tissue-specific phenotypes, and emphasize the value of these data to rare diseases such as SSc. Further analyses of these data from SSc studies, possibly in the context of other related disorders, are likely to continue to provide insight into disease pathology.
Treatment: molecular phenotyping and biomarkers in SSc therapeutic trials
The characterization of molecular heterogeneity in SSc and the identification of intrinsic subsets naturally lead to questions of patient stratification for the purpose of treatment, precision medicine, or prognostication. Indeed, this heterogeneity could explain the failure of most clinical trials in SSc to meet clinical endpoints. If the biological processes underlying different patients’ skin disease are distinct, it is to be expected that a therapeutic agent targeting one particular process (or set of related processes) would fail when that pathway does not contribute to disease.
Early studies investigated whether pre-treatment intrinsic subset was informative about clinically significant improvement during treatment. Hinchcliff et al. [3] considered the immunosuppressive agent MMF, which is believed to suppress lymphocyte proliferation through the inhibition of de novo synthesis of guanine nucleotides [33]. Hinchcliff and coworkers found that patients who improved on MMF were more likely to map to the inflammatory subset. Notably, no improvement was observed in fibroproliferative patients [3]. A strength of this work was the high quality clinical information that accompanied the molecular data from skin biopsies; one physician scored skin disease severity using the modified Rodnan skin score (mRSS), a semi-objective score calculated from evaluating skin thickness at 17 anatomic sites as rated by clinical palpation.
Many drug trial cohorts, particularly pilot studies, are not as large and as extensively phenotyped as the Hinchcliff cohort, which can present challenges in analyzing and interpreting the associated molecular data. In some cases, less than half of the patients in a trial improve while on a particular treatment and placebo arms are uncommon [34••]. Below, we review the expression-based findings from the primary publications (where applicable) for drug trials in SSc that have accompanying molecular data and additional research on those therapeutics in SSc (see also [35] for a recent review).
Rituximab (Rituxan™, anti-CD20) depletes cells expressing CD20, a cell surface marker on pro-B to mature B cell stages but not plasmablasts [36]. Lafyatis and coworkers found that the mean change in mRSS (the primary outcome in most of the studies included herein) between base and 6 mos. of treatment was not significant, although depletion of circulating and dermal B cells was observed [37]. Pendergrass et al. were unable to identify significant differences in gene expression pre- and post-treatment using Significance Analysis of Microarrays (SAM) [38], “consistent with apparent lack of clinical response” [2].
The immunomodulatory biologic abatacept (Orencia™) is a CTLA4-IgG fusion protein designed to preferentially bind to CD80/86 receptors on antigen presenting cells (APCs) and, therefore, block co-stimulation of T cells through CD28. Chakravarty et al. performed a placebo-controlled study of abatacept and found that most treated patients improved and that most improvers mapped to the inflammatory subset pre-treatment [39•]. Abatacept improvers showed a decrease in immune-related pathways post-treatment [39•]. The CD28 signaling pathway was specifically downregulated in improvers, but not in placebo-treated patients or one non-improver [39•]. A multi-center placebo-controlled trial of abatacept is currently underway (NCT02161406).
Nilotinib (Tasigna™) is a tyrosine kinase inhibitor (TKI) with a “narrow” target profile, designed specifically to inhibit Abelson murine leukemia viral oncogene homolog 1 (ABL1) and the platelet-derived growth factor receptor (PDGFR). Gordon et al. reported a significant improvement in mRSS at 12 months in an open-label trial of nilotinib in early diffuse systemic sclerosis. Improvers, as classified by this study, had significantly higher expression of TGFBR and PDGFRB signaling pathways pre-treatment as compared to non-improvers. Expression of these pathways was downregulated post-treatment in improvers [40•].
Fresolimumab (anti-TGF-β) is a human monoclonal antibody that binds all three isoforms of the pro-fibrotic growth factor TGF-β. Consistent with the mechanism of action, Rice et al. found that fresolimumab-treated patients showed evidence of the inhibition of TGF-β induced genes (correlated with biomarker genes first described in [41] and updated in [42•, 43•]. Rice et al. also noted that these changes in gene expression were “generally correlated” with changes in mRSS [43•].
Taken together, these data suggest that pre-treatment (or baseline) activity levels of the pathways or processes targeted by a therapeutic may be predictive of response to that treatment. Molecular phenotyping prior to prescribing treatment may be of value. However, these results should be interpreted with caution due to the small number of patients included in many of these studies. As of this writing, there is still no FDA-approved treatment for SSc. As mentioned above, mRSS is a semi-objective measurement—a robust multi-gene biomarker may be a more reliable measurement of skin disease severity [41]. Prognostic biomarkers would be particularly valuable.
A recent study by Taroni et al. examined gene expression data from the aforementioned studies (MMF, abatacept, rituximab, nilotinib, and fresolimumab) in the context of a skin-specific network [34••]. Taroni et al. made use of the fact that studies typically include samples from patients that meet clinically important criteria for improvement during the course of a trial (termed improvers) and those patients that do not (termed non-improvers). Taroni et al. identified differentially expressed genes pre- and post-treatment in improvers from gene expression data in these trials and then used a skin-specific functional network [19•] to identify subnetworks associated with these response genes. The authors found that the gene modules that were targeted by multiple trial therapies had significant overlap in immune-related processes, with the exception of the fresolimumab trial [34••]. The authors then demonstrated that the gene patterns that were elevated in fresolimumab non-improvers pre-treatment are similar to those patterns that were uniquely altered in MMF improvers, suggesting that these non-improvers may have benefited from treatment with MMF [34••]. This early study establishes the utility of this approach to the analysis of therapeutic studies in SSc and may lead to avenues of investigation that include the prediction of combination therapies.
Conclusions
The systems biology approaches described above can also be applied to the facets of SSc that have eluded study. For example, using gene signatures derived from well-studied or easily assayed SSc affected tissues (e.g., skin), it is now possible to study SSc-affected tissues that are substantially more difficult to acquire in patients or controls (e.g., the GI tract or kidney). This is because, while we might lack SSc tissue from many affected organs, we do have solid estimates of the gene expression abnormalities in many affected tissues, and functional networks can tell us how those genes interact in tissues of interest. Such study could yield deep insight into the most intractable clinical SSc outcomes.
In closing, we emphasize that modern bioinformatics and systems biology are ecosystems of data and code that are growing exponentially. The more publicly available SSc data there are, the more finely tuned all of these tools become. In many cases, the most detailed and sophisticated results from a data set derive from reanalysis by researchers who can integrate across multiple data modalities. Data integration may allow researchers to combine multiple underpowered studies, a concern in a rare disorder such as SSc, for greater gain. In the genomic era, the importance of data release at the time of publication cannot be overstated. Tools such as functional genomic networks operate at data scales that have to be automated. The better the public data, the more we can all learn from it.
The result of these systems biology studies in SSc should be a better understanding of disease mechanisms, thus allowing us to develop better and more targeted therapies to SSc. A second result of these studies will be better patient stratification tools that can be used in clinics to identify the patients most likely to improve on a given therapy. Future clinical trials may use these tools and methods to improve outcomes and better understand both successes and failures in this difficult disease.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3(7):e2696.
Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol. 2012;132(5):1363–73.
Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol. 2013;133(8):1979–89.
Pendergrass SA, Hayes E, Farina G, Lemaire R, Farber HW, Whitfield ML, et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS One. 2010;5(8):e12106.
Risbano MG, Meadows CA, Coldren CD, Jenkins TJ, Edwards MG, Collier D, et al. Altered immune phenotype in peripheral blood cells of patients with scleroderma-associated pulmonary hypertension. Clin Transl Sci. 2010;3(5):210–8.
Cheadle C, Berger AE, Mathai SC, Grigoryev DN, Watkins TN, Sugawara Y, et al. Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients. PLoS One. 2012;7(4):e34951.
Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63(3):783–94.
Christmann RB, Sampaio-Barros P, Stifano G, Borges CL, de Carvalho CR, Kairalla R, et al. Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014;66(3):714–25.
• Taroni JN, Martyanov V, Huang C, Mahoney JM, Hirano I, Shetuni B, et al. Molecular characterization of systemic sclerosis esophageal pathology identifies inflammatory and proliferative signatures. Arthritis Res Ther. 2015;17:194. First report of the intrinsic gene expression subsets first identified in skin in a second organ system in SSc patients.
•• Taroni JN, Greene CS, Martyanov V, Wood TA, Christmann RB, Farber HW, et al. A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med. 2017;9(1):27. This study is the first network analysis of multiple tissues in SSc and finds key similarities in the immune- and fibrotic-related expression patterns across tissues, implicating alternatively activated macrophages in SSc pathogenesis.
Oswald M, Curran ME, Lamberth SL, Townsend RM, Hamilton JD, Chernoff DN, et al. Modular analysis of peripheral blood gene expression in rheumatoid arthritis captures reproducible gene expression changes in tumor necrosis factor responders. Arthritis Rheumatol. 2015;67(2):344–51.
•• Banchereau R, Hong S, Cantarel B, Baldwin N, Baisch J, Edens M, et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell. 2016;165(3):551–65. Report of utilizing molecular networks to stratify patients with systemic lupus erythematosus.
Hudson M, Baron M, Colmegna I, Bernatsky S, Klein Oros K, Pastinen T, et al. Novel approaches to discovery of biomarkers in rheumatoid arthritis: comment on the article by Oswald et al. Arthritis Rheum. 2015;67(8):2276–7.
Johnson ME, Mahoney JM, Taroni J, Sargent JL, Marmarelis E, Wu MR, et al. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PLoS One. 2015;10(1):e0114017.
•• Mahoney JM, Taroni J, Martyanov V, Wood TA, Greene CS, Pioli PA, et al. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol. 2015;11(1):e1004005. Data integration and network analysis of SSc skin cohorts that shows an interconnection of immune and fibrotic pathways. This study connects the genetic risk polymorphisms for SSc with the inflammatory response in these patients.
Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003;100(21):12319–24.
• Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, et al. Dissecting the Heterogeneity of Skin Gene Expression Patterns in Systemic Sclerosis. Arthritis Rheumatol. 2015;67(11):3016–26. doi:10.1002/art.39289. Partial recapitulation of the SSc skin gene expression subsets by a independent group. Importantly, this study shows that the normal-like group likely represents end stage disease.
Bolen CR, Uduman M, Kleinstein SH. Cell subset prediction for blood genomic studies. BMC Bioinformatics. 2011;12(1):258.
• Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, et al. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47(6):569–76. doi:10.1038/ng.3259. Report of the GIANT tissue-specific networks used in Taroni, et al. 2016 and Taroni, et al. 2017.
Chikina M, Zaslavsky E, Sealfon SC. CellCODE: a robust latent variable approach to differential expression analysis for heterogeneous cell populations. Bioinformatics. 2015;31(10):1584–91.
Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, Perry NM, et al. Cell type-specific gene expression differences in complex tissues. Nat Methods. 2010;7(4):287–9.
Nelms BD, Waldron L, Barrera LA, Weflen AW, Goettel JA, Guo G, et al. CellMapper: rapid and accurate inference of gene expression in difficult-to-isolate cell types. Genome Biol. 2016;17(1):201.
Wong AK, Park CY, Greene CS, Bongo LA, Guan Y, Troyanskaya OG. IMP: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res. 2012;40(Web Server issue):W484–90.
Lemaire R, Farina G, Kissin E, Shipley JM, Bona C, Korn JH, et al. Mutant fibrillin 1 from tight skin mice increases extracellular matrix incorporation of microfibril-associated glycoprotein 2 and type I collagen. Arthritis Rheum. 2004;50(3):915–26.
Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503(7474):126–30.
Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68.
Assassi S, Radstake TR, Mayes MD, Martin J. Genetics of scleroderma: implications for personalized medicine? BMC Med. 2013;11:9.
Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;1:15002. doi:10.1038/nrdp.2015.2.
Greene CS, Troyanskaya OG. Chapter 2: data-driven view of disease biology. PLoS Comput Biol. 2012;8(12):e1002816.
Hintze A, Adami C. Evolution of complex modular biological networks. PLoS Comput Biol. 2008;4(2):e23.
Jeong H, Mason SP, Barabási A-L, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–2.
•• Lofgren S, Hinchcliff M, Carns M, Wood T, Aren K, Arroyo E, et al. Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. JCI Insight. 2016;1(21):e89073. Analysis of publicly available data from SSc cohorts to develop a 4S signature of MRSS severity in SSc patients
Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47(2–3):85–118.
•• Taroni JN, Martyanov V, Mahoney JM, Whitfield ML. A functional genomic meta-analysis of clinical trials in systemic sclerosis: towards precision medicine and combination therapy. J Invest Dermatol. 2017;37(5):1033–41. doi:10.1016/j.jid.2016.12.007. First meta-analysis of molecular gene expression data from SSc clinical trials. Study makes the first attempt to predict improvers across treatments.
Martyanov V, Whitfield ML. Molecular stratification and precision medicine in systemic sclerosis from genomic and proteomic data. Curr Opin Rheumatol. 2016;28(1):83–8.
Whitfield ML. Editorial: plasma and B cell gene signatures: quantitative targeting and monitoring of B cell—depleting therapies in autoimmune diseases in the genomic era. Arthritis Rheum. 2014;66(1):10–4.
Lafyatis R, Kissin E, York M, Farina G, Viger K, Fritzler MJ, et al. B cell depletion with rituximab in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2009;60(2):578–83.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–21.
• Chakravarty EF, Martyanov V, Fiorentino D, Wood TA, Haddon DJ, Jarrell JA, et al. Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res Ther. 2015;17:159. Pilot study of abatacept in SSc with molecular data. Suggest patients most likely to improve on abatacept are those with an inflammatory signature.
• Gordon JK, Martyanov V, Magro C, Wildman HF, Wood TA, Huang WT, et al. Nilotinib (Tasigna™) in the treatment of early diffuse systemic sclerosis: an open-label, pilot clinical trial. Arthritis Res Ther. 2015;17:213. Pilot study of nilotinib in SSc. Study finds that patients most likely to improve on nilotinib are the subset of patients that have high TGFβ signaling at baseline.
Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2010;62(2):580–8.
• Rice LM, Ziemek J, Stratton EA, McLaughlin SR, Padilla CM, Mathes AL, et al. A longitudinal biomarker for the extent of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2015;67(11):3004–15. Longitudinal biomarker for MRSS for use in SSc clinical trials.
• Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. 2015;125(7):2795–807. Pilot study of fresolimumab to target TGFβ activation in SSc patients, which included clinical and molecular gene expression data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jaclyn N. Taroni declares that she has no conflict of interest. J. Matthew Mahoney declares that he has no conflict of interest. Michael L. Whitfield declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Scleroderma
Rights and permissions
About this article

Cite this article
Taroni, J.N., Mahoney, J.M. & Whitfield, M.L. The Mechanistic Implications of Gene Expression Studies in SSc: Insights From Systems Biology. Curr Treat Options in Rheum 3, 181–192 (2017). https://doi.org/10.1007/s40674-017-0072-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-017-0072-0