
Abstract
Nitric oxide synthase (NOS) genes (NOS1, NOS2A, and NOS3) may create excess nitric oxide that contributes to neurodegeneration in Parkinson’s disease (PD). NOS genes might also interact with one another or with environmental factors in PD. Coding and tagging single nucleotide polymorphisms (SNPs) (27 NOS1, 18 NOS2A, and five NOS3 SNPs) were genotyped in families with PD (1,065 cases and 1,180 relative and other controls) and were tested for allelic associations with PD using the association in the presence of linkage test and the pedigree disequilibrium test (PDT), allelic associations with age-at-onset (AAO) using the quantitative transmission disequilibrium test, and interactions using the multifactor dimensionality reduction—PDT. Gene–environment interactions involving cigarette smoking, caffeine, nonsteroidal anti-inflammatory drugs, and pesticides were examined using generalized estimating equations in participants with environmental data available. Significant associations with PD were detected for the NOS1 SNPs rs3782218, rs11068447, rs7295972, rs2293052, rs12829185, rs1047735, rs3741475, and rs2682826 (range of p = 0.00083–0.046) and the NOS2A SNPs rs2072324, rs944725, rs12944039, rs2248814, rs2297516, rs1060826, and rs2255929 (range of p = 0.0000040–0.047) in earlier-onset families with sporadic PD, and some SNPs were also associated with earlier AAO. There was no compelling statistical evidence for gene–gene interactions. However, of the significantly associated SNPs, interactions were found between pesticides and the NOS1 SNPs rs12829185, rs1047735, and rs2682826 (range of p = 0.012–0.034) and between smoking and the NOS2A SNPs rs2248814 (p = 0.021) and rs1060826 (p = 0.013). These data implicate NOS1 and NOS2A as genetic risk factors for PD and demonstrate that their interactions with established environmental factors may modulate the environmental effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the United States, at least one million individuals suffer from the neurodegenerative disorder known as Parkinson disease’s (PD) [1]. Progressive depletion of dopaminergic neurons in the substantia nigra of the brain leads to PD, and once approximately 70% of the neurons have been lost, individuals with PD begin to display the primary clinical signs of PD, including resting tremor, muscular rigidity, and bradykinesia (slowness of movement) [2]. Rare non-synonymous mutations in several genes (α-synuclein, parkin, UCH-L1, PINK1, DJ-1, and LRRK2) have been shown to cause familial forms of PD, but taken together, these variants account for only a small portion of the overall prevalence of PD [3, 4]. Far less is known about the genetic variants that are not sufficient on their own to cause disease but increase disease susceptibility for the more common sporadic form of PD. Several environmental factors have been suggested to influence risk of sporadic PD [3], but their biological mechanisms of action in the substantia nigra have eluded investigators. The vast majority of cases with sporadic PD likely results from interactions between susceptibility genes and environmental triggers [5], so identification of gene-environment interactions is a paramount step in deciphering the complex etiology of PD.
Nitric oxide (NO) performs vital physiological functions in the nervous and other systems under normal concentrations, but persistent high levels of NO can create a toxic environment. Three nitric oxide synthase (NOS) isoforms, produced by different genes, have been well characterized in humans: neuronal NOS (nNOS) produced by the gene NOS1, inducible NOS (iNOS) produced by the gene NOS2A, and endothelial NOS (eNOS) produced by the gene NOS3. iNOS normally induces low amounts of NO, but under pathogenic conditions, high levels of NO are generated to combat environmental insults in a wide range of cells upon induction. Conversely, the constitutive NOS isoforms, nNOS and eNOS, control a fluctuating low level of NO to perform normal physiological functions in neurons and vascular endothelial cells, respectively.
Altogether, the highest levels of NO throughout the body are found in the neurons [6]. Given this and the delicate balance between beneficial and toxic consequences of NO synthesis [7], NOS1, NOS2A, and NOS3 are strong candidate susceptibility genes for PD. Studies, including our own, have reported significant associations of NOS1 and/or NOS2A single nucleotide polymorphisms (SNPs) with PD [8–10], but contradictory reports have emerged [11, 12]. The majority of common genetic variation was not captured in most of the studies examining the NOS genes as only one or a few polymorphisms were tested for association with PD [9–11]. A comprehensive evaluation of associations between PD and multiple polymorphisms across each candidate NOS gene is thus warranted. Further, regulatory interactions between the NOS genes have been proposed [13], but no studies have reported such interactions in PD.
Environmental factors implicated in PD potentially interact with one or more of the candidate NOS genes. Numerous epidemiologic studies have reported inverse associations of cigarette smoking and/or caffeine consumption with PD, and meta-analysis suggested that individuals with PD are 50% less likely to report ever smoking and 30% less likely to report caffeinated coffee consumption as compared to controls [14]. We previously corroborated inverse associations of cigarette smoking and caffeine consumption with PD in our family-based case-control sample [15]. Recently, nonsteroidal anti-inflammatory drugs (NSAIDs) have also been suggested to delay or prevent onset of PD [16, 17]. In our study, we did not corroborate previous inverse associations of nonsteroidal anti-inflammatory drugs (NSAIDs) with PD [15], but it remains possible that interactions between NSAIDs and genetic factors may obscure the associations of NSAIDs with PD.
In contrast to these protective environmental factors, pesticide application has been suggested to increase the risk of PD, and meta-analysis of studies conducted in the US suggested that individuals with PD are over two times as likely to report ever being exposed to pesticides as compared to controls [18]. We also found a significant positive association between PD and pesticides, and more specifically, insecticides and herbicides, in our family-based case-control sample [19]. The mechanisms through which these environmental factors exert their effects remain unknown, and we hypothesize that these environmental effects might act in concert with the NOS genes. Significant interactions between cigarette smoking and SNPs in NOS1 and NOS2A have been reported [8, 10], but to our knowledge, no other studies have examined interactions involving the NOS genes and any other environmental factor in PD. To better understand the relationship of the candidate NOS genes and the potentially interacting environmental factors on PD, we conducted a comprehensive examination of SNPs across each NOS gene (NOS1, NOS2A, and NOS3) and interactions among these SNPs and environmental factors (smoking, caffeine, NSAIDs, and pesticides) in PD.
Subjects and methods
Study population
The Morris K. Udall PD Research Center of Excellence at Duke University Medical Center and the 13 centers of the PD Genetics Collaboration ascertained families with PD to identify genetic risk factors. Probands were referred to our study by various sources, including physicians, clinics, local support groups, other participants, media coverage, and the Udall Center website. Once recruited, probands were administered a family history interview to generate at least a three-generation pedigree. Given sporadic PD (i.e., only one individual with PD in the family), the probands were asked to contact siblings, parents, and spouses to request their participation. Given familial PD (i.e., more than one individual with PD in the family), probands also requested participation of other individuals with PD, their siblings, parents, and spouses as well as relatives connecting the individuals with PD. Informed consents were signed by all participants prior to collection of data and a blood sample for a DNA source, in accordance with the study protocols approved by the institutional review boards at the respective centers.
Previously described diagnostic criteria were followed to classify individuals as affected, unaffected, or unclear based on the presence of two out of three cardinal features (resting tremor, rigidity, or bradykinesia) [8]. Individuals with an unclear diagnosis were referred to movement disorder specialists for further evaluation and were removed from analyses to minimize phenotypic misclassification. Age-at-examination (AAE) was recorded for all participants, and age-at-onset (AAO) was recorded for individuals with PD as the age at which they first recalled one of the cardinal features of PD. Self-reported AAO has been shown to be a reliable measure of clinical onset as it correlates strongly with AAO noted in medical records and AAO reported by close relatives of the proband [20]. Participants with detected mutations in parkin, α-synuclein, or LRRK2 were excluded from this study to minimize genetic heterogeneity. To minimize bias due to population stratification, only self-reported white participants were included as this racial subset was the only subset with sufficient statistical power.
SNP selection and genotyping
Genotype data from 30 white European families (Centre d’Etude du Polymorphisme Humain, or CEPH, families) included in the International HapMap project were examined to select single nucleotide polymorphisms (SNPs) [21]. Tagging SNPs across NOS1, NOS2A, and NOS3 were chosen using the Tagger program in Haploview [22, 23]. All common (minor allele frequency ≥5%) phase II HapMap variants were considered, and tagging SNPs that capture these variants with a pairwise r 2 ≥ 0.67 were selected. In addition, previously implicated SNPs (rs2682826 in NOS1 and rs1060826 in NOS2A) [9, 10] were selected for genotyping as well as other validated coding SNPs (rs1047735 in NOS1, rs3730014 and rs16966563 in NOS2A, and rs1549758 in NOS3) identified from dbSNP of the National Center for Biotechnology Information database. A total of 27 NOS1 SNPs, 18 NOS2A SNPs, and five NOS3 SNPs were selected for genotyping.
Genomic DNA was extracted from whole blood using the Puregene DNA purification kit (Gentra Systems, Minneapolis, MN). The selected SNPs were genotyped using the TaqMan allelic discrimination assay (Applied Biosystems [ABI], Foster City, CA) with probes and primers designed through ABI’s assay-on-demand and assay-by-design services. TaqMan polymerase chain reaction (PCR) amplification was performed in 5 μl volumes (3 ng dried DNA, 1x TaqMan master mix from ABI, 900 nM of each primer, and 200 nM of each probe) using GeneAmp PCR system 9700 thermocyclers (ABI) with 50-cycle programs. Fluorescence from PCR amplification was detected using the ABI Prism 7900HT Sequence Detection systems and analyzed with its software.
Stringent quality control measures were implemented to maximize genotype accuracy and precision. Each 384-well plate contained wells with no DNA template to serve as negative controls. Internal control samples included two CEPH samples duplicated across all plates and 24 duplicated subject samples per plate, which were blinded from laboratory technicians. To pass quality control, each DNA plate had to meet 100% matching for duplicated samples as well as 95% overall genotyping efficiency.
Environmental risk factor data collection
Individuals enrolled through the Udall Center were administered a structured telephone questionnaire to collect detailed environmental risk factor data on demographics, health and habits, and pesticide and other chemical exposures. Reference ages were determined to give cases and controls comparable environmental exposure periods prior to age at onset of symptoms in the cases. The reference age was the AAO for cases, while the reference age for controls was the AAE minus the mean disease duration among cases (8 years).
The health and habits section of the questionnaire assessed cigarette smoking, caffeine (coffee, tea, and/or soft drinks) consumption, and NSAIDs (aspirin, ibuprofen, and/or naproxen) use. Participants who reported smoking, consuming any of the caffeine sources, or using any of the NSAID sources for at least once per week for 1 month or longer prior to their reference age were considered ever exposed to the relevant lifestyle factor. Participants were considered never exposed to any factor if they reported never being exposed or their exposure did not fit the specified criteria for being classified as ever exposed.
The pesticide exposure section assessed whether participants applied pesticides at work or in their home, garden, or lawn. Only first-hand exposures were considered. Participants who reported applying pesticides were asked to list the name of any pesticide they remembered using, the number of days it was used, and the years application started and stopped (if applicable). The reported pesticides were classified into specific chemical classes, but this classification scheme did not provide adequate statistical power to examine gene–environment interactions in our sample. Instead, the reported pesticide chemicals were classified more broadly into functional types (e.g., insecticides or herbicides). Those who reported applying any pesticides (or insecticides and herbicides specifically given our previous findings [19]) prior to the reference age were considered ever exposed. Frequency (days per year) and duration (years prior to reference age) of all reported pesticide applications were summed, and cumulative exposure (days) was calculated as the product of frequency and duration. Participants who reported never applying any pesticides were considered never exposed.
Statistical analyses
We used the Genetic Data Analysis (GDA) program to assess deviations from Hardy–Weinberg equilibrium (HWE) [24]. Linkage disequilibrium (LD), as measured by r 2 and D′ values, was assessed using the Haploview program for all pairwise marker combinations in each gene [23]. HWE and LD analyses were conducted in case and control samples separately, with at most one case and one control randomly selected from each family.
Family-based tests of association were used to examine associations of SNPs with PD. The association in the presence of linkage (APL) test [25] and the pedigree disequilibrium test (PDT) [26, 27] are both validated transmission-disequilibrium based methods, which compare the distributions of alleles transmitted to affected offspring to alleles not transmitted, but the power of the two methods varies depending on the pedigree structures in a data set [28]. Given that our data set consists of a variety of nuclear as well as extended pedigree structures, we applied both the APL and PDT methods to test for allelic associations of the 50 SNPs with PD. SNPs with significant allelic association results (p ≤ 0.05) were also tested for genotypic association using the genotype-PDT [29].
Given that family history is considered an important risk factor in PD [30], data were stratified into families with sporadic and familial PD. Within family history subsets, families were further stratified by AAO. Families with a minimum AAO <40 years were considered early-onset, and families with a minimum AAO ≥ 40 years were considered late-onset. Progressively older AAO cutoffs of 45, 50, 55, and 60 were also applied to test for AAO trends in association. SNPs significantly associated with PD in any AAO strata were then tested for association with AAO as the outcome variable. The Monks–Kaplan method of the quantitative transmission disequilibrium test (QTDT) was used to test for allelic associations with AAO in the relevant family history subset [31].
The multifactor dimensionality reduction-PDT (MDR-PDT) method, a merger of the genotype-PDT [29] and MDR [32] methods, was used to test for multi-locus associations between the NOS SNP genotypes in PD [33]. The MDR-PDT method involves calculation of the genotype-PDT test statistic for each multi-locus combination, identification of high-risk multi-locus genotypes, and permutation testing to control type I error in assessing the significance of those combinations predicting disease status [33]. Addition of loci that do not contribute to disease into the MDR-PDT model reduces power to detect truly associated loci [33]. Given this, interactions between the NOS SNP genotypes were first examined two genes at a time and then examined for all three NOS genes. The best models identified by MDR-PDT with two and three SNPs were explored, and if any of these models were statistically significant (p ≤ 0.05), an additional model involving four SNP loci was explored.
When using data-driven methods such as MDR-PDT, it is important to validate significant findings with a traditional regression modeling method [34]. A convergence of statistically significant results across analytic methods would support a true gene–gene interaction rather than a multi-locus association driven by a main association at a single locus or chance findings [34, 35]. Modeling with generalized estimating equations (GEE) is a valid and powerful approach to testing gene–gene and gene–environment interactions in family-based case-control data that can use all sampled relatives regardless of pedigree structure [36]. GEE model building was used to assess the goodness of fit of nested models containing the SNPs identified by significant MDR-PDT models (p ≤ 0.05). The patterns of high-risk genotype combinations identified by MDR-PDT were followed to choose the appropriate genotype coding in the GEE models with PD affection status as the outcome.
A likelihood ratio statistic was calculated by taking two times the difference between the log likelihoods for the nested models, and a chi-square test was performed using this statistic with degrees of freedom being equal to the number of deleted terms. A chi-square test with p ≤ 0.05 provided evidence for an interaction between the SNPs. The model building approach was taken one step further by removing one of the SNPs at a time to assess which SNP provided the most information about the disease affection status. GEE was implemented with the independence correlation matrix using PROC GENMOD of SAS version 8e (SAS Institute, Cary, NC). This matrix was found to be preferable to the exchangeable correlation matrix in a simulated data set of similar structure to our actual PD data set [36].
Gene–environment interaction analyses were conducted using GEE in the subset of the overall data set with environmental risk factor data available consisting mostly of individuals from sporadic PD families. Two types of GEE models were constructed using PD affection status as the outcome variable. Sex and AAE were included as confounding variables in all GEE models given their significant differences between cases and controls. The allele that was overtransmitted to cases for significantly associated SNPs was defined as the risk allele; otherwise, the minor allele was defined as the risk allele. In both GEE models, individuals carrying the risk allele in either homozygote or heterozygote form were considered risk allele carriers, while individuals carrying the wild-type allele in homozygote form were considered risk allele non-carriers. The full GEE model assessed interactions for each NOS SNP and environmental factor combination by including risk allele carrier status (carrier vs non-carrier), environmental exposure history (ever vs never), and an interaction term between carrier status and exposure history. In this model, participants who were risk allele non-carriers for the relevant SNP and never exposed to the relevant environmental factor served as the referent group. P values were calculated for the full GEE model to show the significance of each combination.
For SNP and environmental factor combinations with a significant interaction term (p ≤ 0.05), stratified analyses were performed to examine differential associations of the environmental factor by risk allele carrier status. The stratified GEE models assessed the associations of ever being exposed to the environmental factor (or being exposed at the high and low cumulative exposure levels) in carriers and non-carriers of risk alleles, separately. In each stratum, participants never exposed to the environmental factor served as the referent group. Adjusted odds ratios (ORs) and 95% confidence intervals (CIs) were calculated for stratified GEE models to show any differential patterns of association for stratified analyses.
Several tests were conducted to detect gene–environment interactions, but a multiple testing correction with approaches such as the Bonferroni correction or the false discovery rate would be too conservative given the correlations between intragenic SNPs. Instead, the following criteria were used to select the gene–environment combinations with the strongest statistical evidence for interaction: (1) more than one SNP in the gene must present a significant (p ≤ 0.05) interaction term with the environmental factor, (2) at least one SNP with a significant interaction term must also be significantly associated with PD by itself (p ≤ 0.05), and (3) when stratifying the data by risk allele carrier status, the same pattern of association for the environmental factor must be shown for each SNP.
Results
In total, 695 families (1,065 cases and 1,180 relative and other controls) were analyzed for our genetic study, including 337 families with sporadic PD and 358 with familial PD. Characteristics of the study population as divided into families with sporadic and familial PD are provided in Table 1. There were no significant deviations from HWE in any of the 50 genotyped SNPs for both cases and controls in families with either sporadic or familial PD.
Single-locus genetic associations
The NOS1, NOS2A, and NOS3 SNPs were tested for allelic association with PD using APL and PDT in overall, early-onset (minimum AAO <40 years), and late-onset (minimum AAO ≥40 years) families with sporadic and familial PD. Several NOS1 and NOS2A SNPs were significantly associated with PD in families with sporadic PD, and these results are shown in Table 2. For NOS1, significant associations were detected between PD and the minor alleles of three SNPs (rs12829185 [T], rs3741475 [A], and rs2682826 [A]) using the APL and/or PDT methods in the 40 early-onset families with sporadic PD. For NOS2A, alleles of five SNPs (rs2072324 [A], rs3794764 [A], rs12944039 [G], rs2297516 [A], and rs2255929 [T]) showed significant associations with PD using APL and/or PDT in the 337 overall families with sporadic PD. The allelic associations of three of these SNPs (rs12944039, rs2297516, and rs2255929) became highly significant (p < 0.0001 using APL and p < 0.01 using PDT) in the 40 early-onset families with sporadic PD, and significant genotypic associations were also present for rs12944039 (genotype-PDT p = 0.001), rs2297516 (p = 0.013), and rs2255929 (p = 0.021) in these families. Allelic associations of two additional SNPs (rs2248814 [A] and rs1060826 [A]) were found to be significant using APL (p < 0.01) in these families. The SNP rs12933039 was also significantly associated with PD in the 297 late-onset families with sporadic PD. The major alleles of rs12944039 and rs2255959 were the overtransmitted alleles in the association tests, whereas the minor alleles of the other significantly associated NOS2A SNPs were the overtransmitted alleles. There were no significant associations between PD and any NOS3 SNP in families with either sporadic or familial PD.
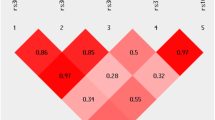
NOS1 and NOS2A SNPs were not significantly associated with PD in familial PD families (data not shown), with the exception of significant associations for the NOS2A SNPs rs1060826 (p = 0.052 using APL and p = 0.0040 using PDT) and rs2255929 (p = 0.052 using APL and p = 0.020 using PDT) in the 32 early-onset familial PD families. Since significant allelic associations were restricted to families with sporadic PD for the NOS1 SNPs and were strongest in these families for the NOS2A SNPs, subsequent analyses were restricted to families with sporadic PD. LD patterns for each gene were similar between the case and control samples from families with sporadic PD. LD measures from this case sample are reported in Fig. 1.

LD patterns in 333 unrelated individuals with PD from families with sporadic PD. r 2 values between (A) NOS1, (B) NOS2A, and (C) NOS3 SNPs are shown with shading becoming darker as the r 2 value increases
Most of the significant allelic associations for NOS1 and NOS2A SNPs were detected in the early-onset families using an AAO cutoff of 40 years, so allelic associations were further tested in early-onset families using progressively older AAO cutoffs (45, 50, 55, and 60 years) to assess the patterns of association by AAO. Allelic association results for NOS1 SNPs using APL in early-onset families at varying AAO cutoffs are shown in Fig. 2. Significance of the previously associated NOS1 SNPs (rs12829185, rs3741475, and rs2682826) was even stronger in the 80 early-onset families using an AAO cutoff of 45 years and in the 114 early-onset families using an AAO cutoff of 50 years. Alleles of five additional NOS1 SNPs became significant when using the higher AAO cutoffs of 45 and 50 years (rs7295972 [G] and rs2293052 [G] at p < 0.01 and rs3782218 [T], rs11068447 [G], and rs1047735 [A] at p < 0.05). The major alleles of rs2293052 and rs11068447 and the minor alleles of rs7295972, rs3782218, and rs1047735 were the overtransmitted alleles.

NOS1 allelic associations with PD using APL with varying AAO cutoffs in early-onset families with sporadic PD. –log10(APL p) are shown for each SNP
The NOS1 SNPs with the most significant associations (rs12829185, rs3741475, and rs2682826) along with rs1047735 exist in fairly strong LD with r 2 values ranging from 0.31 to 0.85 (Fig. 1a) and D′ values ranging from 0.59 to 1. The SNPs rs3782218, rs11068447, rs7295972, and rs2293052 are in low to moderate LD with all other significantly associated SNPs (Fig. 1a).
Allelic association results for NOS2A SNPs using APL in early-onset families at varying AAO cutoffs are shown in Fig. 3. Unlike the NOS1 trend in associations by AAO, the significance of most associated NOS2A SNPs (rs2248814, rs2297516, rs1060826, and rs2255929) decreased when using increasing AAO cutoffs. The significance of the associated SNP rs12944039 increased slightly in the 80 early-onset families with AAO less than 45 years but then decreased with each increasing AAO cutoff. The minor alleles of two additional NOS2A SNPs (rs2072324 [A] and rs944725 [T]) became significant in early-onset families with an AAO less than 45 or 50 years.
The SNPs rs2248814, rs1060826, and rs2255929 exist in strong LD with r 2 values ranging from 0.45 to 0.95 (Fig. 1b) and D′ values ranging from 0.96 to 1. Other pairwise combinations of significantly associated NOS2A SNPs showed only low to moderate LD levels (Fig. 1b).

NOS2A allelic associations with PD using APL with varying AAO cutoffs in early-onset families with sporadic PD. –log10(APL p) are shown for each SNP
Given the AAO trends in association of NOS1 and NOS2A SNPs with PD, QTDT was used to examine associations between AAO as the outcome and the SNPs significantly associated with PD in any early-onset strata. The NOS1 SNPs rs3782218, rs11068447, rs7295972, rs2293052, rs12829185, rs1047735, rs3741475, and rs2682826 were tested for allelic association with AAO of PD, and QTDT analyses revealed significant associations between earlier AAO and the G allele of rs2293052 (p = 0.041) as well as the A allele of rs2682826 (p = 0.0020). In addition, the NOS2A SNPs rs2072324, rs944725, rs12944039, rs2248814, rs2297516, rs1060826, and rs2255929 were tested, and there were significant associations between earlier AAO of PD and alleles at three SNPs: the G allele of rs12944039 (p = 0.010), the A allele of rs2297516 (p = 0.032), and the T allele of rs2255929 (p = 0.017). The NOS1 and NOS2A alleles associated with an earlier AAO are the same alleles as those associated with risk of PD in earlier-onset families.
Gene–gene interactions
In the 40 early-onset families with sporadic PD, the best two-SNP MDR-PDT model revealed a significant association of PD with genotypes at the NOS2A SNP rs2255929 and the NOS3 SNP rs1808593 (p = 0.046). This NOS2A SNP also had a significant single-locus association with PD in early-onset families, whereas the NOS3 SNP was not significantly associated with PD in any stratum. The best three-SNP MDR-PDT model in the 40 early-onset families with sporadic PD showed a significant association of PD with genotypes at the NOS2A SNPs rs2297516 and rs2297515 (r 2 = 0.30, Fig. 1b) and the NOS3 SNP rs1549758 (p = 0.0070). Of these SNPs, only rs2297516 had a significant single-locus association with PD. A four-SNP model was also tested in these families, but the best model was not significant. There were no significant multi-locus associations of any two- or three-SNP models identified in the 337 overall or the 297 late-onset families with sporadic PD, and there were no significant findings when all NOS SNPs were input into MDR-PDT (data not shown).
From the pattern of high-risk genotype combinations identified by MDR-PDT for the best two-SNP model in the early-onset families with sporadic PD, dominant coding was selected as the appropriate genotype coding scheme for GEE model building of the significant two-SNP NOS2A-NOS3 combination. A reduction in the full model containing rs2255929, rs1808593, and an interaction term to a reduced model with no interaction term resulted in a non-significant likelihood ratio test thus failing to provide significant evidence for an interaction between rs2255929 and rs1808593. Further, the goodness of fit of this reduced model was not significantly different from the model containing only rs2255929 but was significantly different from the model containing only rs1808593 (p = 0.021). The model containing rs2255929 thus provided more information about PD affection status than the model without rs2255929. GEE model building could not be applied to the best three-SNP model identified by MDR-PDT in the early-onset families with sporadic PD due to convergence problems when analyzing high-order interactions in small sample sizes. Nonetheless, the trends in high-risk and low-risk genotype distributions for the best two-SNP and three-SNP models involving NOS2A and NOS3 in early-onset families with sporadic PD reflected the distributions of SNPs having significant single-locus associations with PD (data not shown).
Gene-environment interactions
Gene–environment interaction analyses focused on 163 cases and 178 relative and other controls from 168 sporadic PD families with environmental risk factor data available. Of these, 48.5% reported ever smoking cigarettes, 90.3% reported ever consuming caffeinated beverages (coffee, tea, and/or soft drinks), 21.5% reported ever using NSAIDs (aspirin, ibuprofen, and/or naproxen), and 55.1% reported ever applying pesticides. GEE modeling showed significant interactions between these environmental factors and several NOS SNPs when adjusting for the main genetic and environmental factors, AAE, and sex.
Interactions between three NOS1 SNPs and pesticide application (p = 0.034 for rs12829185, p = 0.026 for rs10774910, and p = 0.028 for rs2682826) met all criteria for gene–environment interaction. Moderate LD existed between these SNPs with r 2 values ranging from 0.27 to 0.52 (Fig. 1a) and D′ values ranging from 0.58 to 0.90. In data stratified by risk allele carrier status at each of these SNPs, a significant positive association of pesticides and PD was observed in risk allele non-carriers, but this association diminished in risk allele carriers (Table 3). Further, this pattern of association showed a dose–response pattern, whereby the strongest positive association of pesticides and PD in risk allele non-carriers of rs12829185, rs10774910, or rs2682826 occurred with the high cumulative exposure level (≥ median of 87.5 days) followed by the low cumulative exposure level (< median of 87.5 days). Using rs2682826 to illustrate, there was a strong positive association of being exposed at the high cumulative exposure level (OR = 5.03; 95% CI, 2.28–11.12) and a more moderate association of being exposed at the low cumulative exposure level (OR = 2.87; 95% CI, 1.19–6.94) in risk allele non-carriers (rs2682826 GG). There were no significant associations in risk allele carriers (rs2682826 GA/AA) exposed at either cumulative exposure level.
When limiting pesticide exposure to use of insecticides and herbicides, interactions with the NOS1 SNPs became more significant (p = 0.0079 for rs12829185, p = 0.015 for rs10774910, and p = 0.011 for rs2682826). Five additional SNPs showed significant interaction with insecticide/herbicide exposure (rs547954, rs11611788, rs7139256, rs2293054, and rs1047735 with a range of p = 0.012–0.042) and followed the same pattern of association where the significant positive association of insecticides/herbicides with PD was only observed in risk allele non-carriers (data not shown). Low to moderate LD existed among the SNPs that significantly interacted with insecticide/herbicide use (range of r 2 = 0.03–0.65 from Fig. 1a, range of D′ = 0.46–1), and among these, only rs1047735 showed moderate LD levels with the SNPs that significantly interacted with use of any pesticide (range of r 2 = 0.31–0.78 from Fig. 1a, range of D′ = 0.59–1).
Interactions involving cigarette smoking and the NOS2A SNPs rs2314810 (p = 0.024), rs2248814 (p = 0.021), and rs1060826 (p = 0.013) provided a second combination meeting all specified criteria. There was also a nearly significant interaction between cigarette smoking and rs2255929 (p = 0.060). These SNPs are in strong LD with one another as defined by D′ values of at least 0.96, although r 2 values were low for combinations involving rs2314810 (range of r 2 = 0.04–0.08 from Fig. 1b). In data stratified by risk allele carrier status at each of these SNPs, a significant inverse association between PD and smoking was observed in risk allele non-carriers, but this significant inverse association diminished in risk allele carriers (Table 3).
It is possible to have a significant interaction between genetic and environmental factors that do not confer significant effects on their own. If we remove all criteria, even the one stating that at least one SNP with a significant interaction term must also be significantly associated with PD by itself, then additional gene–environment combinations would be considered in the analysis. This wider search for interaction needs to guard against increasing type I error, and therefore we only consider additional pairwise interactions with a significance level of p ≤ 0.01 (a slight increase in significance level). This analysis detects one additional significant interaction, between caffeine and the NOS2A SNP rs944725 (p = 0.0088). Stratified analyses showed that the significant inverse associations of caffeine was present in risk allele non-carriers of the NOS2A SNP, while there were no significant associations in the risk allele carriers (data not shown).
Discussion
Five independent studies have now examined the associations of NOS1, NOS2A, and/or NOS3 SNPs with PD [9–12], and this study is the largest to date with more than twice the number of participants and more SNPs that capture common variation across the NOS genes than any other study. Multiple SNPs in NOS1 (rs3782218, rs11068447, rs7295972, rs2293052, rs12829185, rs1047735, rs3741475, and rs262826) and NOS2A (rs2072324, rs944725, rs12944039, rs2248814, rs2297516, rs1060826, and rs2255929) were significantly associated with PD, particularly in earlier-onset families with sporadic PD. The effect in earlier AAO was also supported by quantitative trait analysis using AAO, in which we found that the risk alleles of rs2293052, rs2682826, rs12944039, rs2297516, and rs2255929 were associated with an earlier AAO. Further, the NOS genes may interact with one another or with environmental factors in influencing risk of PD. Although our findings did not provide compelling evidence for gene–gene interactions, promising gene-environment combinations with statistical evidence for interaction were revealed. The statistical patterns suggest that NOS1 genotypes may modify the effect of pesticides and NOS2A genotypes may modify the effect of smoking. When considering all pairwise interactions and a slightly more stringent significance level, one additional interaction between NOS2A and caffeine was detected.
Excess NO synthesis is potentially involved in the progressive neuronal loss that characterizes PD. NOS gene variants that alter the regulation or function of NO synthesis could thus influence PD susceptibility. We previously reported significant associations of two NOS2A SNPs located in the flavin adenine dinucleotide (FAD) binding domain with risk in early-onset families and with an earlier AAO [8]. This work extends our previous association findings for NOS2A by implicating additional SNPs and expanding our region of interest beyond the FAD-binding domain. In this study, data were also stratified by family history of PD, and significant associations for NOS2A were strongest in earlier-onset families with sporadic PD. Significant associations for NOS1 were present only in earlier-onset families with sporadic PD. The restriction of significant association findings for NOS1 and NOS2A to families with sporadic PD suggests that variants in these genes may not be sufficient to cause disease but increase the likelihood for PD development, thus implicating NOS1 and NOS2A as susceptibility genes for sporadic PD. The associated NOS1 and NOS2A SNPs do not confer known functional or regulatory consequences, but these SNPs may confer unknown consequences or reside in LD with unidentified variants with true effects.
Prior biological studies have implicated NOS1 (the nNOS gene) and NOS2A (the iNOS gene) in PD development. Mutant mice with an ablated nNOS or iNOS gene are significantly resistant to the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a parkinsonism-inducing agent [37–39]. Mice injected with a pharmacological inhibitor of the nNOS gene also show significant neuroprotection from MPTP [37]. Similarly, pharmacological inhibition of the iNOS gene attenuates inflammatory-induced dopaminergic neuronal loss [40]. In humans, heightened concentrations of both nNOS and iNOS correlating with NO overproduction have been detected in the substantia nigra of post mortem PD brains [41]. The question of whether increased NOS activity contributes to disease onset or accompanies the disease process remains unanswered, but biological links between excess NO synthesis and parkinsonism authenticate the statistical implications of NOS1 and NOS2A in PD.
The case-control studies examining associations of NOS1, NOS2A, and/or NOS3 SNPs with PD have reached differing conclusions. Levecque et al. initially tested the hypothesis that NOS1 and NOS2A influence PD in a French sample and found that the TT genotype of the NOS1 SNP rs2682826 significantly increased the risk of PD and the AA genotype of the NOS2A SNP rs1060826 significantly decreased the risk of PD [10]. Hague et al. replicated the significant inverse association of rs1060826 with PD but found no significant association of rs2682826 in a Finnish sample [9]. In our family-based case-control sample from the US, we reported associations of rs1060826 and other NOS2A SNPs with PD, but we found a significant positive association between the risk of PD and the A allele and AA genotype of rs1060826. This “flip-flop” association may indeed be confirmatory, since unmeasured interactive effects or variable LD patterns with a truly causal variant can lead to opposing directions of association for an investigated variant [42]. Subsequent studies in Spanish and German samples failed to detect associations between PD and NOS SNPs, including rs1060826 and rs2682826 [11, 12]. In this study, we detected significant associations between PD and several NOS1 and NOS2A SNPs, including the previously implicated SNPs. No studies have provided significant evidence to suggest that NOS3 is associated with PD.
There are several differences between our study and others to explain the different association findings. Each independent study has been conducted in a different American or European population. Different LD patterns between associated SNPs and truly causal variants in the various populations could explain the confirmation of association in some samples but not in others. Secondly, we detected associations for NOS1 and NOS2A SNPs in early-onset families, particularly in families with AAO less than 40, 45, or 50. In contrast, other studies examined associations in their overall samples with older onset cases (65 years median AAO for Levecque et al., 67.2 years mean AAO for Hague et al., and 52 years median AAO for Schulte et al.) [9, 10, 12]. The previous association findings could therefore reflect differing effects of NOS1 and NOS2A by AAO. Lastly, evaluation of single locus associations without accounting for potential interactions with other genetic factors or environmental factors could hinder replication.
Each NOS gene individually has the potential of influencing risk of PD, but a regulatory “cross-talk” between the NOS genes has been proposed [13]. The constitutive NOS genes (NOS1 and NOS3) and the inducible NOS2A gene are inversely regulated, such that decreased constitutive NOS expression and consequently low basal levels of NO are required for NOS2A expression. Despite this, no previous studies have reported testing for NOS gene interactions in PD.
Significant multi-locus associations of NOS2A and NOS3 SNPs with PD were detected in 40 early-onset families with sporadic PD, but important methodological concerns weaken confidence that this statistical finding is biologically relevant. First, our data show consistent trends of high-risk combinations that correspond to the high-risk genotypes of SNPs with significant single-locus associations. These trends suggest that the significant associations of the NOS2A SNPs rs2255929 and rs2297516 may be driving the significant two-SNP and three-SNP associations involving NOS2A and NOS3. The GEE model building provided no evidence for an interaction between the two SNPs and also showed that rs2255929 was a more important indicator of PD affection status than rs1808593.
Second, many cells in the MDR model contained either missing or singleton data especially for high-dimension models in relatively small sample sizes, such as our subset of 40 early-onset families with sporadic PD. This complicates the estimation of the prediction error [32].
Lastly, incorporation of all NOS SNPs into MDR-PDT revealed no significant multi-locus associations, meaning that the NOS2A-NOS3 models were not robust enough to overcome additional correction for multiple comparisons. Further exploration of NOS gene interactions in a larger sample size, particularly for early-onset PD, is needed given the biological plausibility and this initial statistical implication of a NOS2A-NOS3 interaction.
Each NOS SNP was also tested for interaction with exposure history of four putative environmental factors for PD (cigarette smoking, caffeine, NSAIDs, and pesticides) in our family-based case-control sample. The use of family-based designs alleviates problems with confounding by ethnic background in genetic association studies and increases efficiency for detecting gene-environment interactions when compared to population-based study designs [43]. The reduced number of families in which gene–environment interactions were evaluated is therefore offset by the increased efficiency of a family-based sample to detect interactions. The use of retrospective environmental exposure data may introduce the potential for recall bias if the accuracy of responses differed between cases and controls. Cases are typically more aware of potential risk factors for disease compared to population controls, and this may create a biased association if cases are more likely to recall exposure due to their increased awareness. However, the family-based study design likely reduces this potential bias as unaffected relatives may also be aware of potential risk factors for disease and be equally likely to recall exposure as their affected relatives. Several potential gene–environment interactions were presented. Although we cannot eliminate the possibility of false-positives in our gene–environment interaction data, we specified criteria to prioritize those interactions with the most consistent evidence for the relevant genetic and environmental factors influencing risk of PD.
Statistical interactions between NOS1 and pesticides in relation to PD were previously unexplored, and our data identify three SNPs (rs12829185, rs10774910, and rs2682826), in only moderate LD with one another, that significantly interact with a history of pesticide application. In stratified analysis, a positive association of PD with use of any pesticide was observed in the risk allele non-carriers of the three significantly interacting SNPs but not in carriers. This interaction was further supported by a dose–response pattern in risk allele non-carriers, showing that the positive association between pesticides and PD was stronger in those with a high cumulative exposure compared to those with a low cumulative exposure. Five additional NOS1 SNPs (rs547954, rs11611788, rs7139256, rs2293054, and rs1047735) showed significant interactions when limiting exposure to insecticide/herbicide application.
Although the delineation of a direct interaction between NOS1 and pesticides remains elusive, increased NO levels have been observed in the brains of rats chronically administrated the pesticide chemical rotenone [44]. For both NO and pesticides, impairment of mitochondrial complex I activity is the proposed mode of detrimental action [6]. One hypothesis proposes that excessive levels of NOS-generated NO and pesticide-generated O2 − react to form peroxynitrite (ONOO−), a potent toxin known to inhibit mitochondrial complex I function [6, 45]. A disruption in NOS1 expression by the NOS1 risk alleles (or a correlated variant not yet identified) could alter NO levels and lead to differing effects of pesticide application on risk of PD. Alternatively, the existence of a significant association of pesticides with PD in NOS1 risk allele non-carriers but not in carriers might indicate a heterogeneity of effects, in which one subset of individuals (individuals from early-onset families) are influenced by NOS1, whereas the risk of PD in another subset are influenced by pesticide application.
We previously proposed an interaction of smoking with the NOS2A SNPs rs1060826 and rs2255929 in PD [8], and the current study identified two additional SNPs showing significant interactions with cigarette smoking (rs2314810 and rs2248814). In risk allele non-carriers of these SNPs, the well-documented inverse association of cigarette smoking was observed, but this association diminished in risk allele carriers. This may suggest that a strong genetic effect by NOS2A overrides the protective effect of smoking. However, prior biological evidence supports a direct interaction between NOS2A and smoking, as an in vitro study suggested that cigarette smoke condensates attenuate iNOS induction and ultimately reduce toxicity [46]. The implicated NOS2A SNPs (or a correlated variant not yet identified) could disrupt this regulatory pathway and lead to the observed disruption in smoking’s association with PD.
Our findings support NOS1 and NOS2A as genetic risk factors for PD and show that these genes might modify the effects of established environmental factors for PD. Despite the epidemiologic evidence, the biological mechanisms of action by pesticides and smoking in PD, if existent, have long eluded investigators. Interactions with genetic risk factors likely hold the key to unraveling these puzzles. This study shows that NOS1 and NOS2A variants associated with PD eliminate the effects of pesticides and cigarette smoking. Individuals who carry the NOS1 risk alleles are not subject to the risk associated with pesticides, and those who carry the NOS2A risk alleles are not subject to the protective effect of smoking. Pesticides and smoking are significantly associated with PD in individuals who do not carry these variations.
Statistical association does not equate to biological mechanism, but our comprehensive examination of interactions of NOS1 and NOS2A SNPs with environmental factors generates plausible hypotheses for how NOS1, NOS2A, cigarette smoking, and pesticide exposure might influence risk of PD. Further biological investigations should be aimed at identifying functional or regulatory NOS1 and NOS2A variants responsible for increasing the risk of PD, decreasing AAO, and modulating the effects of established environmental factors.
References
McDonald WM, Richard IH, DeLong MR (2003) Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 54:363–375 doi:10.1016/S0006-3223(03)00530-4
Fearnley JM, Lees AJ (1991) Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5):2283–2301 doi:10.1093/brain/114.5.2283
Coppede F, Mancuso M, Siciliano G, Migliore L, Murri L (2006) Genes and the environment in neurodegeneration. Biosci Rep 26:341–367 doi:10.1007/s10540-006-9028-6
Wood-Kaczmar A, Gandhi S, Wood NW (2006) Understanding the molecular causes of Parkinson’s disease. Trends Mol Med 12:521–528 doi:10.1016/j.molmed.2006.09.007
Benmoyal-Segal L, Soreq H (2006) Gene-environment interactions in sporadic Parkinson’s disease. J Neurochem 97:1740–1755 doi:10.1111/j.1471-4159.2006.03937.x
Ebadi M, Sharma SK (2003) Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid Redox Signal 5:319–335 doi:10.1089/152308603322110896
Colasanti M, Suzuki H (2000) The dual personality of NO. Trends Pharmacol Sci 21:249–252 doi:10.1016/S0165-6147(00)01499-1
Hancock DB, Martin ER, Fujiwara K, Stacy MA, Scott BL, Stajich JM et al (2006) NOS2A and the modulating effect of cigarette smoking in Parkinson’s disease. Ann Neurol 60:366–373 doi:10.1002/ana.20915
Hague S, Peuralinna T, Eerola J, Hellstrom O, Tienari PJ, Singleton AB (2004) Confirmation of the protective effect of iNOS in an independent cohort of Parkinson disease. Neurology 62:635–636
Levecque C, Elbaz A, Clavel J, Richard F, Vidal JS, Amouyel P et al (2003) Association between Parkinson’s disease and polymorphisms in the nNOS and iNOS genes in a community-based case-control study. Hum Mol Genet 12:79–86 doi:10.1093/hmg/ddg009
Huerta C, Sanchez-Ferrero E, Coto E, Blazquez M, Ribacoba R, Guisasola LM et al (2007) No association between Parkinson’s disease and three polymorphisms in the eNOS, nNOS, and iNOS genes. Neurosci Lett 413:202–205 doi:10.1016/j.neulet.2006.11.044
Schulte C, Sharma M, Mueller JC, Lichtner P, Prestel J, Berg D et al (2006) Comprehensive association analysis of the NOS2A gene with Parkinson disease. Neurology 67:2080–2082 doi:10.1212/01.wnl.0000247672.41736.bd
Persichini T, Cantoni O, Suzuki H, Colasanti M (2006) Cross-talk between constitutive and inducible NO synthase: an update. Antioxid Redox Signal 8:949–954 doi:10.1089/ars.2006.8.949
Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ (2002) A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol 52:276–284 doi:10.1002/ana.10277
Hancock DB, Martin ER, Stajich JM, Jewett R, Stacy MA, Scott BL et al (2007) Smoking, caffeine, and nonsteroidal anti-inflammatory drugs in families with Parkinson disease. Arch Neurol 64:576–580 doi:10.1001/archneur.64.4.576
Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ et al (2005) Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol 58:963–967 doi:10.1002/ana.20682
Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA et al (2003) Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 60:1059–1064 doi:10.1001/archneur.60.8.1059
Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS (2001) Environmental risk factors and Parkinson’s disease: a metaanalysis. Environ Res 86:122–127 doi:10.1006/enrs.2001.4264
Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, Stacy MA et al (2008) Pesticide exposure and risk of Parkinson’s disease: a family-based case-control study. BMC Neurol 8:6 doi:10.1186/1471-2377-8-6
Reider CR, Halter CA, Castelluccio PF, Oakes D, Nichols WC, Foroud T (2003) Reliability of reported age at onset for Parkinson’s disease. Mov Disord 18:275–279 doi:10.1002/mds.10391
The International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796 doi:10.1038/nature02168
de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D (2005) Efficiency and power in genetic association studies. Nat Genet 37:1217–1223 doi:10.1038/ng1669
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265 doi:10.1093/bioinformatics/bth457
Lewis PO, Zaykin D (2000) Genetic data analysis: Computer program for analysis of allelic data, version 1.0 (d15)
Martin ER, Bass MP, Hauser ER, Kaplan NL (2003) Accounting for linkage in family-based tests of association with missing parental genotypes. Am J Hum Genet 73:1016–1026 doi:10.1086/378779
Martin ER, Bass MP, Kaplan NL (2001) Correcting for a potential bias in the pedigree disequilibrium test. Am J Hum Genet 68:1065–1067 doi:10.1086/319525
Martin ER, Monks SA, Warren LL, Kaplan NL (2000) A test for linkage and association in general pedigrees: the pedigree disequilibrium test. Am J Hum Genet 67:146–154 doi:10.1086/302957
Nicodemus KK, Luna A, Shugart YY (2007) An evaluation of power and type I error of single-nucleotide polymorphism transmission/disequilibrium-based statistical methods under different family structures, missing parental data, and population stratification. Am J Hum Genet 80:178–185 doi:10.1086/510498
Martin ER, Bass MP, Gilbert JR, Pericak-Vance MA, Hauser ER (2003) Genotype-based association test for general pedigrees: the genotype-PDT. Genet Epidemiol 25:203–213 doi:10.1002/gepi.10258
Sellbach AN, Boyle RS, Silburn PA, Mellick GD (2006) Parkinson’s disease and family history. Parkinsonism Relat Disord 12:399–409 doi:10.1016/j.parkreldis.2006.03.002
Monks SA, Kaplan NL, Weir BS (1998) A comparative study of sibship tests of linkage and/or association. Am J Hum Genet 63:1507–1516 doi:10.1086/302104
Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF et al (2001) Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet 69:138–147 doi:10.1086/321276
Martin ER, Ritchie MD, Hahn L, Kang S, Moore JH (2006) A novel method to identify gene–gene effects in nuclear families: the MDR-PDT. Genet Epidemiol 30:111–123 doi:10.1002/gepi.20128
Coffey CS, Hebert PR, Ritchie MD, Krumholz HM, Gaziano JM, Ridker PM et al (2004) An application of conditional logistic regression and multifactor dimensionality reduction for detecting gene–gene interactions on risk of myocardial infarction: the importance of model validation. BMC Bioinformatics 5:49 doi:10.1186/1471-2105-5-49
Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley-Koch AE, Mei H et al (2005) Identification of significant association and gene–gene interaction of GABA receptor subunit genes in autism. Am J Hum Genet 77:377–388 doi:10.1086/433195
Hancock DB, Martin ER, Li YJ, Scott WK (2007) Methods for interaction analyses using family-based case-control data: conditional logistic regression versus generalized estimating equations. Genet Epidemiol 31:883–893 doi:10.1002/gepi.20249
Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM (1996) Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A 93:4565–4571 doi:10.1073/pnas.93.10.4565
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB (2000) Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem 74:2213–2216 doi:10.1046/j.1471-4159.2000.0742213.x
Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG et al (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med 5:1403–1409 doi:10.1038/70978
Arimoto T, Bing G (2003) Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dis 12:35–45 doi:10.1016/S0969-9961(02)00017-7
Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y et al (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 72:355–363 doi:10.1016/0306-4522(95)00578-1
Lin PI, Vance JM, Pericak-Vance MA, Martin ER (2007) No gene is an island: the flip-flop phenomenon. Am J Hum Genet 80:531–538 doi:10.1086/512133
Witte JS, Gauderman WJ, Thomas DC (1999) Asymptotic bias and efficiency in case-control studies of candidate genes and gene–environment interactions: basic family designs. Am J Epidemiol 149:693–705
Bashkatova V, Alam M, Vanin A, Schmidt WJ (2004) Chronic administration of rotenone increases levels of nitric oxide and lipid peroxidation products in rat brain. Exp Neurol 186:235–241 doi:10.1016/j.expneurol.2003.12.005
Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS (1999) A mechanism of paraquat toxicity involving nitric oxide synthase. Proc Natl Acad Sci U S A 96:12760–12765 doi:10.1073/pnas.96.22.12760
Mazzio EA, Kolta MG, Reams RR, Soliman KF (2005) Inhibitory effects of cigarette smoke on glial inducible nitric oxide synthase and lack of protective properties against oxidative neurotoxins in vitro. Neurotoxicology 26:49–62 doi:10.1016/j.neuro.2004.07.005
Acknowledgments
We are grateful to the individuals with PD and their families for their participation in our study. This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke grants NS39764 (to J.M.V.) and NS056632 (to D.B.H.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hancock, D.B., Martin, E.R., Vance, J.M. et al. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics 9, 249–262 (2008). https://doi.org/10.1007/s10048-008-0137-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-008-0137-1