
Abstract
At the start of the postgenomics era, most Parkinson’s disease (PD) etiology cannot be explained by our knowledge of genetic or environmental factors alone. For more than a decade, we have explored gene–environment (GxE) interactions possibly responsible for the heterogeneity of genetic as well as environmental results across populations. We developed three pesticide exposure measures (ambient due to agricultural applications, home and garden use, and occupational use) in a large population-based case–control study of incident PD in central California. Specifically, we assessed interactions with genes responsible for pesticide metabolism (PON1); transport across the blood–brain barrier (ABCB1); pesticides interfering with or depending on dopamine transporter activity (DAT/SLC6A3) and dopamine metabolism (ALDH2); impacting mitochondrial function via oxidative/nitrosative stress (NOS1) or proteasome inhibition (SKP1); and contributing to immune dysregulation (HLA-DR). These studies established some specificity for pesticides’ neurodegenerative actions, contributed biologic plausibility to epidemiologic findings, and identified genetically susceptible populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders, with a complex multifactorial etiology attributed to both environmental and genetic risk factors [1, 2•, 3, 4]. Pesticide exposures have received special attention ever since the neurotoxic metabolite of 1-methyl-4phenyl-1,2,3,6-tetrahydropyridine (MPTP) was first described as causing Parkinsonism in humans [5••]. And, additionally, the finding that the herbicide paraquat is not only structurally similar to this neurotoxin but also exhibits similar effects in animal models [6]. Subsequently, many studies have associated pesticides in general with PD risk, but few investigated specific chemicals [7]. Large genome-wide association studies (GWAS) have identified more than two dozen common genetic variants for PD, each with a relatively small effect size; in combination with rare Mendelian genes, genetics account for at best 10–20 % of PD [4, 8]. The remaining unexplained PD etiology has been referred to as “missing genetic heritability” and attributed to undiscovered rare genetic variants conferring larger risks [9], though early studies do not support this explanation [10]. Nonetheless, we argue that gene–environment (GxE) interactions may have an even larger contribution that remains largely underexplored.
Success in both identifying and replicating GxE interactions remains somewhat elusive, since it depends as much on valid and reliable environmental exposure assessment as on pinpointing appropriate genetic markers. Chronic diseases of advanced age, such as PD, are especially challenging because exposures may accumulate over decades or even a lifetime. Thus, short of following humans across the lifespan, exposures in the distant past are not easily addressable and population-based case–control studies often rely on recall of exposure. Recall is more easily prone to bias and study participants are rarely able to reliably report exposures to specific chemicals.
In California, over the past decade, we were able to overcome the challenge of long-term and population-based pesticide exposure assessment by using the unique pesticide use reporting (PUR) system to explore gene–pesticide interactions [11]. The PUR system was mandated by law and initiated in 1974 in California. It provides spatially detailed information on agricultural pesticide use statewide. We developed a sophisticated geographic information system (GIS) computer model to combine pesticide use reports with land use information in combination with residential and occupational addresses to estimate ambient pesticide exposure for specific active chemical ingredients [11, 12]. Our GIS-derived method of exposure estimation has been validated for organochlorines using measurements of serum biomarkers for l,l-dichloro-2,2-bis(P-chlorophenyl)ethylene (DDE), a metabolite of 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane (DDT) [13]. In addition, we identified lifelong household pesticide exposure to specific chemicals and chemical ingredients relying on self-report enhanced with the unique California Department of Pesticide Regulation product label database [14]. Finally, we combined job history information with a job exposure matrix (JEM) generating an objective measure of general occupational pesticide exposures [15].
Even though GxE studies might help explain heterogeneity of results across populations due to variation in susceptibility to pesticide neurotoxicity and contribute to biologic plausibility of associations observed in human studies, only a small number of studies have been able to test these hypotheses. Here, we will summarize, review, and put into broader context the most intriguing insights we have gained from GxE interaction analyses in a central California population enrolled for the Parkinson’s Gene and Environment (PEG) study. Our studies combine well-defined, record-based exposure assessment for specific chemical agents with genetic information from a range of pathways picked for their pathophysiologic relevance to PD as well as pesticide metabolisms and transport (Table 1). The study population and methods of the PEG study are described in more detail in Supplement 1. Briefly, we conducted a population-based case–control study that enrolled patients with incident idiopathic PD (within 3 years of diagnosis) between 2001 through the present and population controls from 2002 to 2011, from three counties in central California (Kern, Fresno, and Tulare). County resident participants were 35 years or older and had lived in California for 5 or more years prior to recruitment. Study participants were predominantly of European ancestry, over the age of 65, and did not report a family history of PD (Supplement 2).
Pesticides and PD
Paraquat is structurally similar to the neurotoxic metabolite 1-methyl-4-phenylpyridinium (MPP+) of MPTP and has become an established animal model of PD [16]. Paraquat treatment leads to incomplete dopaminergic cell death in animals but appears to act as a redox cycler and not through complex I inhibition [17]. Its toxicity is markedly enhanced by coadministration of the fungicide maneb, a manganese-containing ethylene bis-dithiocarbamate fungicide. It has been reported that maneb is toxic due to its inhibition of mitochondrial respiratory chain complex III [18]. Also, maneb toxicity may be mediated by ubiquitin-proteasome system inhibition [19, 20], aldehyde dehydrogenase inhibition [21], and disruption of protein thiol functions analogous to oxidative stress [22].
A so-called organic pesticide, rotenone, is a very potent inhibitor of mitochondrial complex I and has become another established toxicant-based animal model of Parkinsonism [17, 23]. Unlike other pesticide models of PD, low-level rotenone exposure can produce many of the pathological features of PD. This includes the induction of alpha-synuclein misfolding leading to synuclein templating, transneuronal spread, and a widespread synucleinopathy seen in human PD [24]. While pesticide-based animal models recapitulate much of the pathology of human PD, it is impossible to model many of the important aspects of chemical exposures, such as route of entry or low level exposures spread over decades [17]. Thus, as experimental models can only capture some aspects of human PD, it remains important to investigate potential neurotoxicity of specific pesticides to susceptible humans.
A recent meta-analysis of existing human studies summarized across 39 case–control, 4 cohort, and 3 cross-sectional studies and reported an overall 62 % increase in risk for PD with ever/never pesticide exposure (OR 1.62; 95 % confidence interval (CI) 1.40–1.88) [25•]. Yet, the authors strongly recommended that “future studies should focus on more objective and improved methods of pesticide exposure assessment.”
Sources of Pesticide Exposures in Humans
In the USA, the widespread use of pesticides in agriculture and homes and gardens makes it virtually impossible to completely avoid exposure via food intake, treatment of structures, or drift from large-scale applications. Insecticides are purposefully designed to be acutely neurotoxic. They may modulate the function of voltage-gated sodium channels or affect central γ-amino butyric acid, noradrenergic, dopaminergic, and cholinergic neurotransmission [26]. Other pesticide classes, such as herbicide and fungicides, do not purposefully target the nervous system but have been found to affect mechanisms relevant for neurodegeneration such as mitochondrial inhibition, oxidative stress, aldehyde dehydrogenase or proteasome inhibition, and neuroinflammation [17, 18].
Pesticides often expose agricultural communities, sometimes over substantial distances, as they travel in the air through spray drift and postapplication volatilization [27–29]. Some agricultural-use pesticides are highly persistent and can be found in treated soils and dust even decades after application, indoors [30–32] and in ambient air [33, 34]. A California central valley study assessed inhalation risks from airborne agricultural pesticides and concluded that agricultural applications of organophosphates and their oxon products may have substantial volatization and off-field movement [35]. This was confirmed by a monitoring study conducted in a small community, where California Environmental Protection Agency (Cal EPA) placed air monitors on elementary school roofs over a 1-year period and detected 23 pesticides known to be applied in the area according to the CA-PUR system [36]. Additionally, a northern California study found that crops within 1500 m of homes predicted carpet dust levels of pentachlorophenol and DDE [37]. Thus, pesticides from agricultural applications find their way into homes, exposing residents in agricultural communities to pesticides, even those not involved in agricultural work.
Transport of Toxicants: DAT and the Pesticides Paraquat and Maneb
A recent review addressing the neurotoxicity of paraquat concluded that while evidence for paraquat being a risk factor for Parkinson’s disease is accumulating from experimental, clinical, and epidemiological data, genetic variations associated with susceptibility to paraquat may be responsible for risk differences observed across populations [38]. Thus, gene–environment interactions may be crucial for resolving heterogeneity of results reported in human pesticide studies.
We published the first human data showing that paraquat and maneb act synergistically in increasing PD risk [39]. Furthermore, genetic variability in the 5′A clade and the 3′VNTR 9-repeat of the human dopamine transporter (DAT/SLC6A3) locus was shown to increase susceptibility of humans to occupational pesticide exposure [40], especially two evolutionary clades (A and B) suspected to affect gene expression [41, 42]. Risk was strongly increased among pesticide-exposed male farmers who were carriers of multiple DAT susceptibility alleles (Table 2). Relying on exposure assessment most similar to the previous study, our JEM-derived occupational pesticide measure corroborated the following finding: PD risk was found to be increased among pesticide-exposed males who were carriers of DAT/SLC6A3 susceptibility alleles (Table 2) [43]. We also observed an even larger increase in risk for those with high ambient residential exposure to both maneb and paraquat and two or more DAT/SLC6A3 susceptibility alleles (Table 2).
In organotypic midbrain culture, the coadministration of a DAT inhibitor prevents dopaminergic cell death caused by paraquat treatment [53]. While paraquat in its native divalent cation state (paraquat2+ N,N′-dimethyl-4,4′-bipyridinium) is not a substrate for DAT [54], microglia can convert it to the monovalent cation paraquat(+), which is a substrate for DAT and induces oxidative stress and cytotoxicity [55]. Also, impaired DAT function in cultured cells and mutant mice attenuates paraquat neurotoxicity [55]. DAT /SLC6A3 polymorphisms may generate a selective vulnerability to paraquat in dopaminergic neurons. A human imaging study measured striatal DAT availability with [123I]β-CIT SPECT in 79 healthy young adults and genotyped both the 40-base-pair VNTR in the 3′ region of the DAT/SLC6A3 gene as well as the single nucleotide polymorphisms rs2652511 and rs2937639 in the 5′ end of the gene [56]. DAT VNTR 9-repeat carriers exhibited higher striatal DAT availability than 10-repeat homozygotes. The haplotype T-A-9 repeat (rs2652511-rs2937639-VNTR) was associated with higher striatal DAT expression than other haplotypes [56]. Thus, a combination of polymorphisms in both the 3′ and the 5′ ends of the DAT/SLC6A3 gene seems to be associated with in vivo striatal DAT expression in the same direction expected by our GxE results.
Our DAT pesticide results are in agreement with Kelada et al. [40]. They suggest that the gene encoding the dopamine transporter might increase susceptibility for pesticides such as paraquat. Our GIS-derived, record-based residential pesticide exposure measures added specificity to the human exposure assessment. Instead of relying solely on a general occupational measure, we were able to examine the chemical of most interest based on the putative role the dopamine transporter plays in paraquat neurotoxicity.
Transport of Toxicants: the Multidrug Resistance Protein 1 (ABCB1) and Organophosphorus and Organochlorine Pesticides
The P-glycoprotein, encoded by the multidrug resistance protein 1 (MDR1 or ABCB1) gene, is thought to protect the brain against neurotoxicants. ABCB1 genetic variants known to affect transporter function might influence PD risk. Animal and cell studies have suggested that some pesticides are removed from blood–brain barrier endothelial cells by P-glycoprotein [57]. Specifically, many lipophilic and amphipathic xenobiotic compounds, including several organochlorines and organophosphates, are P-glycoprotein substrates and they may stimulate or inhibit its transporter activity or modulate P-glycoprotein expression [58–60]. Genetic variants of importance for transporter function that have been studied most are the following two SNPs: a synonymous mutation in rs1045642—possibly altering substrate specificity [61]—and a missense mutation in rs2032582 [62]. Homozygous TT genotypes at rs1045642 and rs2032582 have been related to lower protein expression [63].
For our population, we created a genetic risk score that included the ABCB1 SNPs rs1045642 and rs2032582 and counted the number of variant or “susceptibility” alleles. Ambient work place and residential address-derived organophosphorus (OP) and organochlorine (OC) pesticide exposure as well as household or occupational OP or OC pesticide use increased PD risk as much as twofold in participants with one or less susceptibility alleles, but three to fourfold in OP or OC exposed carriers of at least two susceptibility alleles (Table 2) [47].
Three prior studies in European populations reported on interactions between pesticides and polymorphisms in ABCB1 and PD [44–46]. While the first two studies used a case-only design and reported over fourfold increased risks in pesticide exposed T allele carriers of rs1045642, both studies were small and only able to generically assess pesticide exposure (Table 2) [44, 45]. The third study [46] focused on an agricultural population mainly with OC exposure from professional farming-related use and found those exposed to OC pesticides in combination with being carriers of the rs1045642 TT genotype to be at highest risk of PD compared with unexposed C allele carriers (OR = 7.2 (95 % CI 2.1, 24.8) [46]. Also, exposed farmers with a TT or TA genotype in rs2032582 exhibited 7.9 times the risk of PD compared with unexposed G allele carriers (95 % CI 2.2, 28.9) (Table 2). In conclusion, experimental data described P-glycoprotein’s role in xenobiotic transport across the blood–brain barrier. Our results support several prior studies that showed that pesticide exposures contributed to PD risk in combination with variants in ABCB1 that may increase susceptibility to xenobiotics and suggest a role specifically for OC or OP pesticides.
Metabolism of Toxicants: the Paraoxonase Enzyme (PON1) Detoxifies Organophosphorus Pesticides
We have previously reported that OP pesticides increase PD risk [64]. The paraoxonase enzyme (PON1) detoxifies organophosphorus compounds and, importantly, enzyme serum activity varies up to 40-fold in populations [65, 66]. This variations has been attributed to a few important genetic polymorphic sites, including the PON1 192QQ, the 55MM, and the C-108T genotypes [67]. Based on functional research, those with an MM genotype at L55M and QQ or QR at Q192R are considered “slower” metabolizers [68]. Polymorphisms in this gene are not marginally associated with PD [69].
In our population, we assessed whether the three PON1 SNPs modified PD risk from three commonly used organophosphate pesticides: diazinon, chlorpyrifos, and parathion. Assessing the risk by genotype separately or jointly using the L55M/Q192R diplotype showed an increased risk for PD with ambient exposures in homozygous variant genotype carriers (slow metabolizers) as compared with fast metabolizer diplotype carriers (OR = 2.43, 95 % CI = 0.78–7.56 to OR = 3.28, 95 % CI = 1.02–10.58) (Table 2) [48]. Without OP exposure, slow metabolizer diplotype carriers were not at increased risk of developing PD. Furthermore, we saw the same for exposures to household pesticide products that contain OPs as active ingredients, i.e., carriers of the PON1 55MM-192QQ diplotype using OPs in homes or gardens were at the highest risk of developing PD relative to low/unexposed non-carriers (Table 2) [14].
Two studies in populations of European ancestry previously investigated pesticide interactions with PON1 SNPs in PD. The first identified only nine pesticide-exposed subjects with the PON1192RR genotype and found no interaction [70]. The second was a large European multicenter study [71]. They did not find interactions between PON1 and occupational pesticide use. However, the mostly clinic-based enrollment of prevalent PD cases in this study might have introduced survival bias in cases and controls. Importantly, including participants from different European countries increased the heterogeneity of the pesticide measure and occupational practices. Thus, no previous studies have investigated OP pesticide–PON1 interactions sufficiently and to date, ours is the only study to suggest that PON1 variants identifying slow metabolizers of OP pesticides contribute to PD risk in OP-exposed subjects.
Toxicants Interacting with Biologic Pathways Relevant for PD: Nitric Oxide Synthase (NOS1) and Organophosphorus Pesticides
The nitric oxide synthase (NOS) enzymes catalyze reactions that generate free radicals including nitric oxide (NO), a chemical messenger used in neurotransmission that can also contribute to oxidative/nitrosative stress and damage dopaminergic neurons [72]. Further, NO readily reacts with superoxides to form peroxynitrite (NO3 −), a potent toxin able to irreversibly inhibit mitochondrial respiration [72–74]. The NOS1 gene encodes neuronal NOS expressed in the brain [75]. While polymorphisms in NOS1 have been associated with PD risk in some studies [49, 76–79], GWAS did not identify a role for the gene in PD [4]. Nevertheless, experimental evidence strongly supports a neurotoxic role for NOS since inhibition or absence of neuronal NOS prevents MPTP-induced Parkinsonism [80, 81]. Furthermore, in the nigrostriatal region of postmortem human PD brains, NO levels were found to be increased [82].
An early study investigated GxE interactions with pesticides relying on a family-based design and described interactions between self-reported home pesticide use and a NOS1 rs2682826 variant for PD [49]. We identified interactions between pesticides and NOS1 rs2682826, though in the opposite direction to what was previously reported. We not only found interactions with household use of OPs but also based on our PUR-derived ambient exposures from agriculturally applied OPs. Concerning household OP use, frequent users carrying the T allele were at highest risk (OR = 2.84, 95 % CI = 1.49–5.40; Table 2); high ambient OP exposures at both residential or workplace addresses increased risk most strongly in T allele carriers (Table 2) [50]. Furthermore, three additional NOS1 SNPs, rs1047735, rs816353, and 3741480, also showed similar interactions with all of our OP exposure measures (data not shown) [50].
Our results suggest that reactive oxygen or nitrogen species (ROS/RNS) from multiple sources may potentiate each other, increasing the stress in dopamine neurons beyond their capacity to cope. We identified gene–pesticide interactions and estimated strong effects for exposure to common home and agricultural use OP pesticides and PD in genetically susceptible subjects carrying NOS1 variant alleles. This human data supports the hypothesis that oxidative/nitrosative stress-inducing mechanisms may be involved in OP pesticide effects on PD [83].
Toxicants Interacting with Biologic Pathways Relevant for PD: ALDH2 and ALDH-Inhibiting Pesticides
Aldehyde dehydrogenase (ALDH) metabolizes biogenic amine-related aldehydes, including the highly toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL), protecting neurons against aldehyde- and oxidative stress-related neurotoxicity. Thus, inhibition or lower function of this enzyme may be a mechanism for dopaminergic neuron loss in PD. ALDH acts in the inner mitochondrial membrane and the enzymatic activity of mitochondrial ALDH was found to be increased in postmortem brains of PD patients [84]. The pesticide rotenone was recently shown to decrease intracellular ALDH [85], while on the other hand, activation of ALDH was neuroprotective in rotenone-induced cell and animal models of Parkinsonism [86]. Cellular ALDH enzymes are sensitive to oxidative stress and lipid peroxidation, and we previously reported that the fungicide benomyl damages dopaminergic neurons by inhibiting ALDH enzyme activity in vitro and in vivo and is associated with increased PD risk [87].
In a laboratory screen, we identified multiple neuronal ALDH-inhibiting pesticides: benomyl, captan, dieldrin, mancozeb, maneb, triflumizole, zineb, and ziram to which our PEG study participants were exposed [21]. We found a trend of increasing risk with ambient exposures at both workplace and residential addresses with a 3.5-fold increase in PD risk (95 % CI 1.51–8.30) for exposure to three or more of these pesticides [21]. The ALDH2 gene haplotypes in our study population clustered into two clades, primarily determined by the genotype at rs737280. The presence of one or two copies of the less common clade (clade 2) modified the association between exposure to ALDH-inhibiting pesticides and PD; there was a 2- to 5-fold increase in PD risk for subjects with high exposure who carried at least one copy of clade 2 in ALDH2 compared with unexposed subjects homozygous for clade 1 (Table 2). This provides the very first evidence for the relevance of ALDH inhibition in PD pathogenesis and identifies new GxE pesticide interactions with relevance for PD.
Toxicants Interacting with Biologic Pathways Relevant for PD: SKP1 Gene and UPS-Inhibiting Pesticides
Parkinson’s disease shares protein misfolding, aggregation, and deposition as a disease mechanism with other neurodegenerative diseases. One of the elaborate pathways neurons use to remove damaged protein is the ubiquitin-proteasome system (UPS). Ever since rare familial PD has been linked to genetic defects in the E3 ubiquitin ligase gene parkin (PARK2) and in ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) [88], the UPS has been a candidate pathway for PD etiology. This hypothesis gained further support from experimental studies in cultured cells and animals as well as human postmortem brain tissue [89]. In Parkinson’s, Lewy bodies are cytoplasmic accumulations of damaged proteins that contain not only α-synuclein protein, but also ubiquitin and UCHL1 [90] as well as other damaged proteins not removed by the UPS or autophagy [91]. Permanently raised levels of α-synuclein impair the UPS, which in turn leads to further α-synuclein accumulation [92]. Chemical agents that induce oxidative stress and damage proteins may contribute to this process.
The UPS breaks down proteins in multiple steps starting with the activation of ubiquitin by E1 enzyme, followed by conjugation of activated ubiquitin to an E2 enzyme, ubiquitination of proteins via an E3 ligase, breaking down of ubiquitinated proteins in the 26S proteasome, and finally, the recycling of ubiquitin by deubiquitinating enzymes such as the UCHL1 [93]. Previous studies also identified pesticides that inhibit the proteasome in model systems [19, 94–96].
Thus, we investigated interactions between UPS-inhibiting pesticides our study participants were exposed to in PEG (propargite, cyanazine, dieldrin, endosulfan, benomyl, carbendazim, triflumizole, ferbam, metam, ziram) and six candidate genes in the UPS pathway and found evidence for interaction with s-phase kinase-associated protein 1(SKP1). Specifically, we found that the combination of high ambient exposure in carriers of at least one T allele in SKP1 rs2284312 resulted in a considerably larger risk of PD (OR of 7.57; Table 2) [51]. The function of rs2284312 is unknown, but the SKP1 gene encodes an E3 enzyme involved in target identification for ubiquitination [97] and expression of SKP1 in PD brains has been reported to be downregulated [98]. In addition, a cell model of murine substantia nigra found a decrease in SKP1 after treatment with MPTP [99].
The increase in PD risk with exposures at both residential and workplace addresses for the UPS-inhibiting pesticides and their interaction with the polymorphism in the SKP1 gene that encodes an important enzyme in the UPS pathway is intriguing but needs to be further investigated.
Toxicants Interacting with Biologic Pathways Relevant for PD: HLA-DR Influenced Immune Response and Pyrethroid Exposures
Inflammation has widely been suspected to contribute to PD etiology, with increased expression of inflammatory cytokines and altered composition of peripheral immune cells found in serum and spinal fluid of PD patients, as well as microglial activation and lymphocyte invasion found in areas of degeneration in postmortem PD brains [52]. The major histocompatibility complex class II (MHC-II) region, specifically HLA-DR rs3129882, implicated in idiopathic PD in multiple populations and in GWAS [2•, 4], is responsible for antigen presentation to the adaptive immune system. It has, however, also been suggested that associations between MCH-II and PD may depend on environmental exposures such as pesticides. Environmental exposures have been found to influence inflammation, for example, by impacting antigen presentation [100].
Thus, we examined interactions between immunomodulatory pesticides, including the OPs, OCs, and pyrethroids, and rs3129882 of HLA-DR in PD. We found positive interactions when comparing groups homozygous at the SNP (AA versus GG; Table 2) and when we used an additive genetic model to include heterozygous individuals among those exposed to pyrethroids. Interestingly, neither the genotype nor pyrethroid exposure alone influenced PD risk, but in those exposed to pyrethroids and carrying the GG genotype, PD risk was increased two and a half fold compared with unexposed AA carriers (Table 2). This pesticide–gene interaction suggests that variation in this MHC-II region together with exposure to pyrethroids, pesticides shown to modulate the immune system, influence PD risk. However, these associations need to be reexamined in other populations.
Conclusions
A review of 38 epidemiologic studies on pesticides and PD published almost a decade ago concluded that while general pesticide–PD association were reported consistently, evidence was lacking for causal relationships with any particular pesticide compound or for combinations of pesticide exposures in humans [7]. This conclusion was recently repeated in several reviews stating that although classes of pesticides have been linked to PD, attention still needs to be directed to identify specific chemicals in human studies [25•]. Further recommendations were that detailed quantitative exposure data for humans should be accompanied by determinations of genetic polymorphisms [101], especially those that increase genetic susceptibility in metabolism, elimination, and transport of pesticides [102]. The epidemiologic studies we conducted in California in the past decade in combination with evidence from other human GxE studies and experimental studies are starting to move us closer to reaching these goals.
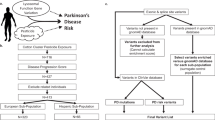
Different from most previous epidemiologic studies of PD, all of our diagnoses were clinically confirmed in one or more examination(s) by movement disorder specialists. Thus, we expect disease misclassification to be minimal. Additionally, population controls were drawn from the same region as the cases, likely providing adequate representativeness of the source population. We estimated pesticide exposure from multiple sources—household, occupational use, and ambient from agricultural uses—and our GxE results mutually corroborated each other when using these various exposure assessment methods. We were able to assess exposures to specific chemicals based on our GIS approach that used the unique CA-PUR records and examined GxE interactions taking existing knowledge about biologic/toxicological pathways into account (Fig. 1). We employed pathway-specific knowledge for pesticides that cause Parkinsonism in animal models and linked DAT/SLC6A3 variants likely affecting transport of specific chemicals into dopaminergic neurons to an increased risk from paraquat and maneb exposures. We learned that maneb is a UPS-inhibiting chemical and that genetic vulnerability in this pathway together with chemicals affecting the UPS contributes to PD risk. We confirmed previous reports that the metabolizing enzyme PON1 increases susceptibility to PD for OP-pesticide exposed individuals who are considered slow metabolizers. We found vulnerability for OP exposures in carriers of NOS1 gene variants that may contribute to the nitrosative stress pathway and—finally—corroborated a previous report that OC effects on PD risk may depend on ABCB1 gene variants that impact blood–brain barrier transport of chemicals. We were able to identify a group of ALDH-inhibiting pesticides and to show associations with PD for some common ALDH2 gene variants that increased susceptibility to these specific agents. Finally, for the latest chemicals to come into widespread agricultural and in-home use, pyrethroid insecticides, we found that they may be responsible for increased immune response among HLA-DR variant carriers putting them at an increased risk of developing PD.

Proposed Parkinson’s disease pathological mechanisms involving discussed GxE reports
Exploring additional gene–pesticide interactions with candidate genes in pathomechanistically relevant pathways for PD and pesticides is possible and needed. However, the past decade of research illustrates that it is unlikely that a single pathway/mechanism or a single pesticide is responsible for the complex etiology of neurodegeneration. The constant change in potentially neurotoxic agents used for pest control worldwide requires continued research and surveillance to protect humans from the long-term effects of neurotoxic agents.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Caudle WM, Guillot TS, Lazo CR, et al. Industrial toxicants and Parkinson’s disease. Neurotoxicology. 2012;33:178–88.
Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet. 2012;8:e1002548. This study provides an exhaustive summary of the status of PD genetics on more than seven million polymorphisms originating from GWAS or smaller-scale PD association studies.
Goldman SM. Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol. 2014;54:141–64.
Nalls MA, Pankratz N, Lill CM, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46:989–93.
Langston JW, Ballard P, Tetrud JW, et al. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–80. This study was the first to describe a specific toxicant as acting selectively on dopamine neurons.
Barbeau A, Dallaire L, Buu NT, et al. Comparative behavioral, biochemical and pigmentary effects of MPTP, MPP+ and paraquat in Rana pipiens. Life Sci. 1985;37:1529–38.
Brown TP, Rumsby PC, Capleton AC, et al. Pesticides and Parkinson’s disease—is there a link? Environ Health Perspect. 2006;114:156–64.
Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–54.
Pihlstrøm L, Toft M. Parkinson’s disease: what remains of the ‘missing heritability’? Mov Disord. 2011;26:1971–3.
Quadri M, Yang X, Cossu G, et al. An exome study of Parkinson’s disease in Sardinia, a Mediterranean genetic isolate. Neurogenetics. 2015;16:55–64.
Goldberg DW, Wilson JP, Knoblock CA, et al. An effective and efficient approach for manually improving geocoded data. Int J Health Geogr. 2008;7:60.
Rull RP, Ritz B. Historical pesticide exposure in California using pesticide use reports and land-use surveys: an assessment of misclassification error and bias. Environ Health Perspect. 2003;111:1582–9.
Ritz B, Costello S. Geographic model and biomarker-derived measures of pesticide exposure and Parkinson’s disease. Ann N Y Acad Sci. 2006;1076:378–87.
Narayan S, Liew Z, Paul K, et al. Household organophosphorus pesticide use and Parkinson’s disease. Int J Epidemiol. 2013;42:1476–85.
Liew Z, Wang A, Bronstein J, et al. Job exposure matrix (JEM)-derived estimates of lifetime occupational pesticide exposure and the risk of Parkinson’s disease. Arch Environ Occup Health. 2014;69:241–51.
Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal. 2005;7:685–93.
Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal. 2012;16:920–34.
Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog Brain Res. 2010;184:17–33.
Wang X-F, Li S, Chou AP, et al. Inhibitory effects of pesticides on proteasome activity: implication in Parkinson’s disease. Neurobiol Dis. 2006;23:198–205.
Zhou Y, Shie F-S, Piccardo P, et al. Proteasomal inhibition induced by manganese ethylene-bis-dithiocarbamate: relevance to Parkinson’s disease. Neuroscience. 2004;128:281–91.
Fitzmaurice AG, Rhodes SL, Cockburn M, et al. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology. 2014;82:419–26.
Roede JR, Jones DP. Thiol-reactivity of the fungicide maneb. Redox Biol. 2014;2:651–5.
Johnson ME, Bobrovskaya L. An update on the rotenone models of Parkinson’s disease: their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology. 2015;46:101–16.
Pan-Montojo F, Schwarz M, Winkler C, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898.
Van der Mark M, Brouwer M, Kromhout H, et al. Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results. Environ Health Perspect. 2012;120:340–7. This is a comprehensive systematic review of PD and exposure to pesticides that investigates methodological differences between studies and heterogeneity in results.
Barr DB, Olsson AO, Wong L-Y, et al. Urinary concentrations of metabolites of pyrethroid insecticides in the general U.S. population: National Health and Nutrition Examination Survey 1999–2002. Environ Health Perspect. 2010;118:742–8.
Glotfelty DE, Seiber JN, Liljedahl LA. Pesticides in fog. Nature. 1987;325:602–5.
USGS. Pesticides in the atmosphere—distribution, trends and governing factors. Sacromento: U.S. Geological Survey; 1995. p. 94–506.
Tiefenbacher J. Mapping the pesticide driftscape: theoretical patterns of the drift hazard. Geogr Environ Model. 1998;2:83–102.
Camann DE, Geno PW, Harding HJ, et al. A pilot study of pesticides in indoor air in relation to agricultural applications. In: Indoor air’93: proceedings of 6th International Conference on Indoor Air Quality and Climate. Helsinki: Finnish Society of Indoor Air Quality and Climate; 1993. p. 207–12.
CPDR. Summary of pesticide use report data. Sacramento: California Department of Pesticide Regulation; 2000.
Teschke K, Chow Y, Bartlett K, et al. Spatial and temporal distribution of airborne Bacillus thuringiensis var. kurstaki during an aerial spray program for gypsy moth eradication. Environ Health Perspect. 2001;109:47–54.
Baker LW, Fitzell DL, Seiber JN, et al. Ambient air concentrations of pesticides in California. Environ Sci Technol. 1996;30:1365–8.
Majewski MS, Foreman WT, Goolsby DA, et al. Airborne pesticide residues along the Mississippi River. Environ Sci Technol. 1998;32:3689–98.
Harnly M, McLaughlin R, Bradman A, et al. Correlating agricultural use of organophosphates with outdoor air concentrations: a particular concern for children. Environ Health Perspect. 2005;113:1184–9.
Wofford P, Segawa R, Schreider J, et al. Community air monitoring for pesticides. Part 3: using health-based screening levels to evaluate results collected for a year. Environ Monit Assess. 2014;186:1355–70.
Ward MH, Colt JS, Metayer C, et al. Residential exposure to polychlorinated biphenyls and organochlorine pesticides and risk of childhood leukemia. Environ Health Perspect. 2009;117:1007–13.
Jones BC, Huang X, Mailman RB, et al. The perplexing paradox of paraquat: the case for host-based susceptibility and postulated neurodegenerative effects. J Biochem Mol Toxicol. 2014;28:191–7.
Costello S, Cockburn M, Bronstein J, et al. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol. 2009;169:919–26.
Kelada SNP, Checkoway H, Kardia SLR, et al. 5′ and 3′ region variability in the dopamine transporter gene (SLC6A3), pesticide exposure and Parkinson’s disease risk: a hypothesis-generating study. Hum Mol Genet. 2006;15:3055–62. doi:10.1093/hmg/ddl247.
Kelada SN, Costa-Mallen P, Checkoway H, et al. Dopamine transporter (SLC6A3) 5′ region haplotypes significantly affect transcriptional activity in vitro but are not associated with Parkinson’s disease. Pharmacogenet Genomics. 2005;15:659–68.
Drgon T, Lin Z, Wang G-J, et al. Common human 5′ dopamine transporter (SLC6A3) haplotypes yield varying expression levels in vivo. Cell Mol Neurobiol. 2006;26:875–89.
Ritz BR, Manthripragada AD, Costello S, et al. Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ Health Perspect. 2009;117:964–9.
Droździk M, Białecka M, Myśliwiec K, et al. Polymorphism in the P-glycoprotein drug transporter MDR1 gene: a possible link between environmental and genetic factors in Parkinson’s disease. Pharmacogenetics. 2003;13:259–63.
Zschiedrich K, König IR, Brüggemann N, et al. MDR1 variants and risk of Parkinson disease. Association with pesticide exposure? J Neurol. 2009;256:115–20.
Dutheil F, Beaune P, Tzourio C, et al. Interaction between ABCB1 and professional exposure to organochlorine insecticides in Parkinson disease. Arch Neurol. 2010;67:739–45.
Narayan S, Sinsheimer JS, Paul KC, et al. Genetic variability in ABCB1, occupational pesticide exposure, and Parkinson’s disease. Environ Res. 2015. Accepted A.
Lee P-C, Rhodes SL, Sinsheimer JS, et al. Functional paraoxonase 1 variants modify the risk of Parkinson’s disease due to organophosphate exposure. Environ Int. 2013;56:42–7.
Hancock DB, Martin ER, Vance JM, et al. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics. 2008;9:249–62.
Paul KC, Sinsheimer JS, Rhodes SL, et al. Organophosphate pesticide exposures, nitric oxide synthase gene variants, and gene-pesticide interactions in a case-control study of Parkinson’s Disease, California (USA). Environ Health Perspect. 2015. doi:10.1289/ehp.1408976. Published Online First.
Rhodes SL, Fitzmaurice AG, Cockburn M, et al. Pesticides that inhibit the ubiquitin-proteasome system: effect measure modification by genetic variation in SKP1 in Parkinson׳s disease. Environ Res. 2013;126:1–8.
Kannarkat GT, Cook DA, Lee J-K, et al. Common genetic variant association with altered HLA expression, synergy with pyrethroid exposure, and risk for Parkinson’s disease: an observational and case–control study. NPJ Park Dis. 2015;1:15002.
Shimizu K, Matsubara K, Ohtaki K, et al. Paraquat leads to dopaminergic neural vulnerability in organotypic midbrain culture. Neurosci Res. 2003;46:523–32.
Richardson JR, Quan Y, Sherer TB, et al. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci. 2005;88:193–201.
Rappold PM, Cui M, Chesser AS, et al. Paraquat neurotoxicity is mediated by the dopamine transporter and organic cation transporter-3. Proc Natl Acad Sci U S A. 2011;108:20766–71.
Van de Giessen EM, de Win MML, Tanck MWT, et al. Striatal dopamine transporter availability associated with polymorphisms in the dopamine transporter gene SLC6A3. J Nucl Med. 2009;50:45–52.
Schinkel AH, Smit JJ, van Tellingen O, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502.
Bain LJ, McLachlan JB, LeBlanc GA. Structure-activity relationships for xenobiotic transport substrates and inhibitory ligands of P-glycoprotein. Environ Health Perspect. 1997;105:812–8.
Lecoeur S, Videmann B, Mazallon M. Effect of organophosphate pesticide diazinon on expression and activity of intestinal P-glycoprotein. Toxicol Lett. 2006;161:200–9.
Sreeramulu K, Liu R, Sharom FJ. Interaction of insecticides with mammalian P-glycoprotein and their effect on its transport function. Biochim Biophys Acta. 2007;1768:1750–7.
Kimchi-Sarfaty C, Oh JM, Kim I-W, et al. A ‘silent’ polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–8.
Cascorbi I, Gerloff T, Johne A, et al. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–74.
Hitzl M, Schaeffeler E, Hocher B, et al. Variable expression of P-glycoprotein in the human placenta and its association with mutations of the multidrug resistance 1 gene (MDR1, ABCB1). Pharmacogenetics. 2004;14:309–18.
Wang A, Cockburn M, Ly TT, et al. The association between ambient exposure to organophosphates and Parkinson’s disease risk. Occup Environ Med. 2014;71:275–81. doi:10.1136/oemed-2013-101394.
Davies HG, Richter RJ, Keifer M, et al. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat Genet. 1996;14:334–6.
Richter RJ, Jarvik GP, Furlong CE. Paraoxonase 1 (PON1) status and substrate hydrolysis. Toxicol Appl Pharmacol. 2009;235:1–9.
Costa LG, Cole TB, Furlong CE. Polymorphisms of paraoxonase (PON1) and their significance in clinical toxicology of organophosphates. J Toxicol Clin Toxicol. 2003;41:37–45.
O’Leary KA, Edwards RJ, Town MM, et al. Genetic and other sources of variation in the activity of serum paraoxonase/diazoxonase in humans: consequences for risk from exposure to diazinon. Pharmacogenet Genomics. 2005;15:51–60.
Liu Y-L, Yang J, Zheng J, et al. Paraoxonase 1 polymorphisms L55M and Q192R were not risk factors for Parkinson’s disease: a HuGE review and meta-analysis. Gene. 2012;501:188–92.
Taylor MC, Le Couteur DG, Mellick GD, et al. Paraoxonase polymorphisms, pesticide exposure and Parkinson’s disease in a Caucasian population. J Neural Transm. 2000;107:979–83.
Dick FD, De Palma G, Ahmadi A, et al. Gene-environment interactions in Parkinsonism and Parkinson’s disease: the Geoparkinson study. Occup Environ Med. 2007;64:673–80.
Kavya R, Saluja R, Singh S, et al. Nitric oxide synthase regulation and diversity: implications in Parkinson’s disease. Nitric Oxide. 2006;15:280–94.
Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909.
Lukaszewicz-Hussain A. Role of oxidative stress in organophosphate insecticide toxicity—short review. Pestic Biochem Physiol. 2010;98:145–50.
Licinio J, Prolo P, McCann SM, et al. Brain iNOS: current understanding and clinical implications. Mol Med Today. 1999;5:225–32.
Hague S, Peuralinna T, Eerola J, et al. Confirmation of the protective effect of iNOS in an independent cohort of Parkinson disease. Neurology. 2004;62:635–6.
Huerta C, Sánchez-Ferrero E, Coto E, et al. No association between Parkinson’s disease and three polymorphisms in the eNOS, nNOS, and iNOS genes. Neurosci Lett. 2007;413:202–5.
Levecque C, Elbaz A, Clavel J, et al. Association between Parkinson’s disease and polymorphisms in the nNOS and iNOS genes in a community-based case-control study. Hum Mol Genet. 2003;12:79–86.
Schulte C, Sharma M, Mueller JC, et al. Comprehensive association analysis of the NOS2A gene with Parkinson disease. Neurology. 2006;67:2080–2.
Przedborski S, Jackson-Lewis V, Yokoyama R, et al. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–71.
Liberatore GT, Jackson-Lewis V, Vukosavic S, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–9.
Hunot S, Boissière F, Faucheux B, et al. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience. 1996;72:355–63.
Tsang AHK, Lee Y-I, Ko HS, et al. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proc Natl Acad Sci U S A. 2009;106:4900–5.
Michel TM, Käsbauer L, Gsell W, et al. Aldehyde dehydrogenase 2 in sporadic Parkinson’s disease. Parkinsonism Relat Disord. 2014;20:S68–72.
Goldstein DS, Sullivan P, Cooney A, et al. Rotenone decreases intracellular aldehyde dehydrogenase activity: implications for the pathogenesis of Parkinson’s disease. J Neurochem. 2015;133:14–25.
Chiu C-C, Yeh T-H, Lai S-C, et al. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp Neurol. 2015;263:244–53.
Fitzmaurice AG, Rhodes SL, Lulla A, et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci U S A. 2013;110:636–41. doi:10.1073/pnas.1220399110.
Yoshii SR, Kishi C, Ishihara N, et al. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–40.
Ebrahimi-Fakhari D, Wahlster L, McLean PJ. Protein degradation pathways in Parkinson’s disease: curse or blessing. Acta Neuropathol. 2012;124:153–72.
Licker V, Kövari E, Hochstrasser DF, et al. Proteomics in human Parkinson’s disease research. J Proteome. 2009;73:10–29.
Wakabayashi K, Tanji K, Odagiri S, et al. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol Neurobiol. 2013;47:495–508.
Chen Q, Thorpe J, Keller JN. Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J Biol Chem. 2005;280:30009–17.
Betarbet R, Sherer TB, Greenamyre JT. Ubiquitin-proteasome system and Parkinson’s diseases. Exp Neurol. 2005;191 Suppl 1:S17–27.
Chou AP, Li S, Fitzmaurice AG, et al. Mechanisms of rotenone-induced proteasome inhibition. Neurotoxicology. 2010;31:367–72.
Chou AP, Maidment N, Klintenberg R, et al. Ziram causes dopaminergic cell damage by inhibiting E1 ligase of the proteasome. J Biol Chem. 2008;283:34696–703.
Wills J, Credle J, Oaks AW, et al. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS ONE. 2012;7:e30745.
Bai C, Sen P, Hofmann K, et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–74.
Grünblatt E, Mandel S, Jacob-Hirsch J, et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm. 2004;111:1543–73.
Fishman-Jacob T, Reznichenko L, Youdim MBH, et al. A sporadic Parkinson disease model via silencing of the ubiquitin-proteasome/E3 ligase component SKP1A. J Biol Chem. 2009;284:32835–45.
Esa AH, Warr GA, Newcombe DS. Immunotoxicity of organophosphorus compounds. Modulation of cell-mediated immune responses by inhibition of monocyte accessory functions. Clin Immunol Immunopathol. 1988;49:41–52.
Freire C, Koifman S. Pesticide exposure and Parkinson’s disease: epidemiological evidence of association. Neurotoxicology. 2012;33:947–71.
Dardiotis E, Xiromerisiou G, Hadjichristodoulou C, et al. The interplay between environmental and genetic factors in Parkinson’s disease susceptibility: the evidence for pesticides. Toxicology. 2013;307:17–23. doi:10.1016/j.tox.2012.12.016.
Acknowledgments
This work was funded in part by NIEHS Grants R01-ES010544, U54-ES012078, P01ES016732; the Michael J. Fox Foundation; The Parkinson Alliance, and the American Parkinson Disease Association. The funding organizations had no role in the design, conduct, interpretation, or publication of this work. Kim Paul also reports support from the Burroughs Wellcome Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
BR Ritz, KC Paul, and JM Bronstein declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
Standard Protocol Approvals and Patient Consents
Written informed consent was obtained from all enrolled subjects. All procedures using animals were approved by the UCLA Human Subjects Committee.
Additional information
This article is part of the Topical Collection on Susceptibility Factors in Environmental Health
Rights and permissions
About this article

Cite this article
Ritz, B.R., Paul, K.C. & Bronstein, J.M. Of Pesticides and Men: a California Story of Genes and Environment in Parkinson’s Disease. Curr Envir Health Rpt 3, 40–52 (2016). https://doi.org/10.1007/s40572-016-0083-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-016-0083-2