
Abstract
Background
While reproductive technologies are increasingly used worldwide, epidemiologic, clinical and genetic data regarding infertile men with combined genital tract and renal abnormalities remain scarce, preventing adequate genetic counseling.
Methods
In a cohort-based study, we assessed the prevalence (1995–2014) and the clinical characteristics of renal disorders in infertile males with genital tract malformation. In a subset of 34 patients, we performed a detailed phenotype analysis of renal and genital tract disorders.
Results
Among the 180 patients with congenital uni- or bilateral absence of vas deferens (CU/BAVD), 45 (25 %) had a renal malformation. We also identified 14 infertile men with combined seminal vesicle (SV) and renal malformation but no CU/BAVD. Among the 34 patients with detailed clinical description, renal disease was unknown before the assessment of the infertility in 27 (79.4 %), and 7 (20.6 %) had chronic renal failure. Four main renal phenotypes were observed: solitary kidney (47 %); autosomal-dominant polycystic kidney disease (ADPKD, 0.6 %); uni- or bilateral hypoplastic kidneys (20.6 %); and a complex renal phenotype associated with a mutation of the HNF1B gene (5.8 %). Absence of SV and azoospermia were significantly associated with the presence of a solitary kidney, while dilatation of SV and necroasthenozoospermia were suggestive of ADPKD.
Conclusion
A dominantly inherited renal disease (ADPKD or HNF1B-related nephropathy) is frequent in males with infertility and combined renal and genital tract abnormalities (26 %). A systematic renal screening should be proposed in infertile males with CU/BAVD or SV disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
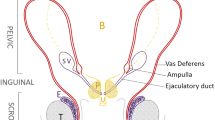
Infertility is recognized in about 7 % of male individuals in Western countries [1]. Malformations of the excretory systems are one of the congenital causes of male infertility and include uni- or bilateral heterogeneous disorders, such as absence, hypotrophy or dilatation of the vas deferens, epididymis or seminal vesicles. Testis hypotrophy or absence is also reported in association with other genital tract disorders. In males investigated for infertility, 1–2 % harbor a congenital unilateral or bilateral absence of the vas deferens (CU/BAVD); this percentage increases to 10 % in patients referred for azoospermia [2–4]. Congenital diseases affecting a component of the seminal tract, but not associated with lumen absence, can also be associated with partial impairment of one of the sperm parameters and subsequent infertility.
Genetic studies of the congenital malformations of the male excretory system are currently limited to the analysis of the CFTR gene in men with CU/BAVD [5, 6]. CU/BAVD linked to CFTR mutations is inherited as an autosomal recessive disorder. In patients with autosomal-dominant polycystic kidney disease (ADPKD, PKD1 or PKD2 mutations), cysts in the seminal tract have been encountered in ~50 % of patients but were not correlated to sperm parameters or fertility [7]. In this last study, renal disease was recognized before genital tract abnormality in all patients.
Because the seminal tract and the kidney/urinary tract share a common embryological origin, disorders that occur at an early stage in the developmental process may lead to combined genital and kidney/urinary tract malformations, occasionally accompanied by male infertility and chronic kidney disease [8, 9]. Most patients with CU/BAVD and renal abnormalities have no or only one variation of CFTR, suggesting genetic heterogeneity or non-inherited mechanisms of the genital/renal disorder in some cases [9, 10]. In addition, the development of new treatments for male infertility (for instance, intra-cytoplasmic sperm injection [ICSI] followed by embryo transfer [ET]) carries the risk of transmission of inherited renal disease [9]. Unfortunately, to date, no study has provided in-depth renal phenotyping of these patients, thus precluding optimal genetic counseling.
Herein, we report the prevalence of renal abnormalities in infertility related to excretory-system abnormalities, as well as the genotypes and phenotypes of 34 men referred for this peculiar association.
Materials and methods
Patients
In this retrospective study, we included adult males with a diagnosis of male infertility related to morphological abnormalities of their genital excretory system and renal malformations detected incidentally (1995–2008) or by systematic screening on abdominal imaging (2009–2014). Patients were followed at the Male Sterility Center and the Nephrology Department of the University Hospital of Toulouse (France). Most patients were cases of primary infertility (n = 27). In seven patients, renal disease was known before infertility. Patients were included irrespective of their CFTR genotype.
While the prevalence of the association of genital tract and kidneys malformations was approximated according to a database including all patients referred between 1995 and 2014, characterization of the genotypes and the phenotypes was based on the subset of patients with sufficient details concerning the malformations. All patients first had a physical examination for the detection of vasal and epididymal (caput, corpus and cauda) abnormalities. The scrotum and kidneys were then examined by ultrasonography (US). Thirteen out of the 16 individuals with azoospermia had epididymal or testicular surgical extraction of sperm, and all parts of the epididymis and vasa deferentia were carefully assessed. Supplementary Table 1 reports the characteristics of the genital tract according to the physical examination or to the surgical exploration.
Clinical, imaging and biological data were collected according to a standardized chart.
Imaging studies
A single experienced physician (I.F.) performed all ultrasonographic abdominal and scrotal examinations of the 34 patients, as well as transrectal ultrasonography (TRUS) in 32. Data from the renal US images and abdominal computed tomography (CT) scans or magnetic resonance imaging (MRI) were obtained for 34 and 25 patients, respectively.
Semen collection
After 3–6 days of recommended sexual abstinence, semen samples were produced by masturbation and collected in sterile containers at the Sperm Laboratory in the Reproductive Medicine service of the Hospital. Semen analyses were performed according to the 1999 and 2010 World Health Organization laboratory manuals. Semen volume was assessed as recommended, and semen pH measured within 1 h of ejaculation using a pH paper (6.5–10.0; VWR International, Fontenay-sous-Bois, France); sperm count was performed on a Malassez cell (Rogo & Cie, Arcueil, France); percentage of live spermatozoa was assessed by identifying those with an intact cell membrane from dye exclusion (using eosin–nigrosin test) within 1 h of ejaculation.
Genetic testing
All patients gave their written informed consent before DNA collection. The institutional Ethical Committee (University Hospital of Toulouse) approved DNA collection and genetic testing and the study complied with the Declaration of Helsinki revised in 2003. CFTR and HNF1B genetic testing were performed in the Department of Medical Genetics at the University Hospital of Toulouse. Screening of the CFTR gene included a search for point mutations and exon deletions, corresponding to a detection rate of 85 to >95 %, according to geographic origin. Screening of the HNF1B gene consisted of a search for exon or whole-gene deletions/duplications using quantitative multiplex polymerase chain reaction of short fragments (QMPSF), followed by direct sequencing of the nine exons and intro-exon boundaries, as previously described [11]. Testing of HNF1B was restricted to patients without ADPKD (n = 7) and with a clinical HNF1B score ≥8 after 2011, as recommended [12].
Results
Prevalence of combined genital tract and renal malformations
From May 1995 to December 2014, congenital bilateral absence of the vas deferens (CBAVD) was diagnosed in 111 patients. Among the 96 patients with renal imaging, 18 (18.8 %) had a solitary kidney (n = 13; 13.5 %) or renal cysts (n = 5; 5.2 %). During the same period, congenital unilateral absence of the vas deferens (CUAVD) was diagnosed in 69 patients. Among the 64 patients with renal imaging, 27 (42.2 %) had a solitary kidney (n = 20; 31.2 %) or renal cysts (n = 7; 10.9 %). Nine out of the 18 CBAVD and 11 out of the 27 CUAVD patients were included in the clinical descriptive cohort. In addition, we also identified 14 patients with renal malformations and seminal vesicle abnormalities (see below), but no CU/BAVD. Overall, detailed clinical data were available in 34 individuals with abnormalities of their genital excretory system (32 years, range: 25–50) and with renal malformations recognized before (n = 7, 18 %) or at the time (n = 27, 82 %) of infertility assessment. The genetic and clinical characteristics are summarized in Table 1. More detailed analyses of individual abnormalities are reported in Supplementary Tables 1 and 2.
Genetic testing
CFTR was analyzed for mutations in all patients with CU/BAVD. Three patients displayed monoallelic CFTR mutations: one patient with ADPKD harbored three CFTR mutations (p.Arg74Trp, p.Val201Met and p.Asp1270Asn) segregating in cis; one patient had an orthotopic solitary kidney (CFTR mutation p.Asp443Tyr); one had a few cortical cysts without family history of ADPKD (CFTR mutation p.Leu206Trp).
A renal disease was reported in at least one first-degree relative in 6 of the 34 patients (17 %). Diagnosis of ADPKD was established in five patients according to consensus criteria [13]. In two additional patients, renal imaging identified bilaterally enlarged multicystic kidneys. In those patients, the lack of a family history of renal and liver cysts suggested a de novo mutation of PKD1 or PKD2.
Since genital tract malformations belong to the spectrum of HNF1B-related disease, in both male and female patients [14–16], HNF1B mutations were analyzed in 20 of the 27 patients without ADPKD. Two patients harbored a HNF1B mutation consisting of a whole-gene deletion in one and an aminoacid substitution (p.Pro437Leu) in the other.
Genital tract abnormalities and semen parameters
In all patients, infertility was associated with genital tract malformations. Abnormality of the seminal vesicles was the most prominent finding (93.7 %) and included mainly a uni- or bilateral absence (28.1 and 34.4 %, respectively). Twenty-seven individuals (79.4 %) had a vas deferens malformation (CU/BAVD 58.8 %, dilatation 20.6 %).
Sixteen of the 34 patients (47.1 %) had an absence of corpus and/or cauda in one or both epididymides, whereas dilatation was identified in ten (29.4 %). Testis hypotrophy was found in 13 individuals (38.2 %), including six with bilateral hypotrophy.
Azoospermia was observed in 16 individuals (47.1 %), oligozoospermia and normozoospermia in 9 (27.2 %). In 8 of the 18 patients with oligo/normozoospermia, necroasthenozoospermia was present. Finally, semen volume was lower than normal in 22/34 patients (64.7 %) and 7 (20.6 %) also displayed an acid pH.
Renal findings
The main renal phenotypes associated with the genital disorders were as follows: (1) solitary kidney (n = 16, 47 %) in orthotopic (n = 11) or pelvic (n = 5) position; (2) polycystic kidney disease suggestive of PKD1 or PKD2 mutations (n = 7, 20.6 %); (3) uni- or bilateral hypoplastic kidneys (n = 7, 20.6 %); and (4) a complex renal phenotype (solitary cystic kidney or bilateral microcystic hyperechogenic kidneys) associated with a mutation of the HNF1B gene (n = 2, 5.9 %) (Fig. 1). Of the two latter patients, one individual had severe unilateral kidney atrophy and a megaureter; the other had some cortical cysts in both kidneys, but no family history of ADPKD and no HNF1B mutation.

Examples of renal imaging of male patients with infertility related to excretory-system abnormalities. a Horseshoe kidney, b solitary normal right kidney, c bilateral polycystic kidney disease and d solitary pelvic kidney
At the time of infertility assessment, one patient was on chronic dialysis, and one had received renal transplantation. In three of the four patients that displayed stage 3 CKD (eGFR <60 ml/min/1.73 m2), renal disease was unrecognized before the characterization of the infertility.
In patients with ADPKD, imaging showed various abnormalities of the vas deferens in four patients: dilatation (n = 2), calcifications (n = 1) and absence (n = 1). Calcifications of the epididymis were identified in two individuals. Seminal vesicles were either bilaterally absent (n = 2), uni/bilaterally dilated (n = 2), or had calcifications (n = 1). Semen analyses showed two kinds of disorders: necro-asthenozoospermia [n = 5/7; with oligo- (n = 3) or normozoospermia (n = 2)], or azoospermia and low semen volume (n = 2/7).
Among the 11 patients with an orthotopic solitary kidney, five had CUAVD (ipsilateral n = 3, contralateral n = 2), two had CBAVD, and three displayed vas deferens dilatation. Bilateral absence of seminal vesicles was observed in three patients. Semen parameters were heterogeneous and included azoospermia in 45 %. In contrast, all five patients with a pelvic solitary kidney had CBAVD and azoospermia. A uni- or bilateral absence of the epididymal corpus and/or cauda was observed in 7/11 (63 %) and in 5/5 (100 %) of patients with non-pelvic or pelvic solitary kidneys, respectively.
In six of the seven patients with hypoplastic kidneys, CUAVD (n = 5) or deferential bulb absence (n = 1) was identified. Among the six patients with unilateral hypoplastic kidneys, three (50 %) had an ipsilateral absence of the epididymal corpus and cauda. Seminal vesicles were absent in five and dilated in the sixth patient.
The patients with a mutation of HNF1B had two kinds of genital tract disorders. The individual with a whole-gene deletion of HNF1B harbored CBAVD, left seminal-vesicle hypotrophy, complete left testis-epididymis dissociation and an absence of the right caput, corpus and cauda of the epididymis. The second patient had bilateral testis hypotrophy, a dilatation of both seminal vesicles and caput epididymides, and calcifications of the genital tract. Both patients had an azoospermia with a low semen volume.
Finally, in this cohort, unilateral or bilateral absence of seminal vesicles was significantly associated with a solitary kidney (5/9 and 8/11, respectively), as well as azoospermia (62.5 % of azoospermic individuals). CUAVD was ipsilateral to the kidney anomaly in 80 % of these patients. In contrast, dilatation of at least one seminal vesicle and necroasthenozoospermia were more suggestive of ADPKD: 2/4 (50 %) and 5/8 patients (62.5 %), respectively.
Other manifestations
Besides kidney, urinary and genital tract abnormalities, seven individuals (20.7 %) had unilateral (n = 4) or bilateral cryptorchidism (n = 3), five (14.8 %) had unilateral or bilateral inguinal hernia, one displayed pancreas atrophy, and two had a polymalformative syndrome, including anal atresia or a vertebral malformation. Last, four patients with a renal phenotype consistent with ADPKD also harbored liver cysts.
Outcomes of pregnancies: spontaneous or following assisted-reproduction technologies
Attempts and outcomes of pregnancies were available for 28/34 couples. The results are summarized in Fig. 2. Among the patients with ADPKD, three completed a pregnancy. No renal cysts were detected in their children (screened at <10 years of age). The two patients with HNF1B mutations became parents with the help of the ICSI-ET method. In the patient with a whole-gene deletion, both female twins harbored the HNF1B deletion and developed ADPKD-like cystic kidney disease, diabetes mellitus of MODY type, liver-enzyme abnormalities, and hypomagnesemia. In the patient with the p.Pro437Leu substitution, the male child developed antenatal bilateral hyperechogenic microcystic kidneys. Among the nine non-ADPKD and non-HNF1B patients with azoospermia, eight benefited from ICSI-ET (seven pregnancies; no overt malformations in the antenatal US). Among the 11 non-ADPKD and non-HNF1B patients with normo- or oligozoospermia, three couples had an uncomplicated spontaneous or assisted pregnancy.

Outcomes of pregnancies and assisted-reproductive technologies in a cohort of 34 male patients with infertility related to excretory-system abnormalities and renal malformations. ART assisted reproductive technologies, ICSI intra-cytoplasmic sperm injection (surgical sperm extraction either from the epididymis or testis), MODY maturity onset diabetes of the young, CRF, chronic renal failure, ADPKD autosomal-dominant polycystic kidney disease
Discussion
Development of assisted reproductive technologies (ART), including ICSI-ET, has led to an increase in the number of successful pregnancies in couples consulting for male infertility. Given the frequency of CFTR variations in patients with CBAVD, genetic testing of CFTR is recommended in male patients with CBAVD-related infertility [17]. However, no firm recommendations as to extra-genital explorations and additional genetic screening have been established for patients with male infertility and genital tract malformations. Moreover, the spectrum of renal malformations, tissue with a common embryological origin, associated with male infertility has been poorly described to date.
In this study including mostly cases of primary infertility, we confirm that renal abnormalities, mostly asymptomatic, can be identified in male patients with infertility and genital malformations, especially when CFTR screening was negative [8, 10]. Prevalence of kidney involvement associated to male infertility related to CU/BAVD could be estimated to be around 25 %, but we show that also the identification of seminal vesicle abnormalities should prompt to search for renal malformation. These features need to be confirmed in a larger and more systematic study, but they confirm the frequent association of renal and genital malformations.
We have confirmed that the simultaneous occurrence of seminal-tract and renal malformations can be identified from fetuses to adulthood [18]. Renal abnormalities were heterogeneous but four main phenotypes emerged: ADPKD; a solitary orthotopic or pelvic kidney; uni- or bilateral hypoplastic kidneys; and a HNF1B-related disease which is a rare dominantly inherited developmental disease. This latter condition has already been recognized [16] and may be associated with clinically detectable genital involvement in 10–30 % of cases, while renal penetrance is almost complete [12]. In our series, one patient had a point mutation of HNF1B not previously described (p. Pro437Leu). The identification of bilateral microcystic hyperechogenic kidneys (antenatal period) in one of his children (also harboring this variant), a phenotype previously associated with HNF1B mutation [19], confirmed the pathogenicity of the mutation. As previously described, the second HNF1B harbored a whole-gene deletion of HNF1B confirming the need to search for both 17q12 micro rearrangement and point mutation [11]. Altogether, in our cohort ~25 % of patients had ADPKD or HNF1B disease. ART may therefore be at risk of renal-disease transmission in unselected male patients, as we have observed for HNF1B mutations.
Among the 25 individuals without proven HNF1B-related nephropathy or ADPKD, none gave birth to children with overt antenatal renal malformations. The lack of family history of renal diseases may suggest recessive inheritance, a dominant disorder with incomplete penetrance, or acquired developmental disorder, related or not to environment. In the latter case, transmission of renal disease would be null or at a very low risk.
In the seven patients with ADPKD included in this cohort, a wide spectrum of genital malformations was identified. These involved all parts of the genital tract, including the absence of seminal vesicles, and this was not restricted to seminal-tract cysts, contrasting with a previous series [7]. Although morphology of the genital tract cannot point specifically to ADPKD, semen analysis showed that necroasthenozoospermia was highly suggestive of ADPKD in patients with normo-/oligozoospermia, a finding previously recognized [7]. Interestingly, three patients with ADPKD had oligozoospermia with necroasthenozoospermia but mild to moderate malformations of the genital tract suggesting a superimposed functional defect. Recent studies showed that both PKD1 and PKD2 control the male reproductive system development but also maintain epithelial integrity [20]. Moreover, PKD2 expression is also required for testis development and directional sperm movement [20, 21]. Altogether, several fundamental studies have pointed to the role of PKD1 and PKD2 in male fertility and provide a molecular rationale to explain the frequent finding of necroasthenozoospermia in patients with ADPKD. Further studies, in a larger cohort of patients, are required to determine if such association between semen parameters, genital-tract malformations, and specific renal diseases can be confirmed.
In our study, all patients were selected when consulting for infertility and in most of them the renal disorder was recognized concomitantly. This finding suggests that minimal renal screening, including abdominal imaging, should be offered to male patients with a seminal-tract malformation. Patients with overt renal disease should be referred to a nephrologist to start a specific follow-up and to address the risk of inherited kidney disease before beginning a pregnancy, either natural or with ART. On the other hand, infertile men with renal malformations should have a transrectal ultrasonography.
In summary, due to the presence of additional renal abnormalities in men with infertility and genital tract abnormalities, and the high incidence of dominantly inherited renal in disease in patients combining both disorders, we recommend systematic renal screening in all infertile men with genital tract abnormality, especially in patients without CFTR alteration.
Abbreviations
- ADPKD:
-
Autosomal dominant polycystic kidney disease
- ART:
-
Assisted reproductive technologies
- CU/BAVD:
-
Congenital unilateral/bilateral absence of vas deferens
- ICSI-ET:
-
Intra-cytoplasmic sperm injection followed by embryo transfer
References
Forti G, Krausz C (1998) Clinical review 100: evaluation and treatment of the infertile couple. J Clin Endocrinol Metab 83:4177–4188
Mak V, Jarvi KA (1996) The genetics of male infertility. J Urol 156:1245–56; discussion 1256–7
Jequier AM, Ansell ID, Bullimore NJ (1985) Congenital absence of the vasa deferentia presenting with infertility. J Androl 6:15–19
Oates RD, Amos JA (1994) The genetic basis of congenital bilateral absence of the vas deferens and cystic fibrosis. J Androl 15:1–8
Dumur V, Gervais R, Rigot J-M et al (1990) Abnormal distribution of CF ΔF508 allele in azoospermic men with congenital aplasia of epididymis and vas deferens. Lancet 336:512
Anguiano A, Oates RD, Amos JA et al (1992) Congenital bilateral absence of the vas deferens. A primarily genital form of cystic fibrosis. JAMA 267:1794–1797
Torra R, Sarquella J, Calabia J et al (2008) Prevalence of cysts in seminal tract and abnormal semen parameters in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 3:790–793
Schlegel PN, Shin D, Goldstein M (1996) Urogenital anomalies in men with congenital absence of the vas deferens. J Urol 155:1644–1648
McCallum T, Milunsky J, Munarriz R et al (2001) Unilateral renal agenesis associated with congenital bilateral absence of the vas deferens: phenotypic findings and genetic considerations. Hum Reprod 16:282–288
Daudin M, Bieth E, Bujan L et al (2000) Congenital bilateral absence of the vas deferens: clinical characteristics, biological parameters, cystic fibrosis transmembrane conductance regulator gene mutations, and implications for genetic counseling. Fertil Steril 74:1164–1174
Bellanné-Chantelot C, Clauin S, Chauveau D et al (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54:3126–3132
Faguer S, Chassaing N, Bandin F et al (2014) The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int 86:1007–1015
Ravine D, Gibson RN, Walker RG et al (1994) Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 343:824–827
Faguer S, Decramer S, Chassaing N et al (2011) Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 80:768–776
Oram R, Edghill EL, Blackman J et al (2010) Mutations in the hepatocyte nuclear factor-1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol 203(364):e1–e5
Bellanné-Chantelot C, Chauveau D, Gautier J-F et al (2004) Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann Intern Med 140:510–517
Jungwirth A, Giwercman A, Tournaye H et al (2012) European Association of Urology guidelines on Male Infertility: the 2012 update. Eur Urol 62:324–332
Nistal M, González-Peramato P, Sousa G et al (2010) Cystic dysplasia of the epididymis: a disorder of mesonephric differentiation associated with renal maldevelopment. Virchows Arch 456:695–702
Decramer S, Parant O, Beaufils S et al (2007) Anomalies of the TCF2 gene are the main cause of fetal bilateral hyperechogenic kidneys. J Am Soc Nephrol 18:923–933
Nie X, Arend LJ (2014) Novel roles of Pkd2 in male reproductive system development. Differentiation 87:161–171
Gao Z, Ruden DM, Lu X (2003) PKD2 cation channel is required for directional sperm movement and male fertility. Curr Biol 13:2175–2178
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding.
Conflict of interest
None of the authors declare competing financial interests.
Ethical approval
Collection of the clinical charts were performed according to the rules of the Institutional Ethical Committee of the University Hospital of Toulouse. The institutional ethical committee (University Hospital of Toulouse) approved DNA collection and genetic testing and the study complied with the Declaration of Helsinki revised in 2003.
Informed consent
All patients gave their written informed consent before DNA collection.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mieusset, R., Fauquet, I., Chauveau, D. et al. The spectrum of renal involvement in male patients with infertility related to excretory-system abnormalities: phenotypes, genotypes, and genetic counseling. J Nephrol 30, 211–218 (2017). https://doi.org/10.1007/s40620-016-0286-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-016-0286-5