
Abstract
Congenital bilateral absence of the vas deferens (CBAVD) involves a complete or partial defect of the Wolffian duct derivatives and is a common cause of obstructive azoospermia. There are different subphenotypes of CAVD such as congenital bilateral absence of vas deferens (CBAVD), congenital unilateral absence of vas deferens (CUAVD), and CBAVD/CUAVD associated with renal anomalies. 95% of men with cystic fibrosis have CBAVD, while 60–70% of men with isolated CBAVD have detectable mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Failure to detect a CFTR abnormality in these men could be due to limitations of the mutation detection methods or due to an etiology other than CFTR gene. The CFTR mutations have been traditionally classified into six classes based on clinical severity of CFTR mutations. Majority of CBAVD men have one severe and one mild CFTR mutation or two mild CFTR mutations. A proportion of CBAVD men (11–20%) suffer from concomitant urogenital abnormalities. CFTR mutations are generally not detected in these men, and other genetic causes are postulated. The type and severity of mutation vary with geography and ethnicity. Current ASRM, ACOG, and ACMG recommendations for genetic diagnosis or risk prediction of CBAVD have limitations, and there is a need to develop regional guidelines based on the ethnic-specific CFTR mutation screening. We describe the clinical presentation, diagnosis, and CFTR abnormalities detected in different subphenotypes of CAVD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Points-
Congenital bilateral absence of the vas deferens (CBAVD) is a common cause of obstructive azoospermia. CBAVD is seen in most men with cystic fibrosis, while isolated CBAVD is frequently associated with mutations in the CFTR gene.
-
CBAVD associated with renal anomalies is often due to causes other than CFTR mutations.
-
There are over 2000 mutations of the CFTR gene, and there is considerable ethnic variation. Population-specific mutation panels and mandatory guidelines are required for Asian and African countries.
-
Unilateral absence of the vas (CBAVD) may be due to CFTR mutations or may be due to other causes when it is associated with ipsilateral renal agenesis.
-
In CBAVD, spermatogenesis is essentially normal, and pregnancy can be obtained through ICSI using sperm aspirated from the epididymis or testes.
Clinical Scenarios
Case 1
VN (29 years old) and SN (25 years old), an unrelated South Indian couple, presented with primary infertility of 2 years duration. There was no family or personal history of cystic fibrosis. The semen analysis showed low volume (<1 ml), azoospermia, and absence of fructose. On examination, testicular volume was normal, and bilateral vasa deferentia were not palpable. Hormone profile was normal. Transrectal ultrasound (TRUS) revealed congenital bilateral absence of seminal vesicles (CASV) with small Mullerian duct cyst. Ultrasound (USG) of the abdomen showed bilateral normal kidneys. After obtaining informed consent and providing counseling, blood samples of both VN and SN were collected and processed for CFTR gene mutations screening. Direct DNA sequencing of essential promoter, entire coding regions, and splice sites of 27 exons of the CFTR gene were carried out in male and female partner. VN was found to carry two mutations: a common mutation associated with classical cystic fibrosis (CF) c.1521_1523delCTT (F508del) and a novel CFTR mutation L578I. There was no family history of CF or past medical history of any respiratory, pancreatic, or gastrointestinal symptoms suggestive of CF. SN was found to be heterozygous carrier of A1285V, a novel CFTR mutation detected in North Indian men with CBAVD [1]. The couple was counseled and advised prenatal genetic diagnosis in view of the risk of having a child with classic CF or CFTR-related disorders such as CBAVD.
Case 2
RS (28 year old) and PS (26 years old), healthy, unrelated, Gujarati Indian couple married since one and half years, were referred for primary infertility. Semen analysis showed azoospermia with absent fructose and volume < 1 ml. On scrotal examination, testes were normal, only the head of the epididymis was palpable, and bilateral vasa were found to be absent. TRUS confirmed CASV. Abdominal USG showed absence of right kidney with left ectopic kidney in pelvis on left side of urinary bladder with mild compensatory hypertrophy. RS was diagnosed as CBAVD with unilateral renal agenesis (URA). Direct DNA sequencing of essential promoter, entire coding regions, and splice sites of 27 exons of the CFTR gene could not detect CF or CBAVD causing mutations except 3 previously reported potential regulatory coding CFTR gene variants (AGA haplotype) c.1540G > A V470 M (heterozygous), c.2694 T > G T854 T (heterozygous), c.4521G > A Q1463Q (homozygous), and TG12-5T/TG11-7T (heterozygous). The female partner was not a CF carrier. Genetic counseling was provided to the couple. Percutaneous epididymal sperm aspiration (PESA) revealed good-quality motile sperm. The couple underwent one cycle of ICSI; however, there was no pregnancy.
Case 3
SS (28 year old) and RS (26 year old), healthy, unrelated Jain couple, presented with 3 years of infertility. Semen analysis indicated low volume, azoospermia with absence of fructose. Scrotal examination revealed a thick, palpable right vas deferens and a non-palpable left vas deferens. TRUS confirmed the absence of left vas deferens and left seminal vesicle. Right seminal vesicle was markedly dilated, filled with intraprostatic fluid. The right terminal vas was not identified. USG abdomen showed absence of the left kidney with compensatory hypertrophy of the right kidney. SS was diagnosed as congenital unilateral absence of vas deferens with unilateral renal agenesis (CUAVD-URA). Blood samples of both SS and RS were collected and processed for CFTR mutation screening. Direct sequencing of essential promoter, entire coding regions, and splice sites of 27 exons of the CFTR gene could not detect CF or CBAVD causing mutations except c.1210–12[5] [5T] variant. The female partner was not a CF carrier. After providing counseling, the couple underwent two cycles of ICSI resulting into live birth of a female child in the second ICSI cycle.
Vas Aplasia
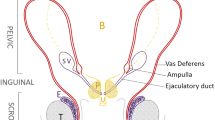
Congenital bilateral absence of the vas deferens (CBAVD) is associated with normal spermatogenesis and obstructive azoospermia, involving a complete or partial defect of the Wolffian duct derivatives [2]. CBAVD affects 2–3% of all male infertility cases and is responsible for 25% cases of obstructive azoospermia [3,4,5]. The etiology of CBAVD is not completely understood; however, there is a well-established linkage between CBAVD, cystic fibrosis, and CFTR gene mutations [3, 6]. The human male internal genitalia originate from the paired Wolffian ducts (WDs), which in the male embryo are stabilized by testosterone. The ducts develop into separate but connecting organs, the epididymis, vas deferens, and seminal vesicles. During development at 6 weeks of embryonic growth, the WD opens to the urogenital sinus at a site adjacent to the ureteral orifice (Fig. 13.1a). At 7–8 weeks, there are discrete differences in the WD position along the mediolateral axis as well as in the morphology of the urogenital sinus (Fig. 13.1b). At 8–9 weeks, the bilateral upper angles of the urogenital sinus start upward growth toward the umbilicus. During the ninth week depending on development of smooth muscles in the bladder as well as rhabdosphincter muscles of the urethra, the descent of the vas deferens becomes evident (Fig. 13.1c). At 10–11 weeks, a radical ascending development of bladder smooth muscles as well as a developing prostate accelerates the descent of the vas [7] (Fig. 13.1d). The effects of the CFTR mutations on the WD may occur after the ninth week of development causing CF- or CFTR-related disorders (CFTR-RD). In the embryo, the penetration of the metanephrogenic blastema by the ureteric bud induces the development of the kidney. Any interruption of this process before the complete separation of the WD and ureteric bud can result in renal agenesis (URA) and CUAVD, whereas interruption in the development of the WD after the separation may lead to an isolated CUAVD [7].

Embryogenesis of male genitalia. (a) 6 weeks: WD Wolffian duct, MD Müllerian duct, K kidney, U ureter, C cloaca. (b) 7–8 weeks: WD Wolffian duct, K kidney, U ureter, UGS urogenital sinus. (c) 8–9 weeks: WD Wolffian duct, K kidney, U ureter. (d) 10–11 weeks: VD vas deferens, E epididymis, T testis, K kidney, U ureter, UB urinary bladder
Genetic Abnormalities in Vas Aplasia
The cystic fibrosis transmembrane conductance regulator (CFTR) gene was first reported as causative factor in cystic fibrosis (CF) [8]. Almost 95% of CF men have CBAVD [6]. CBAVD reported in infertile but otherwise healthy men (without CF) is known as isolated CBAVD and is now classified as CFTR-related disorders [3]. There is a different spectrum of CFTR mutations in CBAVD and classical CF [9]. However, majority of CBAVD men (88%) have one severe and one mild CFTR mutation or two mild CFTR mutations (12%) but never carry two severe CFTR mutations [3, 10]. The CFTR gene is located on the long arm of chromosome 7q31.2 and contains 27 coding exons that spread over 230 kb. Its 6.5-kb mRNA encodes a 1480-amino acid protein that regulates chloride channel in a variety of tissues [11, 12]. Any defect in CFTR gene contributes to abnormal electrolyte transport in the epithelial cells of the respiratory tract, the pancreas, the intestine, the vas deferens, the hepatobiliary system, and the sweat glands [13]. The CFTR mutations have been traditionally classified into six classes based on clinical severity of CFTR mutations (Table 13.1). Class I and II mutations are common, classes III and IV are uncommon (1–5% of CF mutations), and class V and VI mutations are very rare (<1% of CF mutations) [14]. CBAVD is typically caused by a residual function CFTR class IV or V mutation, resulting in less than 10% of wild type CFTR function (Fig. 13.2). Recently, De Boeck and Amaral suggested seven categories wherein classes I, II, and III, and VII are defined as severe mutation, while classes IV, V, and VI are associated with mild phenotypes [15].The traditional class I mutations have been divided into class I (stop-codon mutations) and a new class VII mutation wherein there is no mRNA transcription resulting in absence of the CFTR protein similar to the traditional class I mutation; however, it cannot be altered by drug therapy. Marson’s group proposed that CFTR mutation class VII is important and be retained as class IA, which includes mutations with severe phenotype where corrective therapy is unavailable, followed by classes IB to VI [16].

Cystic fibrosis transmembrane conductance regulator (CFTR)-related male infertility
More than 2000 CFTR gene sequence variants have been reported since the discovery of the CFTR gene [17], and there are three different CFTR databases – http://www.genet.sickkids.on.ca/, http://www.umd.be/CFTR, and http://www.cftr2.org/ [18]. F508del is the most common severe mutation detected in CF and CFTR-RDs with 50–80% frequency among Caucasians, while other known mutations occur with a lower frequency (<6%) [3]. Additionally, mild variants in intron 9 (IVS9) with poly thymidine 5, 7, and 9 affect the splicing of exon 10. The TG repeats (TGm) located upstream to the poly T tract in IVS9 and the polymorphism c.1408G>A, p. (Met470Val) (M470V) rs213950 in exon 11 (HGVS) have been implicated in CBAVD [19, 20]. The frequencies of mutations have been reported to vary with different geographical and ethnic variations [3, 5, 21]. Majority (95%) of CF-CBAVD patients have mutations in the CFTR gene [22] as compared to isolated CBAVD, wherein CFTR gene mutations are detected in 60–70% of patients [23]. Failure to detect CFTR abnormality in isolated CBAVD men could be due to limitations of the mutation detection methods or due to etiology other than CFTR gene. The most common CBAVD genotypes reported in European population are the F508del in trans (located in two different chromosomes) with IVS8-5T (28%) and F508del in trans with R117H (6%) [3, 22]. However, there is a variation in frequency of most common CBAVD causing known CFTR mutations [F508del, c.1210–12[5] (5T)] in CBAVD men residing in different geographical regions (Table 13.2). Although there is significant variability in the frequency of F508del mutation in CBAVD men of different ethnic origins, the 5T variant is found to be present at the same or very similar frequency in CBAVD men from Asia and Europe [Indians, 25–39.4% [24, 25]; Japanese, 30% [26]; Turkish, 19.6% [27]; Iran, 25.9% [28]; Spanish, 23% [29]; Portuguese, 27.4% [30]]. This evidence suggests that the 5T variant plays a role in the pathogenesis of CBAVD even in populations considered to have low CF incidence [3].
The identification of large rearrangements and deletions in the CFTR gene of CBAVD patients with absence of mutations has become possible due to improved techniques of mutation analysis [31,32,33]. Polymorphisms in genes such as Tr2GFB1 (transforming growth factor) and EDNRA (endothelin receptor type A) may increase the penetrance of CBAVD-related mutations [34]. Mutations are usually detected in 80% CBAVD cases; however, failure to detect mutations in the remaining 20% indicates the involvement of genetic etiology other than CFTR gene. Recently, a new pathogenic gene, ADGRG2, encoding the efferent duct and epididymal-specific G protein-coupled receptor with an X-linked inheritance pattern, has been reported in CBAVD patients who were negative for CFTR mutations [2, 35, 36]. A study in a Chinese population indicated differences in the mutations of the promoter region of the CFTR gene as compared to Caucasians. The homozygous c.-966 T >G mutation state had the highest frequency, which reduced the CFTR transcription level [37]. The use of next-generation sequencing (NGS) in evaluating CBAVD men will further improve our understanding of the novel genes that might be involved in the pathogenesis of CBAVD and related phenotypes [38].
Clinical Diagnosis of Vas Aplasia and Associated Subphenotypes
CFTR-related male infertility is subdivided into following subphenotypes:
-
(a)
CBAVD
-
(b)
CUAVD
-
(c)
CBAVD-URA
-
(d)
Congenital absence of seminal vesicles (CASV)
-
(e)
Bilateral ejaculatory duct obstruction (BEDO)
CBAVD
CBAVD is usually detected at adulthood during evaluation of infertility in otherwise asymptomatic males or at the time of a surgical procedure as an incidental finding. In CBAVD men, there is bilateral absence of the vas deferens along with the body and tail of the epididymis and also bilateral or unilateral absence of seminal vesicles. The head of the epididymis is present in all CBAVD cases and has normal function [39]. In some of the CBAVD men, vasa deferentia may be palpable in the scrotum, but during surgical exploration, a fibrous cord or a nonpermeable duct or a blind-ending vas is observed [3]. Isolated CBAVD is now suggested to be a CFTR-related disorder (CFTR-RD) though as per the latest consensus it does not fulfill the diagnostic criteria for CF [3].
CBAVD can be easily diagnosed by a semen analysis showing azoospermia, low seminal volume (<1 ml), low or absent fructose and low semen pH (<6.8) [3], and an impalpable vas deferens on scrotal examination; however, there may be a delay of 4.3 years in correct diagnosis of CBAVD as it can be overlooked by first investigators [40]. Transrectal ultrasonography (TRUS) would usually reveal absent seminal vesicles and ejaculatory ducts, though seminal vesicle-like structures can be observed in 15% of men with vas aplasia and can be a cause of diagnostic confusion. Abdominal and pelvic USG is required to diagnose abnormalities of the upper urinary tract. In majority of CBAVD men, testicular volume and serum gonadotrophins levels are normal, and testicular biopsy shows normal or slightly defective spermatogenesis [40].
CUAVD
CUAVD is a rare entity with a reported incidence of 0.5–1% and is often associated with renal agenesis [41]. Due to the possibility of pregnancy because of the normal function of the other vas deferens, it is likely that the incidence of CUAVD is underestimated [42]. There is a higher incidence of renal anomalies in CUAVD men than in CBAVD men, with an absolute risk increase of 20.1% [43]. Renal anomalies in CUAVD men are reported as malrotation of the solitary kidney, multicystic kidney, ectopic kidney, and horseshoe kidney [44]. CFTR mutations were reported in CUAVD with a lower frequency than in CBAVD [3]. Recently, Klinefelter’s syndrome (KS) cases were reported in association with CUAVD harboring CFTR gene mutations including delta F508 in KS-CUAVD cases [45]. Recent meta-analysis demonstrated a fairly high frequency of overall CFTR variants in CUAVD men with very low frequencies of the heterozygous genotypes F508del/5T and F508del/R117H [43]. Additionally, CUAVD men showed increased of 5T risk allele with a 15.5% frequency [43]. These observations suggest the need for detailed physical examination and genetic screening in CUAVD patients.
CBAVD Men Having Renal Anomalies (CBAVD-URA)
A proportion of CBAVD men (11–20%) suffer from concomitant urogenital abnormalities including unilateral renal agenesis [3]. Therefore, CBAVD or CUAVD men should undergo ultrasound examination of the abdomen and pelvis for detection of renal abnormalities. There is limited information on the exact mechanisms involved in the etiology of CBAVD-URA. The role of CFTR gene mutations in this subset of patients is questionable as majority of studies have failed to detect CFTR gene mutations in CBAVD-URA [3, 46]. Genetic factors other than CFTR gene are suggested to be involved in etiology of CBAVD-URA [3].
Congenital Absence of Seminal Vesicles (CASV) and Ejaculatory Duct Obstruction (BEDO)
CBAVD men usually have bilateral or unilateral absence or hypoplasia of seminal vesicles. CFTR gene mutations were detected in infertile men with BEDO and concomitant seminal vesicle anomalies suggesting CFTR gene abnormalities as molecular basis of the genital tract anomalies and the resulting infertility in this subset of obstructive azoospermia [47]. The authors further suggested that BEDO with concomitant seminal vesicle anomalies to be considered as CFTR-associated disorder confined to the male genital tract as there was considerable overlap of CFTR gene mutations in CBAVD and in BEDO with concomitant seminal vesicle anomalies [47].
Infertile men with BEDO are now able to become biological fathers with the support of assisted reproductive technologies [48]; however, such men and their female partners should be provided genetic counseling and CFTR gene mutation analysis to prevent the transmission of genetic abnormalities to the offspring.
CBAVD, Assisted Reproduction, and Genetic Counseling
Although majority of the CBAVD men show normal spermatogenesis on testicular biopsy and sperm from CBAVD men are capable of fertilizing an egg, these men were deprived of biological fatherhood until 1987. Since the caput of epididymis is always present in CBAVD men, recent advances in assisted reproduction technologies as well as sperm retrieval techniques now allow these obstructive azoospermic men to enjoy biological fatherhood. However, once the clinical diagnosis of CBAVD is confirmed, counseling should be offered to the CBAVD male and female partner to screen for CFTR gene mutations because of the high risk of transmitting CFTR mutation(s) to the offspring. Silber and coworkers [49] documented the first pregnancy for a couple in whom the male partner had CBAVD. They utilized the microsurgical epididymal sperm aspiration (MESA) technique and in vitro fertilization in 1988. Since then, other techniques such as PESA (percutaneous epididymal sperm aspiration), FNA (fine-needle aspiration), and TESA (testicular sperm aspiration) have also been used to obtain sperm from men having CBAVD, and the advent of ICSI (intracytoplasmic sperm injection) dramatically improved fertilization and pregnancy rates [6]. Kamal et al. reported no difference in the rates of fertilization, clinical pregnancy, and miscarriage between CBAVD men (n = 434) and infertile men having other causes of obstructive azoospermia (n = 687) [50]. This study also reported similar rates of fertilization, pregnancy, and miscarriage with use of epididymal spermatozoa and testicular spermatozoa for ICSI [50]. However, another study suggested that CFTR mutations may lead to increased risk of miscarriage and stillbirth and a reduced rate of live birth in CBAVD men compared with non-CBAVD men [51]. Another study in Chinese men found no significant difference in fertilization, implantation, or clinical pregnancy rates between CBAVD and non-CBAVD patients who had PESA followed by ICSI, but there was a significantly lower live birth rate and significantly higher miscarriage rate in CBAVD men as compared to non-CBAVD men. The authors suggested that this increased risk of miscarriage or stillbirth may be associated with the CFTR mutations [23]. Finally, a 10-year ICSI outcomes data analysis evaluating the impact of the quality of testicular spermatogenesis (as determined histopathologically) in CBAVD men suggested that impaired spermatogenesis had a negative impact on early-stage biological outcomes of ICSI [52].
The present American Society for Reproductive Medicine (ASRM), American College of Obstetricians and Gynecologists (ACOG), and American College of Medical Genetics and Genomics (ACMG) recommendation for genetic diagnosis or risk prediction of CBAVD includes either expanded CFTR mutation testing or carrier screening for the 23 most prevalent CFTR mutations [53]. The CFTR mutation panels are well characterized for Caucasians as compared to Asians, Africans, and other populations. The existing CFTR mutation panels were derived from the data of CF patients of Caucasian and Northern European descent and have limited utility for CBAVD men of non-Caucasian origin. Additionally, there are large rearrangements such as exon deletions, insertions, or duplications in 2% of CBAVD men, which are undetected by standard sequencing analysis [54]. The role of modifier genes TGFβ-1 and EDNRA in development of CBAVD has also been reported [34, 55]. Recently, mutations in the ADGRG2 gene are reported in CFTR-negative CBAVD men [2, 36]. Therefore, the current ASRM, ACOG, and ACMG recommendations for genetic diagnosis or risk prediction of CBAVD have limitations, and there is a need to develop regional guidelines based on the ethnic-specific CFTR mutation screening, especially for the people of Asian and African origin.
Future Perspectives
Evidence suggests that majority of CBAVD is associated with CFTR gene abnormalities; however, there are limitations to the currently available CFTR screening panels. Additionally, new information suggesting genetic involvement other than CFTR needs to be taken into consideration while offering the genetic counseling to the CBAVD men and female partners enrolled for an ART program. There is growing evidence suggesting similar incidence of CBAVD in Asian, African, and non-European population to that of Caucasians. Therefore, larger studies should be conducted in Asian, African, and non-European CBAVD populations to determine the CFTR mutation spectrum and other genes involved in etiology of CBAVD. Similarly, female CF carrier frequency should also be determined in these populations which were once considered as having low incidence of CF and CFTR-RD. Regulatory guidelines need to be framed and should be strictly implemented, especially for Asian and African populations, on mandatory CFTR screening and genetic counseling before undergoing ART. This can be achieved with the support of the WHO and international NGOs working for CF and CFTR-RD patients globally.
Also, CUAVD has been given less attention in clinical practice. Recent meta-analysis demonstrated 5T and F508del as the most common CFTR abnormalities in CUAVD men [43]. Additionally, if there is a delay in the diagnosis of CUAVD, it may lead to increased mortality and morbidity due to urogenital defects [56]. Taking into consideration the high frequency of renal anomaly risks in men having CUAVD, it is essential to conduct imaging of urogenital system to improve the quality of life and also provide whole exon/flanking sequencing of CFTR to avoid genetic risks to progeny [43].
Review Criteria
A thorough search of medical literature was done on genetics of vas aplasia using the following search engines: PubMed, Google Scholar, Medline, and Science Direct. The keywords “vas aplasia,” “genetics,” “cystic fibrosis,” “azoospermia,” “CFTR,” “CBAVD,” “CUAVD,” “CFTR and renal anomalies,” and “genetic counseling” were used for the study identification and data extraction.
References
Sachdeva K, Saxena R, Majumdar A, Chadha S, Verma IC. Mutation studies in the CFTR gene in Asian Indian subjects with congenital bilateral absence of vas deferens: report of two novel mutations and four novel variants. Genet Test Mol Biomarkers. 2011;15(5):307–12.
Patat O, Pagin A, Siegfried A, Mitchell V, Chassaing N, Faguer S, et al. Truncating mutations in the adhesion G protein-coupled receptor G2 gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferens. Am J Hum Genet. 2016;99(2):437–42.
Bombieri C, Claustres M, De Boeck K, Derichs N, Dodge J, Girodon E, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10:S86–102.
Claustres M. Molecular pathology of the CFTR locus in male infertility. Reprod Biomed Online. 2005;10(1):14–41.
Yu J, Chen Z, Ni Y, Li Z. CFTR mutations in men with congenital bilateral absence of the vas deferens (CBAVD): a systemic review and meta-analysis. Hum Reprod. 2012;27(1):25–35.
de Souza DAS, Faucz FR, Pereira-Ferrari L, Sotomaior VS, Raskin S. Congenital bilateral absence of the vas deferens as an atypical form of cystic fibrosis: reproductive implications and genetic counseling. Andrology. 2018;6(1):127–35.
Carlson BM. Human embryology and developmental biology. Philadelphia: Mosby; 2004. p. 548.
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–80.
Daudin M, Bieth E, Bujan L, Massat G, Pontonnier F, Mieusset R. Congenital bilateral absence of the vas deferens: clinical characteristics, biological parameters, cystic fibrosis transmembrane conductance regulator gene mutations, and implications for genetic counseling. Fertil Steril. 2000;74(6):1164–74.
Claustres M, Guittard C, Bozon D, Chevalier F, Verlingue C, Ferec C, et al. Spectrum of CFTR mutations in cystic fibrosis and in congenital absence of the vas deferens in France. Hum Mutat. 2000;16(2):143–56.
Hwang K, Yatsenko AN, Jorgez CJ, Mukherjee S, Nalam RL, Matzuk MM, et al. Mendelian genetics of male infertility. Ann N Y Acad Sci. 2010;1214:E1–17.
Moskowitz SM, Gibson RL, Effmann EL. Cystic fibrosis lung disease: genetic influences, microbial interactions, and radiological assessment. Pediatr Radiol. 2005;35(8):739–57.
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891–904.
Ratjen F, Döring G. Cystic fibrosis. Lancet [Internet]. 2003 [cited 2018 Oct 18];361(9358):681–9. Available from: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(03)12567-6/abstract.
De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662–74.
Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. Lancet Respir Med. 2016;4(8):e37–8.
Cftrscience | Clinician’s guide to CFTR [Internet]. [cited 2018 Oct 21]. Available from: https://www.cftrscience.com/.
Gajbhiye R, Gaikwad A. Cystic fibrosis, CFTR gene, and male infertility. In: Singh R, Singh K, editors. Male infertility: understanding, causes and treatment [Internet]. Singapore: Springer Singapore; 2017. p. 131–50. Available from: https://doi.org/10.1007/978-981-10-4017-7_9.
de Meeus A, Guittard C, Desgeorges M, Carles S, Demaille J, Claustres M. Genetic findings in congenital bilateral aplasia of vas deferens patients and identification of six novel mutatations. Mutations in brief no. 138. Online. Hum Mutat. 1998;11(6):480.
Groman JD, Hefferon TW, Casals T, Bassas L, Estivill X, Des Georges M, et al. Variation in a repeat sequence determines whether a common variant of the cystic fibrosis transmembrane conductance regulator gene is pathogenic or benign. Am J Hum Genet. 2004;74(1):176–9.
Xu X, Zheng J, Liao Q, Zhu H, Xie H, Shi H, et al. Meta-analyses of 4 CFTR variants associated with the risk of the congenital bilateral absence of the vas deferens. J Clin Bioinforma. 2014;4:11.
Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332(22):1475–80.
Lu S, Cui Y, Li X, Zhang H, Liu J, Kong B, et al. Association of cystic fibrosis transmembrane-conductance regulator gene mutation with negative outcome of intracytoplasmic sperm injection pregnancy in cases of congenital bilateral absence of vas deferens. Fertil Steril. 2014;101(5):1255–60.
Sharma N, Acharya N, Singh SK, Singh M, Sharma U, Prasad R. Heterogenous spectrum of CFTR gene mutations in Indian patients with congenital absence of vas deferens. Hum Reprod. 2009;24(5):1229–36.
Gaikwad A, Khan S, Kadam S, Kadam K, Dighe V, Shah R, et al. The CFTR gene mild variants poly-T, TG repeats and M470V detection in Indian men with congenital bilateral absence of vas deferens. Andrologia. 2018;50(2):e12858.
Anzai C, Morokawa N, Okada H, Kamidono S, Eto Y, Yoshimura K. CFTR gene mutations in Japanese individuals with congenital bilateral absence of the vas deferens. J Cyst Fibros. 2003;2(1):14–8.
Dayangaç D, Erdem H, Yilmaz E, Sahin A, Sohn C, Ozgüç M, et al. Mutations of the CFTR gene in Turkish patients with congenital bilateral absence of the vas deferens. Hum Reprod. 2004;19(5):1094–100.
Radpour R, Gilani MAS, Gourabi H, Dizaj AV, Mollamohamadi S. Molecular analysis of the IVS8-T splice variant 5T and M470V exon 10 missense polymorphism in Iranian males with congenital bilateral absence of the vas deferens. Mol Hum Reprod. 2006;12(7):469–73.
Casals T, Bassas L, Egozcue S, Ramos MD, Giménez J, Segura A, et al. Heterogeneity for mutations in the CFTR gene and clinical correlations in patients with congenital absence of the vas deferens. Hum Reprod. 2000;15(7):1476–83.
Grangeia A, Niel F, Carvalho F, Fernandes S, Ardalan A, Girodon E, et al. Characterization of cystic fibrosis conductance transmembrane regulator gene mutations and IVS8 poly(T) variants in Portuguese patients with congenital absence of the vas deferens. Hum Reprod. 2004;19(11):2502–8.
Ratbi I, Legendre M, Niel F, Martin J, Soufir J-C, Izard V, et al. Detection of cystic fibrosis transmembrane conductance regulator (CFTR) gene rearrangements enriches the mutation spectrum in congenital bilateral absence of the vas deferens and impacts on genetic counselling. Hum Reprod. 2007;22(5):1285–91.
Taulan M, Girardet A, Guittard C, Altieri J-P, Templin C, Beroud C, et al. Large genomic rearrangements in the CFTR gene contribute to CBAVD. BMC Med Genet. 2007;8:22.
Trujillano D, Ramos MD, González J, Tornador C, Sotillo F, Escaramis G, et al. Next generation diagnostics of cystic fibrosis and CFTR-related disorders by targeted multiplex high-coverage resequencing of CFTR. J Med Genet. 2013;50(7):455–62.
Havasi V, Rowe SM, Kolettis PN, Dayangac D, Sahin A, Grangeia A, et al. Association of cystic fibrosis genetic modifiers with congenital bilateral absence of the vas deferens. Fertil Steril. 2010;94(6):2122–7.
Obermann H, Wingbermühle A, Münz S, Kirchhoff C. A putative 12-transmembrane domain cotransporter associated with apical membranes of the epididymal duct. J Androl. 2003;24(4):542–56.
Yang B, Wang J, Zhang W, Pan H, Li T, Liu B, et al. Pathogenic role of ADGRG2 in CBAVD patients replicated in Chinese population. Andrology. 2017;5(5):954–7.
Bai S, Du Q, Liu X, Tong Y, Wu B. The detection and significance of cystic fibrosis transmembrane conductance regulator gene promoter mutations in Chinese patients with congenital bilateral absence of the vas deferens. Gene. 2018;672:64–71.
Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15(6):369–84.
Hannema SE, Hughes IA. Regulation of Wolffian duct development. Horm Res Paediatr. 2007;67(3):142–51.
Weiske WH, Sälzler N, Schroeder-Printzen I, Weidner W. Clinical findings in congenital absence of the vasa deferentia. Andrologia. 2000;32(1):13–8.
Mo B, Garla V, Wyner LM. A case of congenital unilateral absence of the vas deferens. Int Med Case Rep J. 2013;6:21–3.
Baydilli N, Gökçe A, Karabulut SY, Ekmekcioglu O. Klinefelter’s syndrome with unilateral absence of vas deferens. Fertil Steril. 2010;94(4):1529.e1–2.
Cai H, Qing X, Niringiyumukiza JD, Zhan X, Mo D, Zhou Y, et al. CFTR variants and renal abnormalities in males with congenital unilateral absence of the vas deferens (CUAVD): a systematic review and meta-analysis of observational studies. Genet Med. 2019;21(4):826.
Khan ZAJ, Novell JR. A missing vas. J R Soc Med. 2001;94(11):582–3.
Akinsal EC, Baydilli N, Imamoglu H, Ekmekcioglu O. Three cases of Klinefelter’s syndrome with unilateral absence of vas deferens. Andrologia. 2017;49(9):e12844.
Gajbhiye R, Kadam K, Khole A, Gaikwad A, Kadam S, Shah R, et al. Cystic fibrosis transmembrane conductance regulator (CFTR) gene abnormalities in Indian males with congenital bilateral absence of vas deferens & renal anomalies. Indian J Med Res. 2016;143(5):616–23.
Meschede D, Dworniczak B, Behre HM, Kliesch S, Claustres M, Nieschlag E, et al. CFTR gene mutations in men with bilateral ejaculatory-duct obstruction and anomalies of the seminal vesicles. Am J Hum Genet. 1997;61(5):1200–2.
Silber SJ, Nagy Z, Liu J, Tournaye H, Lissens W, Ferec C, et al. Genetics: the use of epididymal and testicular spermatozoa for intracytoplasmic sperm injection: the genetic implications for male infertility. Hum Reprod. 1995;10(8):2031–43.
Silber SJ, Balmaceda J, Borrero C, Ord T, Asch R. Pregnancy with sperm aspiration from the proximal head of the epididymis: a new treatment for congenital absence of the vas deferens. Fertil Steril. 1988;50(3):525–8.
Kamal A, Fahmy I, Mansour R, Serour G, Aboulghar M, Ramos L, et al. Does the outcome of ICSI in cases of obstructive azoospermia depend on the origin of the retrieved spermatozoa or the cause of obstruction? A comparative analysis. Fertil Steril. 2010;94(6):2135–40.
Attardo T, Vicari E, Mollica F, Grazioso C, Burrello N, Garofalo MR, et al. Genetic, andrological and clinical characteristics of patients with congenital bilateral absence of the vas deferens. Int J Androl. 2001;24(2):73–9.
Llabador MA, Pagin A, Lefebvre-Maunoury C, Marcelli F, Leroy-Martin B, Rigot JM, et al. Congenital bilateral absence of the vas deferens: the impact of spermatogenesis quality on intracytoplasmic sperm injection outcomes in 108 men. Andrology. 2015;3(3):473–80.
Dialog F and S. Cystic fibrosis genetic testing in congenital bilateral absence of the vas deferens (CBAVD) [Internet]. Fertil Steril Dialog. 2017 [cited 2018 Oct 17]. Available from: https://www.fertstertdialog.com/users/16110-fertility-and-sterility/posts/19299-foyouzi-consider-this.
Hantash FM, Milunsky A, Wang Z, Anderson B, Sun W, Anguiano A, et al. A large deletion in the CFTR gene in CBAVD. Genet Med. 2006;8(2):93–5.
Sharma H, Mavuduru RS, Singh SK, Prasad R. Heterogeneous spectrum of mutations in CFTR gene from Indian patients with congenital absence of the vas deferens and their association with cystic fibrosis genetic modifiers. Mol Hum Reprod. 2014;20(9):827–35.
Salwan A, Abdelrahman A. Congenital absence of vas deferens and ectopic kidney. Int J Surg Case Rep. 2017;34:90–2.
Dörk T, Dworniczak B, Aulehla-Scholz C, Wieczorek D, Böhm I, Mayerova A, et al. Distinct spectrum of CFTR gene mutations in congenital absence of vas deferens. Hum Genet. 1997;100(3–4):365–77.
Lissens W, Mahmoud KZ, El-Gindi E, Abdel-Sattar A, Seneca S, Van Steirteghem A, et al. Molecular analysis of the cystic fibrosis gene reveals a high frequency of the intron 8 splice variant 5T in Egyptian males with congenital bilateral absence of the vas deferens. Mol Hum Reprod. 1999;5(1):10–3.
Wu CC, Hsieh-Li HM, Lin YM, Chiang HS. Cystic fibrosis transmembrane conductance regulator gene screening and clinical correlation in Taiwanese males with congenital bilateral absence of the vas deferens. Hum Reprod. 2004;19(2):250–3.
Li H, Wen Q, Li H, Zhao L, Zhang X, Wang J, et al. Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) in Chinese patients with congenital bilateral absence of vas deferens. J Cyst Fibros. 2012;11(4):316–23.
Acknowledgments
Dr. Smita Mahale, Director, ICMR-NIRRH, Mumbai; Dr. Vijay Kulkarni, Consultant Andrologist, ICMR-NIRRH; Mr. Vaibhav Shinde, Technician A, ICMR-NIRRH; and Ms. Aishwarya Bhurke, Project Assistant are sincerely acknowledged.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gajbhiye, R.K., Khan, S., Shah, R. (2020). Genetics of Vas Aplasia. In: Arafa, M., Elbardisi, H., Majzoub, A., Agarwal, A. (eds) Genetics of Male Infertility. Springer, Cham. https://doi.org/10.1007/978-3-030-37972-8_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-37972-8_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-37971-1
Online ISBN: 978-3-030-37972-8
eBook Packages: MedicineMedicine (R0)