
Abstract
Acute kidney injury (AKI) is considered to be a major public health problem around the globe, and it is associated with major adverse clinical outcomes and significant health care costs. There is growing evidence suggesting that AKI is associated with the subsequent development of chronic kidney disease (CKD). While recovery of kidney function occurs in the majority of patients surviving an AKI episode, a large number of patients do not recover completely. Similarly, CKD is a well-known risk factor for the development of AKI. Recent studies suggest that both AKI and CKD are not separate disease entities but are in fact components of a far more closely interconnected disease continuum. However, the true nature of this relationship is complex and poorly understood. This review explores potential relationships between AKI and CKD, and seeks to uncover a number of “missing links” in this tentative emerging relationship.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute Kidney Injury (AKI) is a term that is used to describe an acute reduction in glomerular filtration rate (GFR) occurring over hours to days, with a subsequent rise in serum creatinine concentrations. It is an umbrella term with a variety of diverse causes that precipitate and propagate acute kidney injury with a rapid reduction in kidney function. This abrupt loss of GFR is usually sufficient to result in the retention of metabolic waste products, and in the disruption of fluid, electrolyte, and acid-base homeostasis. AKI was previously referred to as acute renal failure (ARF) but during the past decade this terminology has changed from ARF, for which the focus generally was limited to the most severe episodes with complete or near complete loss of kidney function, to the current terminology of AKI, which is more representative of the full spectrum of acute kidney dysfunction and includes both injury and/or kidney impairment [1–6].
Acute kidney injury is associated with major adverse clinical outcomes and significant health care costs, and is now considered to be a major public health problem around the globe. Several observational studies have suggested that AKI, even if mild and transient, is associated with the subsequent development of chronic kidney disease (CKD) [7–12]. Although recovery of kidney function occurs in the majority of patients surviving an AKI episode, a large number of patients do not recover completely, and some will remain dialysis-dependent as a consequence [13–16]. CKD is a well-known risk factor for AKI, because chronically impaired kidneys lose their ability to auto-regulate and therefore become susceptible to acute injury whenever exposed to a sufficiently severe insult [17]. Recent studies suggest that both AKI and CKD are not separate disease entities but are in fact components of a far more closely interconnected disease continuum. Accordingly, considerable conceptual overlap may exist between these two “separate conditions” with regard to underlying pathology and pathophysiology, definitions, risk factors, and indeed clinical outcomes [18]. However, the true nature of this relationship is complex and poorly understood. This review explores potential relationships between AKI and CKD, and seeks to uncover a number of “missing links” in this tentative emerging relationship.
Epidemiology of AKI and CKD
The epidemiology of acute kidney injury
The prevalence and incidence of AKI vary by definition and among populations. Prior to the adoption of newer and more precise classification systems for AKI, the prevalence ranged widely from 1 to 26% [19–21]. Over the last 15 years, substantive efforts have been made to reach a consensus on definitions and classification of AKI. In 2002, the Acute Dialysis Quality Initiative (ADQI) developed the RIFLE definition (Risk, Injury, Failure, Loss of kidney function, and End-stage kidney disease) and published it in May 2004 [1]. Three years later, in September 2007, the Acute Kidney Injury Network (AKIN) working group made further modifications [3–5]. Finally, in March 2012, KDIGO combined and modified the RIFLE and the AKIN definitions to develop a new definition of AKI in order to establish a single unifying classification system for AKI in clinical practice and in research [6].
In the United States, approximately 1% of patients have AKI at the time of admission to the hospital [22]. The prevalence of AKI is >40% at the time of admission to the intensive-care unit (ICU) if sepsis is present [23] and >60% during the stay in ICU [24]. Large epidemiologic studies from the last quarter century, confirm that the incidence of AKI continues to rise in hospitalized-patients despite substantial advances in healthcare delivery, technology, and research [7, 25]. Current estimates suggest that the incidence ranges from 295 cases per million people per year for dialysis-requiring AKI, to more than 5000 cases per million people per year for non-dialysis-requiring AKI [26].
The estimated incidence rate of AKI during hospitalization is between 2 and 5%. Postoperatively, AKI develops in approximately 1% of general surgical cases [21], and the highest incidence is seen in ICU, where it occurs in up to two-thirds of patients [27].Twenty-one percent of kidney transplant recipients develop AKI within the first 6 months after transplantation [28]. The incidence of community-acquired AKI among all hospital admissions has been shown to be as high as 4.3% [29]. However, this is probably an underestimate of the true incidence because many patients, with community-acquired AKI, will neither have blood tests performed nor be admitted to the hospital. This ascertainment bias has been described in other community-acquired AKI studies [30]. The economic burden of AKI is substantial and increasing [31]. In England, AKI is estimated to cost between £434 and £620 million per year (excluding costs in the community) and this exceeds the overall costs associated with lung and skin cancer combined [31].
The epidemiology of chronic kidney disease
Chronic kidney disease (CKD) and End-stage renal disease (ESRD) are major global public health problems [32–34]. They are the ninth leading cause of death in the United States [35]. It is reported that one in ten American adults have some degree of CKD, and end-stage renal disease (ESRD) affects more than one million individuals worldwide [33]. The incidence of CKD is at least 30-fold higher than that of ESRD [36, 37]. At the current rate of growth, it is expected that the incidence rate of new ESRD cases in the U.S. will be over 400,000 per year in 2030, with an estimated prevalence of over two million [33]. Hsu et al. concluded that the growth in ESRD incidence in USA has far outpaced the growth in CKD prevalence, and the increase in CKD prevalence had a minor contribution to the ESRD epidemic [37]. This sentinel observation was supported by several studies over the next decade that has linked AKI with CKD, ESRD and other adverse outcomes [7, 14, 38–47].
Studies suggesting a link between AKI and CKD
Whether AKI directly causes CKD is unknown. However, the body of evidence from observational studies suggests the presence of a strong independent association of AKI with the development of new-onset CKD and ESRD (Table 1). Ishani et al. analysed the outcomes of over 200,000 hospitalised elderly patients who were followed for a period of 24 months [7]. Compared to patients with no AKI or CKD, they found that that the hazard ratios for ESRD were highest for patients with pre-existing CKD who experienced a single AKI episode. The rates of new onset ESRD were also higher for patients who had an AKI event but without previous CKD.
Wald et al. provided further evidence that AKI is linked to an elevated risk of ESRD [14]. In a population-based study of hospitalised patients with severe AKI-requiring dialysis in Ontario Canada, they found that found that the risk of new ESRD was increased by almost threefold. Evidence has since accumulated that patients with relatively mild kidney impairment at baseline, and who sustain a severe dialysis-requiring AKI event, incur an elevated risk of ESRD [13]. Using the Kaiser Permanente database, Lo et al. found that dialysis-requiring AKI was independently associated with a 28-fold higher risk of developing stage four or five CKD after adjustment for confounding factors [13]. Newsome et al. demonstrated an increased ESRD risk after acute myocardial infarction in a cohort of patients with pre-existing CKD (evident by reduced GFR) and a higher burden of co-morbidities (e.g. diabetes mellitus and hypertension) that are well known to exert an adverse impact on CKD progression even in the absence of AKI [40]. One might argue that those who eventually progressed to ESRD after AKI did so because of the higher burden of co-morbidities rather than the AKI itself. It is noteworthy that not all observational studies have shown significant independent associations of AKI with ESRD. For example, in a study by Hsu et al. AKI events did not predict subsequent development of ESRD (adjusted HR 1.47; 95% CI 0.95–2.28) when the model was adjusted for age, gender, race, diabetes, diagnosed hypertension, proteinuria and baseline estimated Glomerular Filtration Rate (eGFR) [43]. Moreover, this lack of independence of this association persisted even after a sensitivity analysis using alternative definitions of baseline renal function.
Amdur et al. concluded that ATN (acute tubular necrosis) and ARF independently increased the risk of CKD using observational data from the United States Department of Veterans Affairs database [12]. In their analysis of over 113,000 veterans, patients with a recorded AKI or ATN episode, but without CKD, experienced significantly higher rates of progression to Stage 4 CKD. The adverse impact of an acute AKI event on CKD progression has also been examined in detail within other high-risk patient subgroups. For example, a study by James et al. examined the association between AKI and long-term changes in kidney function following coronary angiography [41]. These investigators described a significant dose-dependent association between the severity of AKI and the subsequent likelihood of kidney disease progression with a very comprehensive adjustment for confounding factors. A similar study by the same investigators that included over 900,000 patients from the Alberta Kidney Disease Network found that proteinuria further magnified both the risk of an AKI event (by over fourfold) and its attendant consequences on mortality and kidney disease progression [42]. These studies add credence to the hypothesis that both AKI and CKD represent a continuum of disease rather than separate disease entities [42]. Table 1 lists the principal studies that link AKI with CKD.
Despite the large number of studies that demonstrated associations of AKI with the development of CKD, one cannot state for certain that AKI is truly in the causal pathway. The lingering doubt that remains is in part due to inherent weaknesses in the design of published studies. First, many studies that have explored the association of AKI with CKD have utilised large administrative data sets which are often based on clinical codes and therefore lack the precision required in defining the presence and severity of AKI. Furthermore, these studies are also limited by the presence of a code-creep bias, a phenomenon that is inherently associated with these types of data sets [48]. Consequently, there is a continued risk of confounding secondary to variation in the availability and degree of recording of follow-up measures of kidney function. In these datasets, patients with less severe AKI are likely to be under-represented due to the low sensitivity of administrative clinical coding [49]. Second, it is noteworthy that a number of AKI outcome studies have not included information on classification of CKD by stage at study inception, or the rates of CKD progression prior to the AKI event. These are potentially serious flaws in that it is uncertain whether patients with CKD at baseline had pre-existing mild or severe degree of kidney impairment which would in any event predict more rapid progression irrespective of intercurrent AKI episodes. Furthermore, without accurate information on the trajectory of GFR decline prior to the AKI event, it is very difficult to ascertain whether those who developed ESKD did so as a result of a pre-ordained CKD trajectory or due to the sole effect of an acute AKI event. Third, it is unclear whether the findings from published studies are generalizable to all patients given inherent differences in the type of and severity of exposure [13], and differences in populations studied [39, 41, 46]. Finally, the risk for developing CKD and ESRD associated with an AKI exposure could not be determined in many studies due to lack of follow-up of appropriate non-AKI controls, an essential component in establishing causality. In fairness, these limitations are common and seem unavoidable in many of the published observational studies. Nevertheless, their presence does reduce the quality and strength of the evidence base linking AKI to ESRD. In truth, prospective cohort studies of sufficient size and diversity with internal or external controls, and with prolonged follow-up periods, are required to examine the true nature of the AKI-exposure ESKD relationship, and to identify subgroups of patients who are at greatest risk.
The pathophysiology of AKI and progression to CKD: What do animal models tell us?
The pathophysiology of AKI represents a very complex interplay between the immune system, the accompanying inflammatory response, tubular injury and the extent of any associated vascular insult [50]. Regardless of the initial insult and aetiology of AKI, there is a rapid loss of proximal tubular cell polarity and cytoskeletal integrity [51]. Adhesion molecules and other membrane proteins undergo mislocalisation [52, 53]. These changes result in disruption of normal cell to cell interactions and when severe enough cell necrosis and apoptosis ensue [53]. With severe AKI injury, viable and non-viable cells are sloughed off and the basement membrane, in some regions of injury, remains as the only barrier between the peri-tubular interstitium and the glomerular filtrate. As cellular debris, from epithelial injury, accumulates in the lumen, obstruction ensues and leads to an increase in the intra-tubular pressure and the back-leakage of the filtrate. As a consequence, a greater inflammatory response is generated which will lead to further injury [50].
Within the injured kidney, the damaged endothelium is a major contributor to the pathophysiology of AKI. Dissipation of the glomerular filtration pressure results in the alteration of vascular tone and reactivity, as well as a loss of vascular autoregulation [54]. There is an enhanced vascular response to vasoconstrictors with impaired response to, and production of, vasodilators [50]. An unbridled vasoconstrictive response to angiotensin II, prostaglandin H2, thromboxane A2, leukotrienes C4, D4, endothelin-1, and other vasoconstrictors occur [55]. At the same time, the effect of nitric oxide, bradykinin, acetylcholine and other vasodilators is diminished [56, 57]. The interactions of Toll-like receptors (TLRs) with their ligands result in the release of cytokines and chemokines, and the attraction of inflammatory cells [58–60]. Cytokines upregulate expression of leucocytes integrins and their ligands, namely intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule (VCAM), resulting in increased adherence of leukocytes to endothelial cells and therefore an in increase in interstitial leucocytes [60, 61]. These activated leukocytes generate pro-inflammatory cytokines and these in turn produce a number of injurious changes in proximal tubular epithelial cells and disrupt cell-matrix adhesion therefore resulting in cell shedding into the lumen [61]. Macrophages contribute to kidney fibrosis and are an important source of the C3 component of complement [50]. The complement system may potentiate leukocyte endothelial interactions and mediates injury [62]. Dendritic cells can activate naive T cells, thus linking the innate immune response to adaptive immunity [63].
The permeability of the damaged endothelium following an AKI event increases with a subsequent development of interstitial oedema and reduction of blood flow. This will result in greater ischaemic injury to vulnerable regions, like the outer medulla [64]. Consequently, inhibitors of angiogenesis are upregulated and angiogenic factors are downregulated, causing a reduction in the number of microvessels, tissue capillaries and chronic hypoxia [65]. T-regulatory cells are involved in this process of inhibition of angiogenesis in mouse models [66]. Chronic hypoxia causes tubulointerstitial fibrosis which reduces the delivery of oxygen and nutrients to tubules resulting in their death leading to further fibrosis [50]. The loss of microvessels has been implicated in post-AKI alteration in the urine concentrating ability of the kidney and the development of salt-sensitive hypertension [50]. It is conceivable that this vicious circle of hypoxic injury and subsequent tubulointerstitial fibrosis, together with the development of salt-sensitive hypertension, may be the driver of future progression to CKD.
Normally, the human proximal tubule cells divide at a low rate [50]. It has been suggested that non-fibrotic healing and recovery from AKI depend on the integrity of the following processes involving the epithelial cells: (1) Spreading and migration to cover exposed areas of the basement membrane. (2) Rapid proliferation to restore cell number. (3) Differentiation to restore functional integrity of the nephron [50, 67]. When the injury is severe or on a background of underlying CKD the recovery process can often be suboptimal. Subsequently, this can result in incomplete repair and persistent tubulointerstitial inflammation, with proliferation of fibroblasts and excessive deposition of extracellular matrix [68, 69]. Tubulointerstitial fibrosis is a characteristic feature of the maladaptive repair that accompanies AKI [70]. In addition, it is well known that tubulointerstitial fibrosis is a hallmark of CKD and it is often much more severe than the glomerular pathology that may have been integral to the initiation of the renal disease [51]. The injured epithelial cells play an important role in the process of fibrosis by generating profibrogenic cytokines [51]. This induces the generation of myofibroblasts, many of which are perivascular fibroblasts, or pericytes, and they contribute directly to the fibrotic process [51]. Yang et al. demonstrated a casual association between the development of fibrosis and epithelial cell cycle arrest at G2/M in mice [71].
In this section, we summarize the findings from several studies that implicate specific pathophysiological mechanisms between AKI and CKD. However, the reliability and predictive value of animal modelling for human outcomes and for understanding human pathophysiology is a contentious issue. Indeed, important concerns have been raised by a growing body of scientific literature which had critically assessed the validity of this type of modelling [72].
The arbitrary definition of CKD
The development of a definition and a staging system for CKD by the Kidney Disease Outcomes Quality Initiative (KDOQI) in 2002 was a landmark event and a major step forward in improving the evaluation and management of CKD [73]. It established a common platform for CKD nomenclature worldwide. Since then the concept of renal disease has undergone a transition from a state of a somewhat neglected ‘nephrologist-only’ life-threatening disorder to a “common disease process with a spectrum of severity that warrants attention by primary care physicians” before referral to kidney specialists. This transition has had a major influence the development of effective preventive strategies and therapeutic interventions. It has also had a huge positive impact on public health. However, there are several major limitations associated with this definition and these have become more obvious and recognizable whenever an attempt is made to link CKD with AKI.
KDOQI defined CKD as the presence of kidney damage (with or without a reduction in GFR) for ≥3 months, or the presence of GFR < 60 mL/min/1.73 m2 for ≥3 months (with or without kidney damage). The choice of a cut-off value of greater than 3 months for the definition of CKD was arbitrary. Similarly, the cut-off levels between different stages were arbitrary, and were based on the categorization of a continuous measure of kidney function (i.e. GFR). A decade after KDOQI guidelines were introduced, the Kidney Disease: Improving Global Outcomes (KDIGO) CKD guidelines were developed and published [74]. They were based on KDOQI definitions and staging with some further modifications to improve accuracy, utility and quality of care. Unfortunately, many limitations of the KDOQI definition/staging system were inherited by KDIGO CKD guidelines. The 3 months threshold that is used to define CKD creates a new and important complexity, when taken in the context of linking AKI and CKD together. First, it would imply that all CKD cases are potentially classified as an AKI at the onset of GFR decline (fulfilling the AKI definition) and during the first 3 months. This creates an obvious overlap between CKD and AKI although the underlying disease process leading to CKD, in this case, is by default a chronic process from the outset rather than an acute insult to the kidney. Second, some cases of AKI, (e.g. acute interstitial nephritis) resolve completely but slowly and may take >3 months to do so [75]. These slowly-resolving AKI episodes may be incorrectly mislabelled as CKD because they persisted for ≥90 days, despite the obvious recovering trend in the creatinine trajectory. Third, it is well known that most cases of CKD are irreversible, persistent and progressive. However, CKD may be entirely reversible in some cases, either spontaneously or with treatment. Whether labelling these cases as CKD rather than a slowly-resolving AKI, remains to be fully debated. More importantly, these limitations of the existing CKD definition makes us wonder as to whether AKI cases that progress to CKD were in fact CKD from the outset but detected early? Were these AKI cases “smouldering CKD” that were detected incidentally for the first time? If these assumptions are true, then the concept of ‘AKI progression to CKD’ should be challenged and carefully redefined.
Imperfect biomarkers of AKI
The Acute Kidney Injury Network (AKIN) and Kidney Disease Improving Global Outcomes (KDIGO) guidelines use small absolute changes in serum creatinine concentrations, 0.3 mg/dL (26.4 µmol/L), to define the presence of AKI [3–6]. This definition was based on findings of several studies that showed a strong association between adverse outcomes and minor changes in serum creatinine level [38, 76–78]. Evidence has now emerged which suggests that this may not be true to the same extent in people with pre-existing CKD [79] as variations in serum creatinine concentration are more prominent in individuals with CKD. As with all other laboratory tests, serum creatinine measurements are affected by within- and between-sample coefficients of variation, intra-individual variation and biologic variation. Biological variation may result from variations in diet, muscle mass and breakdowns, tubular secretion, variability in volume homeostasis and from medications use [80]. The variation in measured serum creatinine level could be as high as 9% [1–11, 13, 15, 16]. Because only a small increase in serum creatinine is needed to meet AKI criteria, random variation in creatinine level may be a significant contributor to AKI diagnosis in the absence of a true reduction in GFR. This is called a false-positive AKI. It has been shown that a high variation in serum creatinine concentrations, in the days preceding the development of AKI, was not associated with inpatient mortality or dialysis [80]. In their study, Lin et al., using the KDIGO definition, found an 8% overall false-positive rate for AKI diagnosis. This false-positive rate was substantially higher at 30.5% for the subgroup of CKD patients with serum creatinine ≥1.5 mg/dL [80]. Therefore, an absolute change in serum creatinine of 0.3 mg/dL may represent a relatively inconsequential change in GFR among CKD patients compared to those without CKD. This fact might partially explain why most randomized trials for AKI interventions have been unsuccessful in improving clinical outcomes [81–84]. Under these circumstances, non-AKI events are likely to be misclassified as AKI events as there is no true reduction in GFR. Consequently, patients with false-positive AKI may be included in clinical trials of AKI and therefore dilute the observed effect size. These imperfections in the definition of AKI have potentially led to the generation of false conclusions regarding the efficacy or lack of efficacy of specific therapeutic interventions. The question of whether small changes in serum creatinine concentrations reflect clinically meaningful variations in kidney function which are then causally linked to adverse outcomes or are merely a marker of underlying severe disease or diminished renal reserve (which could be the actual mediators of the adverse outcomes) remains a major conundrum for the experts. Moreover, an AKI definition using small increments in serum creatinine concentrations has not been validated for use among patients with CKD.
A second major limitation of serum creatinine concentration is the inability to detect early changes in GFR. It takes a day or two for creatinine to rise after the reduction in GFR [85]. Therefore, the serum creatinine level at a point in time reflects a clinical event (kidney injury) that has happened in the past. It is now obvious that serum creatinine is an imperfect AKI biomarker; especially as it is being used on the basis of a relative change in value of a continuous variable instead of crossing a specific threshold [86–90]. Creatinine is a continuous variable that gets dichotomized to define a binary outcome i.e. AKI versus No AKI, using thresholds that are completely arbitrary. Using small changes in the serum creatinine to define AKI increases the sensitivity of the diagnostic criterion at the expense of specificity. The vice-versa happens when larger changes in creatinine are used.
An ideal AKI biomarker should accurately reflect the true level of kidney impairment, predict relevant outcomes, and be detectable early in the course of disease to allow for timely intervention. One of greatest challenges faced in the current evaluation of novel AKI biomarkers is that serum creatinine is used as the gold standard against which all other biomarkers are compared. The validation process is largely hindered by the use of an imperfect gold standard, and the downstream consequence of this action is misinterpretation of the diagnostic performance of the biomarker. To identify a more reliable biomarker for AKI, we first need to find a perfect gold standard for the validation process.
A third limitation in AKI research is the use of urine output for the diagnosis AKI. Research studies in AKI that rely on oliguria as a surrogate end-point lack precision and are likely to result in false conclusions. Urine output constitutes a major component of the diagnostic criteria for AKI [1–6]. Disappointingly, the correlation between the urine output criteria and the serum creatinine criteria remains quite poor [91]. In many occasions, oliguria may well reflect the response to hypovolaemia and suboptimal resuscitation rather than a true AKI event. Consequently, transient oliguria will lead to a diagnosis of AKI. It is also well known that the urine output is a physiological variable that may be affected by several types of medications. For example, patients treated with diuretics or dopamine could well have an increase in urine output independent of a true augmentation of kidney function, and this will alter the urine output criteria for diagnosing an AKI [10, 92]. Similarly, due to the non-linear relationship between body weight and urine output in obese patients, the use of a weight-based definition for AKI is an additional major limitation in this situation. Under the current definition, urine output of 50 mL per hour in a 110-kg patient for 12 h would result in misclassification as AKI stage-2 [10, 92].
Despite the relative advantages of the current AKI criteria, the adoption of serum creatinine as the gold standard for AKI diagnosis limits future advances in AKI research. It is likely that current AKI criteria will require further modification driven in part by development of more sensitive and specific biomarkers of kidney injury. It would be unrealistic to expect that we can develop the perfect de-novo AKI biomarker that can simultaneously diagnose AKI, stratify risk, and predict clinical outcomes over a short period of time. Indeed, it is very possible that the perfect AKI biomarker is in fact a composite set of several biomarkers that measure both kidney function & kidney damage. The development and clinical implementation of such biomarkers will help re-design the AKI classification system in a way that is not solely dependent upon serum creatinine.
The mystery of the unknown: baseline GFR and baseline creatinine
The glomerular filtration rate (GFR) varies under normal physiological conditions and during illness [92]. A popular example is that of a low GFR in vegetarians and higher GFR in consumers of large quantities of animal protein, even when they have a similar renal mass [93]. It is not clear what the maximum GFR can be, but it can be approached by subjecting subjects to high animal protein load [92–97]. The difference between baseline and maximal (i.e. stress or peak) GFR is called the Renal Functional Reserve (RFR) [93]. The maximum capacity of a functioning renal mass is not reflected by the baseline GFR of a given individual. Bellomo et al. explained this using an example of four different patients [93]. Patient A (animal protein consumer) and B (vegetarian) have the same renal mass but different baseline GFRs owing to different basal protein in-takes levels. Patient A has a GFR of 120 mL/min that can be stimulated to 170 mL/min. Patient B has a baseline GFR of 65 mL/min that also can be stimulated to 170 mL/min. Therefore, the RFR differs in these two patients because their GFR capacity is different. Patient C had a unilateral nephrectomy. The baseline GFR corresponds to his maximal GFR under unrestricted dietary conditions. If a moderate protein restriction is applied the baseline GFR may decrease and some degree of RFR become evident. Patient D, a vegetarian with a history of unilateral nephrectomy, will have a lower baseline compared to patient C but a higher RFR. Therefore, in general, restoring some RFR requires severe protein restriction, and hence baseline GFR does not always correspond to the extent of functioning renal mass unless we place it in the context of maximal capacity. Bellomo et al. espoused the view that the baseline GFR for an individual does not give us insight into the true renal function reserve, and needs to be determined: “In this regard GFR is not unlike a resting ECG for the kidney. When it is grossly abnormal, renal function is impaired, but when it is normal, a stress test is required.”
One of the challenges in accurately diagnosing AKI is the absence of a baseline serum creatinine concentration from laboratory records or when available the time lag between its measurement and the current serum creatinine value. The conundrum is whether the patient has a “true” AKI event or a progression of pre-existing CKD. KDIGO recommends that in the absence of a known baseline serum creatinine level prior to AKI, an estimated creatinine should be determined based upon an MDRD (Modification of Diet in Renal Disease) GFR of 75 mL/min per 1.73 m2 [6]. While this clinical construct has some benefits, there are residual deficiencies. Estimating the baseline serum creatinine using the KDIGO approach will almost certainly result in more patients with undiagnosed CKD patients being mislabelled as AKI.
Physiological variations in GFR, in the context of unknown RFR, and the situation of unknown baseline in presumed AKI patients remain as a major challenge in AKI studies (see Fig. 1). In such cases patients may be labelled as having an AKI event (in reality a pseudo-AKI event) when in fact, changes in serum creatinine concentrations are really due to biologic variation, measurement error and inter-laboratory differences.

An illustration of the limitations of the current AKI and CKD definitions, and the resulting ambiguity of the renal diagnosis. Availability of previous serum creatinine level reading have a huge impact on the final renal diagnosis for the same patient. Mr. X is a Caucasian who have an underlying progressive CKD, the onset of which occurred when he was 50 year old. At the age of 57, he was hospitalized with an acute illness and got his serum creatinine level checked. In scenario (1), the final renal diagnosis during hospitalization is progressive CKD, based on the availability of several previous creatinine readings and a recent reading i.e. 155. This is the correct diagnosis in his case. In scenario (2), during hospitalization, the same Mr. X will be misdiagnosed with AKI because of the absence of a recent creatinine reading and the presumed baseline creatinine of 82. In scenario (3), again he will be misdiagnosed with AKI due to the absence of a known creatinine baseline. In scenario (4), the same patient will be misdiagnosed as an AKI on CKD because he is a known case of CKD but did not have any creatinine level checked for several years before his acute hospitalization. In these scenarios, although the patient is the same and the underlying renal disease process did not change, the diagnosis was different in each scenario. It’s unknown how many similar cases were miscoded and labelled as AKI and subsequently got included in epidermiological studies linking AKI and CKD. AKI acute kidney injury, CKD chronic kidney disease, ESRD end stage renal disease
Assessing AKI in the community
The term community acquired-AKI (CA-AKI) has gained popularity with increased attention over the last few years. It is used to define AKI that occurs outside of the hospital setting [98–103]. CA-AKI is reported to account for 1% of all hospital admissions and is two to three times more prevalent than hospital-acquired AKI [29, 99–103]. The true incidence of CA-AKI, however, remains difficult to determine due to differences in AKI definitions, populations studied, timing of diagnosis and differences in inclusion criteria. One particular challenge relates to the classification of AKI as either a community-based event or a hospital-acquired event. It is widely known that changes in serum creatinine level lag behind changes in GFR, therefore, an elevation in creatinine level 48 h following a hospitalization may in truth be a reflection of a CA-AKI event rather than a hospital-acquired event [85]. This renders accurate differentiation between CA-AKI and hospital-acquired AKI very difficult, and thus there is a degree of overlap between these two terms. This is a major limitation of CA-AKI studies and reflects our inability to define the true time frame for the event. Nevertheless, CA-AKI is an important subgroup to study given the high frequency in clinical practice and the potential for early intervention at a primary care level. It is widely known that angiotensin converting enzyme inhibitors (ACE), angiotensin receptor blockers (ARBs) and diuretics are commonly implicated in pre-renal cases of CA-AKI [98]. With rising prevalence of hypertension (29–31 percent) in clinical practice, these medications are commonly prescribed to treat hypertension and reduce cardiovascular complications [104]. Similarly, their use is common among patients with heart failure patients and diabetic nephropathy. Primary prevention programmes for CA-AKI that recommend temporary withdrawal of these agents in the setting of volume depletion and sepsis may prove useful in limiting the severity of kidney injury and indeed longer term consequences.
It is quite possible that the frequency of AKI is far greater in the community than previously considered. A recent study by Xu and colleagues, have demonstrated higher rates of CA-AKI than previously shown using novel informatics software that permits greater detection of discrete AKI events [105]. Taking this into consideration, one might speculate that higher rates of AKI in the community may in part account for the increasing prevalence of CKD. First, undiagnosed CA-AKI events in the community may partly or fully explain cases of CKD that are labelled as having an unknown aetiology? Second, higher rates of CKD progression may in some patients be accounted for by higher frequency of AKI events? Third, is it possible that the high rates of community AKI may be responsible for the rising prevalence of hypertension in the general population? These are important research questions for the clinical and scientific community that require further detailed investigations.
Shared risk factors for AKI and CKD: a single disease continuum versus separate entities
Advanced age, black race, diabetes mellitus, hypertension, metabolic syndrome, atherosclerotic disease, and cardiac failure have all been hypothesised as potential risk factors for developing AKI [7, 11, 106]. Similar risk factors have been identified for CKD [107, 108]. It has been shown that patients with CKD have a 10-fold higher risk for AKI compared to patients without CKD [7, 25, 109]. These observations would suggest a significant overlap in risk factors for both AKI and CKD. This complex relationship between AKI and CKD makes it almost impossible to accurately adjust for confounding factors and hence limits the investigation of AKI as a risk factor for CKD, and vice versa. The complex interplay of AKI and CKD with regard to pathophysiology, shared risk factors and adverse outcomes remains an intriguing relationship as highlighted by Bedford et al. in a recent editorial [18].
There are several prospective studies underway to elucidate the clinical epidemiology of AKI in at-risk populations. Examples of these include the US-based Assessment, Serial Evaluation, and Subsequent Sequelae of Acute Kidney Injury (ASSESS-AKI) study and the UK-based AKI Risk in Derby (ARID) study [110, 111]. These studies aim to delineate risk factors that identify those at highest risk of adverse long-term outcomes. The ASSESS-AKI study is a prospective study that includes patients with and without CKD [110]. The goals of this study are to evaluate long-term outcomes of AKI in hospitalized patients, determine the natural history of AKI and to delineate the risk factors for progression and for complications. Similarly, the ARID study is a prospective, case-control study that aims to determine natural history, risk factors and long term outcomes of AKI in a general hospitalized population, including those with less severe AKI and pre-renal azotaemia [111]. The results of these observational studies are likely to shed new light on the relationships between AKI and CKD.
Biochemical versus histological recovery
There is substantial variation in nephron number among individuals, and this may reach up to 10-fold within select populations [112–122]. It has been suggested that a lower number of nephrons, acquired in utero, markedly increases susceptibility to future kidney disease [122]. Currently, low birth weight is the strongest clinical surrogate marker for an adverse intrauterine environment and the association with a reduced nephron number [123]. Kidneys with fewer nephrons have a smaller filtration surface area and hence diminished capacity for sodium excretion, with a subsequent development of hypertension and kidney disease [112, 113, 124–128]. A meta-analysis of 31 observational studies found a 70% increase in relative risk of CKD in individuals who had with low birth weight [129]. Similar findings were found in patients with acquired intrinsic renal disease and after surgical nephrectomy [122]. Studies have shown that low birth weight increases the prevalence of microalbuminuria and proteinuria in adulthood; likely a consequence of reduced nephron number [130–137].
Following an AKI event which results in nephron loss, the surviving nephrons undergo both structural and functional hypertrophy [138]. The mean driving force for glomerular filtration increases in the surviving glomeruli leading to an increase in filtration rate [139]. The greater the nephron loss the greater the increase in the glomerular filtration rate of the surviving nephrons [140]. In animal models, alterations in the glomerular structure are detected as early as 2 weeks following nephrons loss, and involve over half of the remaining nephrons by 7 weeks [141]. While this adaptive process is typically considered beneficial as it minimizes the reduction in total GFR that would otherwise occur, it nevertheless is detrimental in the long-term. The reduction in nephron number eventually leads to a pathological sclerotic process involving the surviving nephrons [141–144]. This progressive sclerosis represents a breakdown of the adaptation mechanism, and ultimately manifests as proteinuria and long term deterioration in renal function [145].
AKI may results from an injury to one or more segments within the kidney: the renal tubules, the interstitium, blood vessels or glomeruli. The damage that ensues is commonly secondary to a wide variety of insults, which may be ischemic, nephrotoxic, inflammatory or vascular-mediated (injury and/or occlusion). AKI may or may not result in nephron loss. It is well known that nephron loss from acute kidney injury is irreversible [146]. However, there is a paucity of studies that have evaluated kidney biopsy findings after complete recovery from AKI. Patients are usually labelled as ‘recovered from AKI’ when their serum creatinine level is back to the baseline following an AKI episode. It is unclear however if this putative recovery is purely biochemical, histological or both. It is expected that with a histological recovery a full regenerative process took place and was successful in restoring the damage tissues back to normal (i.e. to the pre-injury state). Unfortunately, serum creatinine level cannot be used as a surrogate marker for complete histological recovery. This is because if nephrons were lost during the event, then the surviving nephrons are likely to have undergone an adaptive process described above (i.e. structural and functional hypertrophy with increased filtration rate). These adaptive mechanisms may result in restoration of kidney function to the baseline serum creatinine level and GFR (i.e. a biochemical recovery) although there is no accompanying histological recovery. One might hypothesize that the absence of histological recovery and the resultant adaptive processes in the remaining nephrons may be responsible for the progression of AKI to CKD and ESRD later in life. In these circumstances, where histological recovery did not occur, AKI was responsible for the CKD process from the very outset (at least pathologically) rather than an AKI progressing to CKD. It is understandable that obtaining kidney biopsies from patients who recovered completely, at least biochemically, from AKI is not practical and nor ethical, but testing this hypothesis may help provide greater clarity and better understanding of the complex link between AKI and CKD.
Current perspective and future directions in AKI research
The term AKI is not a single entity but an umbrella term that encompasses a wide variety of renal disorders which lead to the acute reduction of GFR and the subsequent rise in serum creatinine concentration. The causes are diverse and vary in intensity and duration, and the resultant pathobiology and clinical outcomes are likely to reflect the type, magnitude and duration of the exposure. Indeed, despite our growing understanding of this complex condition, it is impossible to describe a standard AKI insult with a well-defined pathogenesis and pathophysiology. Consequently, it remains quite challenging to robustly and scientifically link AKI and CKD and vice versa. Most epidemiological studies that have associated AKI with CKD have investigated populations using the umbrella term “AKI”, but in each case, the cause of AKI was not defined or poorly defined. It is well known that some renal diseases have a progressive course that culminate in ESRD (e.g. acute rapidly progressive glomerulonephritis, acute interstitial nephritis, previously undiagnosed diabetic nephropathy) and that the rates of progression can vary widely. These may be inadvertently mislabelled as AKI in the absence of a baseline serum creatinine.
Regardless of the underlying kidney insult, an AKI reflects an acute deterioration in the functional status of the kidney. It is likely that the underlying kidney insult that causes the AKI is the principal determinant of the long-term prognosis & renal outcome rather than the AKI itself. This is supported by the fact that that current AKI staging system lacks predictive capability [147]. It would appear that the magnitude of change in serum creatinine level does not predict outcome. Furthermore, one must contend that the current AKI staging system, although an improvement on previous systems, remains a purely an artificial system and has not been sufficiently validated for use in the clinical management of patients [148]. The correlation between serum creatinine level and GFR is poor in the acute setting, and hence a rising creatinine level results in progression in AKI stage, despite improvement in GFR. Nevertheless, using the term AKI as a diagnosis is very useful to the practicing clinician in guiding investigations and management of patients. Unfortunately, this might not be the case when it is used for clinical research. In studies of AKI, classifying AKI patients by the underlying cause rather than using the generic term AKI may be more scientifically valid. This will assist clinical investigators in phenotyping patients with greater accuracy. Such studies of AKI may provide results that have greater validity and reliability.
If scientific studies in the future demonstrate that AKI and CKD represent a continuum rather than separate entities then it is likely that a paradigm shift will develop in the classification, diagnosis and management of acute and chronic kidney diseases. The use of AKI and CKD terms may be abolished then, and the underlying disease process may become the principal diagnosis with reference to the resultant change in GFR as a secondary effect that may or may not predict the outcome. It is reasonable to argue that the AKI aetiology may not be known early on until more definitive diagnostic tests are performed (e.g. kidney biopsy). As mentioned above, it is totally acceptable to use the term AKI as a temporary diagnosis in this setting, but it is better avoided when designing research studies and trials. Ideally, patients with AKI should be clustered according to the underlying aetiology. This may be the only scientifically valid method to obtain robust results about kidney disease progression and outcomes, and test various therapeutic and preventive interventions.
Conclusion
Despite significant advancements in our understanding of AKI and CKD, the precise nature of this relationship remains unclear. Several links are missing and without them it is very difficult to ascertain if these two conditions represent a disease continuum or separate distinct entities (see Fig. 2). A major paradigm shift in the classification and staging of AKI and CKD might need to happen first before these missing links can be found.

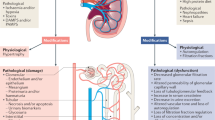
Factors contributing to the missing link between AKI and CKD. AKI acute kidney injury, CKD chronic kidney disease
References
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P (2004) Acute renal failure–definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 8(4):R204
Kellum JA, Bellomo R, Ronco C (2007) The concept of acute kidney injury and the RIFLE criteria. Contrib Nephrol 156:10–16
Mehta RL, Kellum JA, Shah SV et al (2007) Acute Kidney Injury Network (AKIN): report of an initiative to improve outcomes in acute kidney injury. Crit Care 11(2):R31
Levin A, Warnock DG, Mehta RL et al (2007) Improving outcomes from acute kidney injury: report of an initiative. Am J Kidney Dis 50(1):1–4
Molitoris BA, Levin A, Warnock DG, Joannidis M, Mehta RL, Kellum JA, Ronco C, Shah S (2007) Improving outcomes from acute kidney injury. J Am Soc Nephrol 18(7):1992–1994
Kidney Disease Outcomes Quality Initiative (2012) KDIGO Clinical practice guidelines for acute kidney injury. Kidney Int Suppl 2:1–38
Ishani A, Xue JL, Himmelfarb J et al (2009) Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20(1):223–228
Triverio P-A, Martin P-Y, Romand J, Pugin J, Perneger T, Saudan P (2009) Long-term prognosis after acute kidney injury requiring renal replacement therapy. Nephrol Dial Transplant 24(7):2186–2189
Mehta RL, Pascual MT, Soroko S et al (2004) Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 66(4):1613–1621
Mehta RL, Pascual MT, Soroko S, Chertow GM (2002) Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA 288(20):2547–2553
Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE (2011) The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79(12):1361–1369
Amdur RL, Chawla LS, Amodeo S, Kimmel PL, Palant CE (2009) Outcomes following diagnosis of acute renal failure in U.S. veterans: focus on acute tubular necrosis. Kidney Int 76(10):1089–1097
Lo LJ, Go AS, Chertow GM et al (2009) Dialysis-requiring acute renal failure increases the risk of progressive chronic kidney disease. Kidney Int 76(8):893–899
Wald R, Quinn RR, Luo J et al (2009) Chronic dialysis and death among survivors of acute kidney injury requiring dialysis. J Am Med Assoc 302(11):1179–1185. (Erratum, JAMA 2009;302:1532)
Bucaloiu ID, Kirchner HL, Norfolk ER, Hartle JE II, Perkins RM (2012) Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int 81(5):477–485
Wald R, Quinn RR, Adhikari NK et al (2012) Risk of chronic dialysis and death following acute kidney injury. Am J Med 125(6):585–593
Chawla LS, Eggers PW, Star RA, Kimmel PL (2014) Acute kidney injury and chronic kidney disease as interconnected syndromes. New Engl J Med 371(1):58–66
Bedford M, Farmer C, Levin A, Ali T, Stevens P (2012) Acute kidney injury and CKD: chicken or egg?. Am J Kidney Dis 59(4):485–491
Bellomo R, Kellum JA, Ronco C (2012) Acute kidney injury. The Lancet 380:756–766
Chawla LS, Kimmel PL (2012) Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 82:516–524
Star RA (1998) Treatment of acute renal failure. Kidney Int 54:1817–1831
Kheterpal S, Tremper KK, Heung M, Rosenberg AL, Englesbe M, Shanks AM et al (2009) Development and validation of an acute kidney injury risk index for patients undergoing general surgery: results from a national data set. Anesthesiology 110(3):505–515
Bagshaw SM, George C, Bellomo R (2008) and the ANZICS database management committee. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care 12:R47
Hoste EA, Clermont G, Kersten A et al (2006) RIFLE criteria for acute kidney injury are associated with hospital mortality in critically ill patients: a cohort analysis. Crit Care 10:R73
Xue JL, Daniels F, Star RA et al (2006) Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol 17:1135–1142
Hsu CY, McCullough CE, Fan D, Ordonez JD, Cherow GM, Go AS (2007) Community-based incidence of acute renal failure. Kidney Int 72:208–212
Goldberg R, Dennen P (2008) Long-term outcomes of acute kidney injury. Adv Chronic Kidney Dis 15(3):297–307
Panek R, Tennankore KK, Kiberd BA (2016) Incidence, etiology, and significance of acute kidney injury in the early post-kidney transplant period. Clin Transplant 30(1):66–70
Wonnacott A, Meran S, Amphlett B, Talabani B, Phillips A (2014) Epidemiology and outcomes in community-acquired versus hospital-acquired AKI. Clin J Am Soc Nephrol 9(6):1007–1014
Susantitaphong P, Cruz DN, Cerda J, Abulfaraj M, Alqahtani F, Koulouridis I, Jaber BL (2013) Acute kidney injury advisory group of the american society of nephrology: world incidence of AKI: a meta-analysis. Clin J Am Soc Nephrol 8:1482–1493
Thomas M, Davies A, Dawnay A (2013) Acute kidney injury prevention, detection and management of acute kidney injury up to the point of renal replacement therapy. NICE Clinical Guidelines
Kidney Disease Statistics for the United States (2016) National kidney and urologic diseases information clearinghouse (NKUDIC). http://kidney.niddk.nih.gov/kudiseases/pubs/kustats/#17. Accessed 11 Feb 2016
Saran R, Li Y, Robinson B et al (2016) US renal data system 2015 AnnuaL DATA REPORT: EPIDEMIOLOGY OF KIDNEY DISEAse in the United States. Am J Kidney Dis 67(3 suppl 1):S1–S434
Brück K, Stel VS, Gambaro G, Hallan S, Völzke H, Ärnlöv J, Kastarinen M, Guessous I, Vinhas J, Stengel B, Brenner H, Chudek J, Romundstad S, Tomson C, Gonzalez AO, Bello AK, Ferrieres J, Palmieri L, Browne G, Capuano V, Van Biesen W, Zoccali C, Gansevoort R, Navis G, Rothenbacher D, Ferraro PM, Nitsch D, Wanner C, Jager KJ; European CKD Burden Consortium (2016) CKD prevalence varies across the european general population. J Am Soc Nephrol 27(7):2135–2147
Centers for Disease Control and Prevention (2016) Deaths and mortality. http://www.cdc.gov/nchs/fastats/deaths.htm. Accessed 18 Feb 2016
Eknoyan G, Levin NW (2002) K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification-foreword. Am J Kidney Dis 39(2):S14–266
Hsu CY, Vittinghoff E, Lin F, Shlipak MG (2004) The incidence of end-stage renal disease is increasing faster than the prevalence of chronic renal insufficiency. Ann Intern Med 141(2):95–101
Coca SG, Singanamala S, Parikh CR (2012) Chronic kidney disease after acute kidney injury: a systematic review and metaanalysis. Kidney Int 81:442–448
Ishani A, Nelson D, Clothier B, Schult T, Nugent S, Greer N et al (2011) The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med 171:226–233
Newsome BB, Warnock DG, McClellan WM, Herzog CA, Kiefe CI, Eggers PW, Allison JJ (2008) Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Inter Med 168(6):609–616
James MT, Ghali WA, Tonelli M, Faris P, Knudtson ML, Pannu N, Klarenbach SW, Manns BJ, Hemmelgarn BR (2010) Acute kidney injury following coronary angiography is associated with a long-term decline in kidney function. Kidney Int 78(8):803–809
James MT, Hemmelgarn BR, Wiebe N et al (2010) Glomerular filtration rate, proteinuria, and the incidence and consequences of acute kidney injury: a cohort study. Lancet 376:2096–2103
Hsu CY, Chertow GM, McCulloch CE et al (2009) Nonrecovery of kidney function and death after acute on chronic renal failure. Clin J Am Soc Nephrol 4:891–898
Lafrance JP, Djurdjev O, Levin A (2010) Incidence and outcomes of acute kidney injury in a referred chronic kidney disease cohort. Nephrol Dial Transplant 25(7):2203–2209
Choi AI, Li Y, Parikh C, Volberding PA, Shlipak MG (2010) Long-term clinical consequences of acute kidney injury in the HIV-infected. Kidney Int 78(5):478–485
James MT, Ghali WA, Knudtson ML, Ravani P, Tonelli M, Faris P, Pannu N, Manns BJ, Klarenbach SW, Hemmelgarn BR (2011) Associations between acute kidney injury and cardiovascular and renal outcomes after coronary angiography. Circulation 123(4):409–416
Thakar CV, Christianson A, Himmelfarb J, Leonard AC (2011) Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin J Am Soc Nephrol 6:2567–2572
Steinwald B, Dummit LA (1989) Hospital case-mix change: sicker patients or DRG creep?. Health Affairs 8(2):35–47
Vlasschaert ME, Bejaimal SA, Hackam DG, Quinn R, Cuerden MS, Oliver MJ et al (2011) Validity of administrative database coding for kidney disease: a systematic review. Am J Kidney Dis 57:29–43
Bonventre JV (2010) Pathophysiology of AKI: injury and normal and abnormal repair. In: Cardiorenal syndromes in critical care 2010 Apr 20, vol 165. Karger Publishers, Berlin, pp 9–17
Yang L, Humphreys BD, Bonventre JV (2011) Pathophysiology of acute kidney injury to chronic kidney disease: maladaptive repair. In: Controversies in acute kidney injury 2011 Sep 9, vol 174. Karger Publishers, Berlin, pp 149–155
Zuk A, Bonventre JV, Brown D, Matlin KS (1998) Polarity, integrin, and extracellular matrix dynamics in the postischemic rat kidney. Am J Physiol 275:C711–C731
Thadhani R, Pascual M, Bonventre JV (1996) Acute renal failure. New Engl J Med 334:1448–1460
Kwon O, Hong SM, Ramesh G (2009) Diminished NO generation by injured endothelium and loss of macula densa nNOS may contribute to sustained acute kidney injury after ischemia–reperfusion. Am J Physiol Renal Physiol 296(1):F25–F33
Conger J (1997) Hemodynamic factors in acute renal failure. Adv Ren Replace Ther 4(suppl 1):25–37
Conger JD (1983) Vascular abnormalities in the maintenance of acute renal failure. Circ Shock 11:235–244
Linas S, Whittenburg D, Repine JE (1997) Nitric oxide prevents neutrophil-mediated acuterenal failure. Am J Physiol 272:F48–F54
Pulskens WP, Teske GJ, Butter LM, Roelofs JJ, van der Poll T, Florquin S, Leemans JC (2008) Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PloS One 3:e3596
Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S (2005) Renal-associated TLR2 mediates ischemia/ reperfusion injury in the kidney. J Clin Invest 115:2894–2903
Kelly KJ, Williams WW, Colvin RB, Bonventre JV (1994) Antibody to intercellular adhesion molecule-1 protects the kidney against ischemic injury. Proc Natl Acad Sci USA 91:812–816
Kelly KJ, Williams WW, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos JC, Bonventre JV (1996) Intercellular adhesion molecule-1-deficient mice are protected against renal ischemia. J Clin Invest 97:1056–1063
Homeister JW, Lucchesi BR (1994) Complement activation and inhibition in myocardial ischemia and reperfusion injury. Annu Rev Pharmacol Toxicol 34:17–40
Reis e Sousa C (2006) Dendritic cells in a mature age. Nat Rev Immunol 6:476–483
Park KM, Chen A, Bonventre JV (2001) Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem 276:11870–11876
Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR (2008) Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol 294(4):F928–F936
Zouggari Y, Ait-Oufella H, Waeckel L, Vilar J, Loinard C, Cochain C, Recalde A, Duriez M, Levy BI, Lutgens E, Mallat Z, Silvestre JS (2009) Regulatory T cells modulate postischemic neovascularization. Circulation 120:1415–1425
Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV (2008) Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2:284–291
Forbes JM, Hewitson TD, Becker GJ, Jones CL (2000) Ischemic acute renal failure: long- term histology of cell and matrix changes in the rat. Kidney Int 57:2375–2385
Macedo E, Bouchard J, Mehta RL (2008) Renal recovery following acute kidney injury. Curr Opin Crit Care 14:660–665
Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK (2010) Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298(5):F1078–F1094
Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV (2010) Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16(5):535–543
Akhtar A (2015) The flaws and human harms of animal experimentation. Cambr Q Healthc Ethics 24(04):407–419
National Kidney Foundation (2002) K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 39(2):S1–S266
Levey AS, Eckardt KU, Tsukamoto Y, Levin A, Coresh J, Rossert J, Zeeuw DD, Hostetter TH, Lameire N, Eknoyan G (2005) Definition and classification of chronic kidney disease: a position statement from kidney disease: improving global outcomes (KDIGO). Kidney Int 67(6):2089–2100
Preddie DC, Markowitz GS, Radhakrishnan J, Nickolas TL, D’Agati VD, Schwimmer JA, Gardenswartz M, Rosen R, Appel GB (2006) Mycophenolate mofetil for the treatment of interstitial nephritis. Clin J Am Soc Nephrol 1(4):718–722
Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW (2005) Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16(11):3365–3370
Hobson CE, Yavas S, Segal MS et al (2009) Acute kidney injury is associated with increased long-term mortality after cardiothoracic surgery. Circulation 119:2444–2453
Waikar SS, Liu KD, Chertow GM (2008) Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol 3:844–861
Lafrance JP, Miller DR (2010) Defining acute kidney injury in database studies: the effects of varying the baseline kidney function assessment period and considering CKD status. Am J Kidney Dis 56:651–660
Lin J, Fernandez H, Shashaty MG, Negoianu D, Testani JM, Berns JS, Parikh CR, Wilson FP (2015) False-positive rate of AKI using consensus creatinine-based criteria. Clin J Am Soc Nephrol 10:1723–1731
Ejaz AA, Dass B, Lingegowda V, Shimada M, Beaver TM, Ejaz NI, Abouhamze AS, Johnson RJ (2013) Effect of uric acid lowering therapy on the prevention of acute kidney injury in cardiovascular surgery. Int Urol Nephrol 45:449–458
Wilson FP, Shashaty M, Testani J, Aqeel I, Borovskiy Y, Ellenberg SS, Feldman HI, Fernandez H, Gitelman Y, Lin J, Negoianu D, Parikh CR, Reese PP, Urbani R, Fuchs B (2015) Automated, electronic alerts for acute kidney injury: a single-blind, parallel-group, randomised controlled trial. Lancet 385:1966–1974
Tumlin JA, Finkel KW, Murray PT, Samuels J, Cotsonis G, Shaw AD (2005) Fenoldopam mesylate in early acute tubular necrosis: a randomized, double-blind, placebo-controlled clinical trial. Am J Kidney Dis 46:26–34
Berger MM, Soguel L, Shenkin A, Revelly JP, Pinget C, Baines M, Chiólero R (2008) Influence of early antioxidant supplements on clinical evolution and organ function in critically ill cardiac surgery, major trauma, and subarachnoid hemorrhage patients. Crit Care 12:R101. doi:10.1186/cc6981
Herget-Rosenthal S, Pietruck F, Volbracht L, Philipp T, Kribben A (2005) Serum cystatin C–a superior marker of rapidly reduced glomerular filtration after uninephrectomy in kidney donors compared to creatinine. Clin Nephrol 64(1):41–46
Bagshaw SM, Mortis G, Doig CJ, Godinez-Luna T, Fick GH, Laupland KB (2006) One-year mortality in critically ill patients by severity of kidney dysfunction: a population-based assessment. Am J Kidney Dis 48:402–409
Chertow GM, Soroko SH, Paganini EP, Cho KC, Himmelfarb J, Ikizler TA, Mehta RL (2006) Mortality after acute renal failure: models for prognostic stratification and risk adjustment. Kidney Int 70:1120–1126
Lafrance JP, Miller DR (2010) Acute kidney injury associates with increased long-term mortality. J Am Soc Nephrol 21:345–352
Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C (2005) Beginning and ending supportive therapy for the kidney (BEST kidney) Investigators: acute renal failure in critically ill patients: A multinational, multicenter study. J Am Med Assoc 294:813–818
Waikar SS, Betensky RA, Emerson SC, Bonventre JV (2012) Imperfect gold standards for kidney injury biomarker evaluation. J Am Soc Nephrol 23:13–21
Ricci Z, Cruz D, Ronco C (2008) The RIFLE criteria and mortality in acute kidney injury: a systematic review. Kidney Int 73(5):538–546
Friedrich JO, Adhikari N, Herridge MS, Beyene J (2005) Metaanalysis: low-dose dopamine increases urine output but does not prevent renal dysfunction or death. Ann Intern Med 142(7):510–524
Bellomo R, Kellum JA, Ronco C (2004) Defining acute renal failure: physiological principles. Intensive Care Med 30:33–37. doi:10.1007/s00134-003-2078-3
Bosch JP, Lauer A, Glabman S (1984) Short-term protein loading in assessment of patients with renal disease. Am J Med 77:873–879
Bosch JP, Saccaggi A, Lauer A, Ronco C, Belledonne M, Glabman S (1983) Renal functional reserve in humans. Effect of protein intake on glomerular filtration rate. Am J Med 75:943–950
Bosch JP, Lew S, Glabman S, Lauer A (1986) Renal hemodynamic changes in humans. Response to protein loading in normal and diseased kidneys. Am J Med 81:809–815
Ronco C, Brendolan A, Bragantini L, Chiaramonte S, Fabris A, Feriani M, Dell Aquila R, Milan M, Mentasti P, La Greca G (1988) Renal functional reserve in pregnancy. Nephrol Dial Transplant 3:157–161
Der Mesropian P, Othersen J, Mason D, Wang J, Asif A, Mathew RO (2016) Community acquired acute kidney injury: a challenge and opportunity for primary care in kidney health. Nephrology (Carlton) 21(9):729–735. doi:10.1111/nep.12751
Kaufman J, Dhakal M, Patel B, Hamburger R (1991) Community-acquired acute renal failure. Am J Kidney Dis 17(2):191–198
Liano F, Pascual J (1996) Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid acute renal failure study group. Kidney Int 50(3):811–818
Talabani B, Zouwail S, Pyart RD, Meran S, Riley SG, Phillips AO (2014) Epidemiology and outcome of community-acquired acute kidney injury. Nephrology (Carlton) 19(5):2827
Obialo CI, Okonofua EC, Tayade AS, Riley LJ (2000) Epidemiology of de novo acute renal failure in hospitalized African Americans: comparing community-acquired vs hospital-acquired disease. Arch Intern Med 160(9):1309–1313
Schissler MM, Zaidi S, Kumar H, Deo D, Brier ME, McLeish KR (2013) Characteristics and outcomes in community-acquired versus hospital-acquired acute kidney injury. Nephrology (Carlton) 18(3):183–187. doi:10.1111/nep.12036
Der Mesropian PJ, Kalamaras JS, Eisele G, Phelps KR, Asif A, Mathew RO (2014) Long-term outcomes of community-acquired versus hospital-acquired acute kidney injury: a retrospective analysis. Clin Nephrol 81(3):174–184
Xu G, Player P, Shepherd D, Brunskill NJ (2016) Identifying acute kidney injury in the community—a novel informatics approach. J Nephrol 29(1):93–98
Egan BM, Zhao Y, Axon RN (2010) US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. J Am Med Assoc 303(20):2043–2050
Pascual J, Liaño F, Ortuño J (1995) The elderly patient with acute renal failure. J Am Soc Nephrol 6(2):144–153
U.S. Renal Data System (2016) USRDS 2007 annual data report. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, 2007. https://www.usrds.org/atlas07.aspx. Accessed 1 Mar 2016
Levey AS, Coresh J (2012) Chronic kidney disease. Lancet 379:165–180
Go AS, Parikh CR, Ikizler TA et al (2010) The Assessment, Serial Evaluation, and Subsequent Sequelae of Acute Kidney Injury (ASSESS-AKI) study: design and methods. BMC Nephrol 11:22
Horne KL, Packington R, Monaghan J, Reilly T, McIntyre CW, Selby NM (2014) The effects of acute kidney injury on long-term renal function and proteinuria in a general hospitalised population. Nephron Clin Pract 128:192–200
Hoy WE, Hughson MD, Bertram JF, Douglas-Denton R, Amann K (2005) Nephron number, hypertension, renal disease, and renal failure. J Am Soc Nephrol 16:2557–2564. doi:10.1681/ASN.2005020172
Keller G, Zimmer G, Mall G, Ritz E, Amann K (2003) Nephron number in patients with primary hyper-tension. N Engl J Med 348:101–108. doi:10.1056/NEJMoa020549
McNamara BJ, Diouf B, Douglas-Denton RN, Hughson MD, Hoy WE, Bertram JF (2010) A comparison of nephron number, glomerular volume and kidney weight in Senegalese Africans and African Ameri-cans. Nephrol Dial Transplant 25:1514–1520. doi:10.1093/ndt/gfq030
Hinchliffe SA, Sargent PH, Howard CV, Chan YF, van Velzen D (1991) Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and cavalieri principle. Lab Invest 64:777–784
Hughson M, Farris AB, Douglas-Denton R, Hoy WE, Bertram JF (2003) Glomerular number and size in autopsy kidneys: the relationship to birth weight. Kidney Int 63:2113–2122. doi:10.1046/j.1523-1755.2003.00018.x
Manalich R, Reyes L, Herrera M, Melendi C, Fundora I (2000) Relationship between weight at birth and the number and size of renal glomeruli in hu-mans: a histomorphometric study. Kidney Int 58:770–773. doi:10.1046/j.1523-1755.2000.00225.x
McNamara BJ, Diouf B, Hughson MD, Douglas-Denton RN, Hoy WE, Bertram JF (2008) Renal pathology, glomerular number and volume in a West African urban community. Nephrol Dial Transplant 23:2576–2585. doi:10.1093/ndt/gfn039
McNamara BJ, Diouf B, Hughson MD, Hoy WE, Bertram JF (2009) Associations between age, body size and nephron number with individual glomerular volumes in urban West African males. Nephrol Dial Transplant 24:1500–1506. doi:10.1093/ndt/gfn636
Nyengaard JR, Bendtsen TF (1992) Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat Rec 232:194–201. doi:10.1002/ar.1092320205
Rodriguez MM, Gomez AH, Abitbol CL, Chandar JJ, Duara S, Zilleruelo GE (2004) Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr Dev Pathol 7:17–25. doi:10.1007/s10024-003-3029-2
Zimanyi MA, Hoy WE, Douglas-Denton RN, Hughson MD, Holden LM, Bertram JF (2009) Nephron number and individual glomerular volumes in male Caucasian and African American subjects. Nephrol Dial Transplant 24:2428–2433. doi:10.1093/ndt/gfp116
Brenner BM, Garcia DL, Anderson S (1988) Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens 1:335–347
Luyckx VA, Shukha K, Brenner BM (2011) Low nephron number and its clinical consequences. Rambam Maimonides Med J 2(4):e0061
Vehaskari VM, Aviles DH, Manning J (2001) Prenatal programming of adult hypertension in the rat. Kidney Int 59:238–245. doi:10.1046/j.1523-17552001.00484.x
Baum M (2010) Role of the kidney in the prenatal and early postnatal programming of hypertension. Am J Physiol Renal Physiol 298:F235–F247. doi:10.1152/ajprenal.00288.2009
Bhathena DB, Julian BA, McMorrow RG, Baehler RW (1985) Focal sclerosis of hypertrophied glomeruli in solitary functioning kidneys of humans. Am J Kidney Dis 5:226–232
McMillen IC, Robinson JS (2005) Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85:571–633. doi:10.1152/physrev.00053.2003
White SL, Perkovic V, Cass A et al (2009) Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am J Kidney Dis 54:248–261. doi:10.1053/j.ajkd.2008.12.042
Hoy WE, Rees M, Kile E, Mathews JD, McCredie DA, Pugsley DJ, Wang Z (1998) Low birthweight and renal disease in Australian aborigines. Lancet 352:1826–1827
Hoy WE, Wang Z, VanBuynder P, Baker PR, Mathews JD (2001) The natural history of renal disease in Australian aborigines: part 1. Changes in albuminuria and glomerular filtration rate over time. Kidney Int 60:243–248
Hoy WE, Mathews JD, McCredie DA, Pugsley DJ, Hayhurst BG, Rees M, Kile E, Walker KA, Wang Z (1998) The multidimensional nature of renal disease: rates and associations of albuminuria in an Australian aboriginal community. Kidney Int 54:1296–1304
Hoy WE, Rees M, Kile E, Mathews JD, Wang Z (1999) A new dimension to the Barker hypothesis: low birthweight and susceptibility to renal disease. Kidney Int 56:1072–1077
Nelson RG, Morgenstern H, Bennett PH (1998) Birth weight and renal disease in Pima Indians with type 2 diabetes mellitus. Am J Epidemiol 148:650–656
Yudkin JS, Martyn CN, Phillips DI, Gale CR (2001) Associations of micro-albuminuria with intra-uterine growth retardation. Nephron 89:309–314
Painter RC, Roseboom TJ, van Montfrans GA, Bossuyt PM, Krediet RT, Osmond C, Barker DJ, Bleker OP (2005) Microalbuminuriain adults after prenatal exposure to the Dutch famine. J Am Soc Nephrol 16:189–194
Hayslett JP (1979) Functional adaptation to reduction in renal mass. Physiol Rev 59:137–164
Deen WM, Maddox DA, Robertson CR, Br en ner B (1974) Dynamics of glomerular ultrafiltration in the rat. VII: response to reduced renal mass. Am J Physiol 227:556–562
Kaufman JM, Siegel NJ, Hayslett JP (1975) Functional and hemodynamic adaptation to progressive renal ablation. Circ Res 36:286–293
Purkerson ML, Hoffsten PE, Klahr S (1976) Pathogenesis of the glomerulopathy associated with renal infarction in rats. Kidney Int 9(5):407–417
Morrison AB (1966) Experimental chronic renal insufficiency. Methods Achiev Exp Pathol 1:455–475
Shea SM, Raskova J, Morrison AB (1978) A stereologic study of glomerular hypertrophy in the subtotally nephrectomized rat. Am J Pathol 90(1):201
Shimamura T, Morrison AB (1975) A progressive glomerulosclerosis occurring in partial five-sixths nephrectomized rats. Am J Pathol 79(1):95
Brenner BM, Lawler EV, Mackenzie HS (1996) The hyperfiltration theory: a paradigm shift in nephrology. Kidney Int 49(6):1774–1777
Lam AQ, Bonventre JV (2015) Regenerating the nephron with human pluripotent stem cells. Curr Opin Organ Transplant 20(2):187–192
Coca SG, King JT Jr, Rosenthal RA, Perkal MF, Parikh CR (2010) The duration of postoperative acute kidney injury is an additional parameter predicting long-term survival in diabetic veterans. Kidney Int 78(9):926–933
Palevsky PM, Liu KD, Brophy PD, Chawla LS, Parikh CR, Thakar CV, Tolwani AJ, Waikar SS, Weisbord SD (2013) KDOQI US commentary on the 2012 KDIGO clinical practice guideline for acute kidney injury. Am J Kidney Dis 61(5):649–672
Schreuder MF, Langemeijer ME, Bokenkamp A, Delemarre-Van de Waal HA, Van Wijk JA (2008) Hypertension and microalbuminuria in children with congenital solitary kidneys. J Paediatr Child Health 44:363–368. doi:10.1111/j.1440-1754.2008.01315.x
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors have no relevant financial relationships to disclose.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
For this type of study formal consent is not required.
Rights and permissions
About this article

Cite this article
Kaballo, M.A., Elsayed, M.E. & Stack, A.G. Linking acute kidney injury to chronic kidney disease: the missing links. J Nephrol 30, 461–475 (2017). https://doi.org/10.1007/s40620-016-0359-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-016-0359-5