
Abstract
Iron is a redox active metal which is abundant in the Earth’s crust. It has played a key role in the evolution of living systems and as such is an essential element in a wide range of biological phenomena, being critical for the function of an enormous array of enzymes, energy transduction mechanisms, and oxygen carriers. The redox nature of iron renders the metal toxic in excess and consequently all biological organisms carefully control iron levels. In this overview the mechanisms adopted by man to control body iron levels are described.
Low body iron levels are related to anemia which can be treated by various forms of iron fortification and supplementation. Elevated iron levels can occur systemically or locally, each giving rise to specific symptoms. Systemic iron overload results from either the hyperabsorption of iron or regular blood transfusion and can be treated by the use of a selection of iron chelating molecules. The symptoms of many forms of neurodegeneration are associated with elevated levels of iron in certain regions of the brain and iron chelation therapy is beginning to find an application in the treatment of such diseases. Iron chelators have also been widely investigated for the treatment of cancer, tuberculosis, and malaria. In these latter studies, selective removal of iron from key enzymes or iron binding proteins is sought. Sufficient selectivity between the invading organism and the host has yet to be established for such chelators to find application in the clinic.
Iron chelation for systemic iron overload and iron supplementation therapy for the treatment of various forms of anemia are now established procedures in clinical medicine. Chelation therapy may find an important role in the treatment of various neurodegenerative diseases in the near future.
Please cite as: Met. Ions Life Sci. 13 (2013) 229–294
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
1.1 Aqueous Iron Solution Chemistry
Iron, element 26 in the periodic table, is the fourth most abundant element in the Earth’s crust. It has the maximum value of nuclear binding energy of all elements and so tends to accumulate as a result of star based fusion reactions. It is 1000 times more abundant than both copper and zinc. Its position in the middle of the first transition metal row of the periodic table leads to incompletely filled d orbitals and thus the possibility of various oxidation states, the most common being (II), d6, (III), d5, and (IV), d4. Iron(IV) is highly reactive and is restricted to intermediates formed during enzyme catalytic cycles, whereas iron(II) and iron(III) are widely distributed in solution, in the solid phase, and bound to proteins.
When iron salts (both iron(II) and iron(III)) are dissolved in water, they undergo hydrolysis (equation 1), the process effectively being the deprotonation of coordinated water molecules.
 (1)
(1)
With iron(II) (ferrous iron), the hydrolysis of micromolar solutions tends to take place at pH values greater than 7.0 (Figure 1). In contrast, with iron(III) (ferric iron), the charge density on the metal ion is much higher, thereby polarizing the water molecule more effectively than iron(II). As a result, dissociation of the proton from hexa-aqua iron(III) (1) occurs more easily, hydrolysis initiating at much lower pH values, typically pH 2.0 (Figure 1). Whereas it is possible

Iron(III) and iron(II) speciation plots; [iron]total, 1 μM; K H2O solubility products, iron(III): 2.79 × 10–39 M4, iron(II): 4.87 × 10–17 M3.
to prepare a 10 μM iron(II) sulfate solution at pH 7.0, the maximum concentration of iron(III) sulfate at this pH values will be less than 10–18M. Hydroxide ions are also able to crosslink hydrated iron(III) species forming dimers and oligomeric anions (2, 3) [1], finally generating an insoluble polymer of ferrihydrate [2]. The condensation reaction occurs rapidly at

neutral pH values, the initial phase of oligomer formation producing species between 16 and 30 iron atoms. In the presence of suitable ligands, for instance HEIDI (4) and citrate (5), these complexes can be stabilized [3]. As a direct consequence of this high affinity for oxygen donors, iron(III) must be coordinated by ligands in order to maintain appreciable aqueous solubility at most physiological pH values.

The two most common geometries for iron(II) and iron(III) complexes are octahedral for 6-coordinate complexes (1) and tetrahedral for 4-coordinate complexes (6). Octahedral stereochemistry is more frequently observed. As iron(III) possesses a smaller ionic radii than iron(II) (0.65 and 0.78 Å, respectively) and a higher net charge (3+ and 2+), the charge density on the iron(III) ion is much greater than that on the iron(II) ion (0.59 versus 0.27 eÅ–2). This large difference leads to a markedly different ligand selectivity. Iron(III) favors coordination by charged oxygen atoms such as hydroxide, phenoxide, phosphate, and carboxylates, whereas iron(II) favors interaction with aromatic amines, imidazoles, and sulfhydryl groups (see Table 1 [4] for a direct comparison of affinity constants with a range of ligands). As a result of this difference in ligand selectivity, the addition of complexing agents has a major influence on the FeIII/FeII reduction potential. tris-Phenanthroline iron(II) is exceptionally stable, shifting the reduction potential between Fe(Phen) 3+3 and Fe(Phen) 2+3 in favor of the iron(II) complex (E0 = +1.1 volt). In contrast, desferrioxamine-B (7), an iron(III)-selective siderophore binds iron(III) much more

tightly than iron(II) resulting with a reduction potential of −0.45 volt. The range of reduction potentials for iron complexes spans the range +1.1 v to −0.8 v and it is significant to note that a large proportion of this range is covered by iron-proteins (+0.4 v to −0.5 v) [5].
The redox activity of the FeIII/FeII pair can, depending on the coordination of the metal, reduce molecular oxygen to form the superoxide anion (equation 2) and reduce hydrogen peroxide to the hydroxyl radical (equation 3), the latter equation corresponding to the Fenton reaction. As superoxide can be further
converted to the highly damaging hydroxyl radical, rapid generation of these species is damaging to cells [6]. If the reduction potential of an iron complex is either high (> +0.2 v) or low (< −0.2 v) then redox cycling does not occur readily because, under the former condition iron(II) is preferentially stabilized and under the latter, iron(III) is preferentially stabilized. However with a ligand possessing a reduction potential close to zero, for instance EDTA (Eo = +0.12 v) both the iron(III) and iron(II) complexes are relatively stable and so the coordinated iron can readily redox cycle between the two oxidation states.
As with ligand selectivity, the kinetic lability of a metal complex is heavily dependent on the charge density of the cation, the higher the value, the slower the rate of exchange. One method adopted to compare the lability of a series of metal ions is to monitor the rate of exchange of water molecules on the corresponding aqueous ions (equation 4). For ions such as Ca2+, which have a low charge density,
the water-exchange rates approach the diffusion limit (k = 5 × 108 s–1); this renders Ca2+ ideal for a rapid signalling role in cells. The value for iron(II) is 4.4 × 106 s–1 and for iron(III) it is 1.6 × 102 s–1, iron(III) exchanging much more slowly than iron(II). Clearly, iron(II) is kinetically labile and is able to exchange rapidly between binding sites. In contrast iron(III) is much less labile, the rate of exchange between multidentate ligands being particularly slow; for instance the half-life of the donation of iron(III) from FeIII∙EDTA (1mM) to desferrioxamine-B (7, 1 mM) is 42 h [5].
1.2 Iron-Dependent Proteins. The Nature of the Iron Binding Sites
Man has over 500 iron-containing metalloproteins [2].
1.2.1 Heme-Containing Proteins
In this protein class, iron is bound to a porphyrin molecule by the 4 aromatic nitrogen atoms in octahedral fashion, the axial ligands are typically provided by the protein, but can also provide a binding site for gaseous molecules such as dioxygen (Figure 2). There are three main classes of heme proteins: oxygen carriers, as typified by hemoglobin and myoglobin; activators of molecular oxygen, as typified by peroxidases, catalases, and cytochrome P450s. Peroxidases and catalases oxidize a range of compounds using H2O2 as a substrate; cytochrome P450s hydroxylate a wide range of xenobiotic and endogenous substrates, including steroids, using oxygen as a substrate. Electron transport proteins, as typified by cytochromes a, b, and c, are components of the respiratory chain, they accept electrons from reduced donor molecules, transferring them to appropriate acceptors, thereby linking substrate oxidation to cytochrome c oxidase [2].

Heme center in hemoglobin.
1.2.2 Iron-Sulfur Proteins
In this protein class, iron is bound to sulfur in tetrahedral fashion. Two predominate core structures are found in man, 2Fe-2S clusters and 4Fe-4S clusters (Figure 3). Many of the proteins that contain these clusters are involved in electron transfer. Thus ferredoxins are located in electron transfer chains and can also act as donors to enzymes where they exchange electrons between redox centers which are physically separated. The valence state of 2Fe-2S clusters oscillates between 2+ and 3+ and with 4Fe-4S clusters, between 1+ and 2+. The activity of these proteins is not limited to one-electron transfer and iron-sulfur clusters are found in dehydratases and S-adenosylmethionine-dependent enzymes [2].

2Fe-2S and 4Fe-4S clusters:  S;
S;  Fe.
Fe.
1.2.3 Non-heme, Non-sulfur, Iron-Dependent Enzymes
These enzymes fall into two broad categories, those containing either mononuclear or dinuclear iron.
A large number of mononuclear non-heme iron enzymes are involved with the activation of dioxygen to catalyze the hydroxylation of a wide range of substrates. With the α-oxoglutarate-dependent enzymes, iron(II) is bound to the enzyme via two histidines and an aspartate residue (Figure 4) and is involved with the hydroxylation of amino acids and the demethylation of histones [6]. Pterin-dependent hydroxylases fall into a related enzyme group and these are responsible for the hydroxylation of aromatic amino acids [6].

Proposed mechanism of Fe(II)- and 2-oxoglutarate-dependent dioxygenase-catalyzed reactions. In the example of asparaginyl hydroxylation, the reaction proceeds through a radical mechanism involving an iron-oxo intermediate, which adopts both iron(III) and iron(IV) oxidation states during the process. The decarboxylation of 2-oxoglutarate occurs simultaneously. Reproduced with permission from [6]; copyright 2007, Nature Publishing Group.
The dinuclear iron proteins typically contain a four helix bundle protein fold surrounding a (μ-carboxylato)diiron core. The two iron atoms are typically separated by less than 0.4 nm and have one or more bridging carboxylate ligands, together with bridging oxo or hydroxo ligands [7]. The diiron center is bound to the protein via imidazole and carboxylate functions (Figure 5). Such centers are found in the H chains of ferritin, where iron(II) is oxidized to iron(III) and deposited in a core of ferrihydrite and in ribonucleotide reductase where the diiron center generates a tyrosyl radical which is utilized in the conversion of ribosides to deoxyribosides [7].

The metal binding sites of cytoplasmic and nuclear iron(II)-dependent enzymes. (a) ribonucleotide reductase; (b) ferroxidase center on the H-ferritin subunit.
1.2.4 Transport and Iron Storage Proteins
In view of the potential redox activity of many simple iron complexes, the levels of such labile iron forms are maintained at carefully controlled limits, both extracellularly and intracellularly. In the extracellular compartments iron is almost entirely transported between cells, tightly bound to transferrin. Within cells, ferritin acts as an iron sink and is in equilibrium with the labile iron(II) pool, which depending on the cell type, is limited to the range 0.5–2 μM [8].
Serum transferrin is a globular protein which possesses two high affinity iron(III) binding sites (Figure 6a) [9]. Each site consists of two tyrosines, one histidine, one aspartate, provided by the protein, together with a synergistically bound carbonate anion. These ligands create an octahedral coordination sphere (Figure 6b) with a high selectivity for iron(III) and an affinity of 10–20 M–1 (conditional stability constant at pH 7.4) [9]. Serum transferrin is typically circulating in the blood at a level of 35 μM, with between 25 and 35% of the iron binding sites occupied. Under such conditions the levels of the so called non-transferrin bound iron are vanishingly small. Transferrin delivers iron to cells which express transferrin receptors [10,11].

Ferritin is a protein composed of 24 homologous subunits, designed to create a large aqueous enclosure for the storage of iron atoms. Each ferritin molecule can accommodate up to 4500 iron atoms [12] (Figure 7). Ferritin is the major iron storage protein for all mammalian tissues and consists of a mixture of two subunits referred to as L- and H-subunits. In general, L-rich ferritins are characteristic of organs storing iron for long periods (e.g., liver) and these molecules tend to possess a high iron content. H-rich ferritins, which are characteristic of the heart, have a major role in iron detoxification, they possess a ferroxidase site (see Figure 5b) and tend to contain a lower iron content. Iron(II) enters the various pores of the oligomeric structure (Figure 7b) and is rapidly oxidized to iron(III) either by the growing core of ferrihydrite or at the ferroxidase site of the H-subunit [13]. Ferritin molecules are constantly being synthesized and removed to lysosomes, where they are degraded, thereby releasing the entrapped iron [14]. This iron enters the cytosolic labile iron pool under the tight control of iron(II) transport proteins, which are located in the lysosomal membrane.

1.3 Iron Transport
In mammalian cells there is a labile iron pool which supplies iron to the mitochondrion for incorporation into heme and iron-sulfur cluster proteins (Figure 8). It is also the source of iron for many cytoplasmic iron-dependent enzymes. As non-coordinated iron salts can catalyze the formation of toxic oxygen-containing radicals, the levels of this labile iron pool must be tightly controlled. Cells must be able to sense iron levels and regulate homeostasis in such a manner as to maintain nontoxic levels. The concentration of this iron pool is determined by the rates of iron uptake, utilization for incorporation into iron proteins, storage in ferritin and iron export from the cell (Figure 8). The concentration of the pool falls into the 0.5–2 μM range and a likely structure is iron(II) bound to a single molecule of glutathione (8) [15]. This structure ensures protection against autoxidation of iron(II) and offers a mechanism for the introduction of iron into protein-bound iron sulfur clusters via glutaredoxins [8]. The selection of iron(II) in preference to iron(III) for this role is logical in view of the kinetic lability of iron(II) being 5 × 104 times higher than that of iron(III).

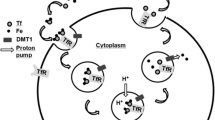
Cytoplasmic iron fluxes. DMT1, divalent metal transporter-1; IRP, iron responsive protein. FBXL5, F-box and leucine-rich repeat protein 5.

1.3.1 Cellular Iron Transport
Within the circulation, transferrin-bound iron is the principle iron source for all mammalian tissues; although under iron overload conditions, transferrin becomes saturated and non-transferrin bound iron appears in the serum and is taken up by cardiac and endocrine tissue by uncharacterized transporters.
1.3.1.1 Transport of Iron-Loaded Transferrin
There are at least three classes of transferrin receptors in mammalian cells; TfR1, which is expressed on many cell types, TfR2, which is restricted to hepatocytes and erythroid cells, and a third type, only located in epithelial kidney cells. The basic mechanism of iron transport is the same for each class. Fe2-transferrin-TfR complexes bind to clathrin-coated pits which are internalized into endosomes. The endosome lumen is acidified to pH 5.5, whereupon both transferrin and TfR undergo conformation changes to release transferrin-bound iron. A ferrireductase reduces the released iron(III) to iron(II), which is a substrate for the divalent metal-ion transporter 1 (DMT1) (Figure 9). Both apo transferrin and TfR are recycled to the cell surface. It has been estimated that a transferrin molecule experiences over 200 such cycles during its life time [10].

Macrophage iron fluxes.
1.3.1.2 Absorption of Dietary Iron
Inorganic iron present in the diet is predominantly iron(III) and this is reduced on the surface of duodenal enterocytes by the ferrireductase DCYTB (Figure 10). The resulting iron(II) enters the cell through the divalent metal transporter which is proton-coupled [16]. DMT1 has been shown to transport other divalent metals, such as Zn(II), Mn(II), and Co(II), but the physiological relevance of this property is unclear.
Heme iron is transported from the lumen of the duodenum by the transport protein HCP1 [17]. Once absorbed, heme is degraded by heme oxygenase to form iron(II) (Figure 10). Thus both DMT1 and HCP1 supply iron to the labile iron pool (possibly as FeII. Glutathione, 8).

Duodenal enterocyte located on intestinal villae. Associated with the unidirectional movement of iron.
1.3.1.3 Mitochondrial Iron Transport
Mitochondria are major sites of iron consumption in mammalian cells as they accommodate the enzymes involved in heme and iron-sulfur cluster synthesis. The uptake of iron is dependent on membrane potential and facilitated by the transport proteins mitoferrin-1 and mitoferrin-2 [18]. Mitoferrin-2 is widely distributed in different tissues, whereas mitoferrin-1 is restricted to erythroid cells. Ferrochelatase, the terminal enzyme in heme synthesis receives iron(II) directly from the inner mitochondrial membrane and has been demonstrated to form a oligomeric complex with mitoferrin-1 [19]. It has been suggested that frataxin shuttles iron(II) from the same membrane, directing iron to enzymes involved in iron sulfur cluster synthesis.
1.3.1.4 Ferroportin-Mediated Iron Efflux
Duodenal enterocytes, macrophages, hepatocytes, and CNS cells release iron in a controlled manner by use of the iron efflux transporter ferroportin (FPN1). This transporter was independently discovered by three groups and was originally termed Ireg1 [20], ferroportin [21], and MTP [22]. The term ferroportin has been widely adopted. The gene encodes a highly hydrophobic membrane protein with 10–12 transmembrane spanning domains, which bears little sequence identity with any other transporter family. Indeed, ferroportin is specific for iron(II) and is the only iron exporter identified to date. Ferroportin is located on the basolateral side of duodenal enterocytes and in the cytoplasmic membranes of both macrophages and hepatocytes (Figure 10).
For efficient transfer of freshly effluxed iron(II) to apo transferrin, ferroportin is frequently closely coupled with a multi-copper ferroxidase, for instance, hephaestin or ceruloplasmin, whereupon the iron(II) is rapidly oxidized to iron(III), without the generation of oxygen-containing free radicals.
1.3.1.5 Iron Metabolism Facilitated by the Macrophage
Macrophages acquire iron via the transferrin receptor, hemoglobin, via erythrophagocytosis and the haptoglobin receptor and heme, sequestered by hemopexin via the lipoprotein receptor, CD91 (Figure 9). Changes in the surface glycoproteins of red blood cells take place as they age. These modified proteins are recognized by receptors on the macrophage surface and trigger phagocytosis.
Haptoglobin and hemopexin scavenge hemoglobin and heme respectively. Both are released in the circulation as a result of hemolysis (Figure 9) [23]. On entry to the cytoplasm, the heme moiety is degraded by heme oxidase and the resulting iron(II) enters the labile iron pool. On exposure to the lumen of a phagolysosome, hemoglobin is degraded, releasing heme which is able to penetrate the membrane also gaining access to heme oxidase. Iron(II) is exported from the macrophage via ferroportin and the effluxed iron(II) is oxidized to iron(III) by ceruloplasmin.
1.3.2 Regulation of Iron Metabolism
Iron homeostasis is regulated by balancing iron uptake with intracellular storage and use. In mammalian cells, this is largely achieved at the level of protein synthesis. Regulatory sequences in mRNA molecules are located in the noncoding regions situated at 5’ and 3’ extremities of the coding regions. Location in the 5’ region is usually associated with initiation of translation, whereas location in the 3’ region is associated with enhanced mRNA stability [24]. The regulatory sequences relating to iron metabolism are termed iron responsive elements (IRE) and their structural features are well established [25]. Iron responsive proteins (IRP) bind to IREs and thereby control the translation of the related mRNA. An IRE consists of an upper stem of five base-paired residues that assume a helical structure which is separated from a lower stem of base-paired structures by an unpaired cytosine, thereby creating a bulge in the structure (Figure 11). At the top of the structure is a six membered loop which includes the sequence AGU. These residues constitute the major recognition feature for IRPs, both the bulged C and the AGU region fit into binding pockets of IRP molecules. The affinity constant for this interaction falls in the range 10–30 pM [2,26].

The iron responsive element is an RNA stem-loop structure. The upper and lower stems are composed of base-pairs which are held in a helical conformation. In the six-membered loop AsafNAsafdasdasdasd, the C at position 1 of the loop forms a base-pair with the G at position 5. This C-G base-pair structures the loop, allowing the A, G, and U residues to form a region that can form multiple hydrogen bonds with proteins. The upper and lower stems are separated by an unpaired “bulge” C that confers flexibility on the structure by interrupting the helix. Reproduced with permission from [25]; copyright 1997, Elsevier.
In man there are two IPRs and when iron levels are low they both bind to mRNA. When iron levels are adequate, IRP1 acquires a [4Fe-4S] cluster converting it to the enzyme aconitase (Figure 8) and IRP2 is degraded by proteasomes. The binding of a IPR to the transferrin receptor mRNA enhances the stability of RNA and hence increases protein synthesis (Figure 12a). This in turn promotes receptor mediated iron uptake. The effect on ferritin mRNA is to reduce protein synthesis (Figure 12b), thereby increasing the availability of intracellular iron. A number of proteins with a central role in iron metabolism are controlled in analogous fashion (Table 2).

Mechanism of iron-regulatory protein 1 (IRP1) in the translational regulation of (a) transferrin receptor and (b) ferritin.
1.4 Iron Physiology
While the body levels of other dietary metals can be regulated by excretion in the feces and urine, humans do not possess the ability to remove excess iron from the body. Consequently, several proteins have evolved which regulate iron homeostasis, their location being primarily in duodenal endothelial cells, hepatocytes and reticuloendothelial macrophages. It is essential for these cells to function in concert. Regulation of total body iron is achieved by feedback mechanisms that operate on iron absorption. Approximately 4 mg of iron circulates bound to transferrin (this is about 0.1% of total body iron). Seventy five percent of body iron is in the form of heme proteins, mainly hemoglobin, the remainder being located in cells either in non-heme enzymes or ferritin. Although only 1–2 mg of iron is absorbed each day by normal individuals, 20 mg is transferred daily to the bone marrow for erythropoiesis and is efficiently recycled by macrophages (Figure 13). There is no active excretion mechanism for iron; loss is uncontrolled and results mainly from bleeding and epithelial desquamation. It has been estimated that the average iron atom survives in a human for approximately ten years.

Iron homeostasis in a normal adult man. Loss is uncontrolled (epithelial desquamation and in women, menstruation).
1.4.1 The Role of Hepcidin
Hepcidin is a 25 residue peptide containing four disulfide bridges (Figure 14). It has an amphipathic nature and possesses a net positive charge. The structure is highly conserved within mammals and fish [27]. Removal of the N-terminal pentapeptide leads to loss of activity. Hepcidin regulates iron export from a range of cells by binding to ferroportin, causing internalization and degradation of the iron-efflux protein. The mechanism of this internalization is similar to that of other receptors and involves hepcidin-induced phosphorylation and subsequent ubiquitination of ferroportin. Thus, under conditions of high hepcidin serum levels, ferroportin located in the duodenal epithelial cells and macrophages will be internalized and such cells are unable to efflux iron. Under conditions of low hepcidin serum levels, the opposite holds and thus, high iron fluxes enter the serum originating from both duodenal epithelial cells and macrophages.

Human hepcidin.
Hepcidin is produced primarily in the hepatocyte, the synthesis being regulated by both the iron and the inflammatory status of the organism. High concentrations of fully saturated transferrin trigger the involvement of bone morphogenetic protein (BMP), which in turn facilitates the phosphorylation of the transcription factor SMAD4. SMAD4 controls hepcidin synthesis. Hepcidin synthesis is also regulated by erythropoietic signals such as GDF15. In addition, hepcidin synthesis is regulated by inflammatory stimuli, thus, interleukin 6 activates the JAK/STAT signaling pathway which again stimulates the hepcidin promoter [28].
Thus, under conditions where man is replete in iron (high saturation of transferrin), hepcidin synthesis is increased and there is an overall retention of iron by the duodenum, hepatocyte, and macrophage. The converse also holds, thus conditions, where a low transferrin saturation exists, will lead to the inhibition of hepcidin production and a concomitant enhanced release of iron from the same group of cells. Significantly, elevation of interleukin 6 occurs when man is infected by microorganisms and parasites. Such an action triggers hepcidin synthesis, reducing the extracellular availability of iron and thereby enhancing the host defense [29].
2 Iron Deficiency and Anemia
2.1 Iron Requirements of Man
Healthy term infants with a normal birth weight are born with high hemoglobin levels and sufficient iron stores to support growth for the first six months of life [30]. Babies are born with body iron loads, typically in the region of 80 mg/kg; the corresponding value for adult men is 55 mg/kg. Thus, the low iron content in human milk does not present a problem to the suckling infant and renders infection to be less of a problem.
Preterm and low birth weight infants exhaust their iron stores at an earlier age than normal children and thus, there is a greater chance of anemia with such children. The transfer of iron from maternal blood to the fetus occurs mainly in the third trimester (Figure 15). Thus premature infants are born with lower iron stores. At six months, solids and cereals should be given to babies, with the goal of beginning to provide appreciable levels of iron in the diet. Iron requirement for the rest of life remains close to 1 mg/day (Figure 15) [31], only exceeding this requirement during the adolescent growth spurt, with menstruating females and during pregnancy. Iron requirements increase dramatically during the second and third trimesters of pregnancy, reaching a requirement of 5–6 mg/day. Iron requirement during lactation does not increase. It is estimated that most pregnant women in developing countries and between 30 and 40% of pregnant women in developed countries are iron-deficient [32].

Daily iron requirements. Information taken from [31].
Blood donations (500 mL/year) require an additional 0.6–0.7 mg iron per day, a significant addition to the normal adult requirement of 1.1 mg/day. In developing countries intestinal parasitic infections can cause appreciable blood loss which in turn leads to increased iron requirements. A list of causes of iron deficiency is presented in Table 3.
2.2 The Influence of Anemia on Human Physiology
After birth, the erythron has the priority for transferrin-bound iron as compared to other tissues. In general, erythrocyte production is unperturbed until the body iron stores are depleted. When the stores become limiting, the saturation of transferrin decreases and patients then show signs of iron-deficient erythropoiesis; protoporphyrin and zinc protoporphyrin appear in the erythrocytes and these changes are followed by the onset of microcytosis.
Iron deficiency is known to be associated with decreased physical activity as demonstrated by exercise tolerance and work performance [33]. These effects have been thoroughly investigated in rodent models. As hemoglobin levels decrease, so do those of myoglobin and the cytochromes. Thus, there is diminished oxygen transport in the blood and diminished oxygen diffusion in muscle. There is also a decrease in the mitochondrial iron-sulfur content which is associated with a decrease of mitochondrial oxidative capacity. These changes have a major influence on physical activity; the Harvard Stop Test score registers impaired performance even with mild anemia (Figure 16a) [34], as does the worktime in workers performing a sub maximal exercise test (Figure 16b) [35].

The adverse influence of anemia on mental behavior is more difficult to demonstrate, but as 25% of the body oxygen is utilized by the brain, adverse effects can be anticipated. Iron is required for the myelination of the spinal cord, for the synthesis of chemical transmitters, particularly dopamine and serotonin and for mitochondrial function. The influence of anemia on mental development has been reviewed and details from 18 studies described [36]. Taken together, they support the hypothesis that iron deficiency is associated with developmental delay. With longer term intervention studies, again there is evidence of a causal link between iron deficiency anemia and poor developmental performance [36].
2.3 Dietary Sources of Iron
There are two major classes of iron in the diet, heme iron and non-heme iron. The former is derived from hemoglobin, myoglobin, and cytochromes and is found in animal sources such as red meat and seafood. Non-heme iron is derived mainly from plant sources such as lentils, beans, rice and cereals (Table 4). The absorption of heme iron is relatively efficient, ranging from 15 to
35%; whereas the absorption of non-heme iron is less efficient (2–20%) and can be inhibited by the presence of other components in the diet. Heme iron, which is strongly chelated by protoporphyrin IX, is absorbed by the HCP-1 transporter (Section 1.3.1.2), and is not subject to competition from other iron-binding compounds which are present in the lumen of the gut. In contrast, non-heme iron is absorbed in the iron(II) state by DMT-1 (Section 1.3.1.2) and so is susceptible to the presence of iron chelating ligands in the gut lumen.
Compounds such as phytates, polyphenols, and tannins, which are widely distributed in plant foods, are capable binding to both iron(II) and iron(III), rendering the iron non-bioavailable [37]. When such compounds bind iron(II), they autoxidize the metal to iron(III). Phytate is present in legumes, rice, and cereals. Tannic acid is present in tea and coffee. Their presence can inflict a powerful inhibitory influence of non-heme iron absorption. Conversely, the presence of vitamin C (ascorbic acid) enhances the absorption of non-heme iron, the reduced form of the vitamin reducing iron(III) to the more bioavailable iron(II) (equation 5).
 (5)
(5)
A typical iron intake for an adult male is 16–18mg/day, whereas for women it is lower, typically 12 mg/day. Thus, for most situations there should be sufficient iron in the diet, although the consumption of energy-restricted diets or those rich in poorly bioavailable iron can contribute to inadequate iron absorption. Selected food sources and their iron contents are presented in Table 4.
2.4 Iron Fortification
Iron fortification of commonly used foods is a practical and cost-effective strategy to improve the iron nutrition of a large population. The concept has been widely adopted in developed countries over the past 50 years, typically with the iron fortification of breakfast cereals. The efficacy of iron fortification in the developing countries has not been so successful and improved guidelines and materials are required. The following iron preparations have been used; ferrous sulfate (promotes oxidation of fats and so leads to rancidity), ferrous fumarate (likely to be similar to ferrous sulfate), elemental iron (finely divided metallic iron), ferric pyrophosphate and ferric EDTA. The bioavailability of the ferric salts and elemental iron is lower than that of ferrous sulfate.
The World Health Organization (WHO) has issued recommendations for a range of iron compounds to be used and the level to be adopted in developing countries. Good efficiency has been reported from South Africa, Morocco, Vietnam, Chile, Thailand, and Kenya [32]. In principle, adverse effects are possible with respect to iron overloading diseases and in regions with a high prevalence of malaria, however, this is less likely to occur with iron fortification when compared with iron supplementation (Section 2.5).
2.5 Oral Iron Supplementation
Another approach to control iron deficiency is iron supplementation, which unlike fortification delivers a relatively large dose of iron in the absence of food. The majority of patients with iron deficiency anemia respond to oral iron therapy. A wide range of iron preparations are available for oral application, the majority being iron(II) by virtue of its greater bioavailability. Thus ferrous sulfate, ferrous fumarate, and ferrous gluconate are widely marketed, indeed ferrous sulfate was introduced as a therapy for anemia by Blaud in 1832 [38]. By virtue of its low cost and good bioavailability ferrous sulfate is still widely used for oral therapy, however, its use is associated with a range of gastrointestinal side effects that frequently lead to poor compliance. There are “slow release” formulations available which can reduce side effects, but typically they also reduce iron absorption when compared to the parent compound. Another problem with iron(II) salts in oral formulations is that they can inhibit the absorption of a wide range of other therapeutics, including antibiotics, L-dopa, and thyroxine [39].
Up to 40% of patients have symptoms associated with the oral administration of iron(II) salts. These symptoms can be avoided by utilizing iron(III) complexes, although the bioavailability of simpler iron(III) salts is lower than the analogous iron(II) salts due to the extreme low solubility of iron(III) hydroxide (Section 1.1). However, if the iron(III) complex is sufficiently stable and water-soluble, then the bioavailability can reach that achieved by ferrous sulfate. Iron(III) maltol [40] and iron(III) polymaltose [41] are two such compounds reported to possess excellent bioavailability, although there is controversy as to the effectiveness of such compounds [42]. Presumably the iron complexes are reduced by DCYTB, thereby generating iron(II) which is the main substrate for DMT1 (Section 1.3.1.2).
2.6 Anemia of Chronic Disease
The anemia of chronic disease is an acquired disorder of iron homeostasis which may be associated with infection, malignancy, organ failure or trauma (Table 5). The anemia is typically mild to moderate and the erythrocyte size being close to normal. Macrophages, which normally recycle iron (Section 1.3.1.5), retain it and intestinal iron absorption is reduced under these pathological conditions [43]. An association with proinflammatory cytokines was suspected for many years and it has recently been established that this modified cellular behavior is associated with elevated levels of hepcidin (Section 1.4.1) [44,45]. Expression of hepcidin that is inappropriately high for the normal body iron status results in interruption of intestinal iron absorption and macrophage iron recycling. Thus, there is less iron available for erythropoiesis. IL-6 is the cytokine that induces hepcidin expression [45] and its levels have been shown to correlate with the severity of anemia in rheumatoid arthritis, cancer, and aging [43].
Treatment options for patients with anemia of chronic disease depends on the severity of the anemia and since the anemia is derived from the innate immune response to infection, there is little clinical support for the use of exogenous iron or erythroid-stimulating agents, such as erythropoietin. In principle it is possible to mobilize macrophage iron by chelation and to redistribute it to the transferrin iron pool [46,47], however, to date no such compounds have been introduced into the clinic.
In contrast to the above general statements, in the specific cases of autoimmune disorders and end-stage renal disease, iron treatment in conjunction with erythroid-stimulating agents can be effective [48,49]. With the use of erythropoietin, the rate of iron mobilization from stores is unable to match the iron requirements of the expanding red cell mass and consequently iron supplementation is required. In many patients oral supplements will not provide an optimal supply of iron and such patients are frequently intolerant to oral iron therapy, in such cases intravenous iron preparations can be introduced. These preparations consist of a carbohydrate ligand and either ferric hydroxide or ferric oxide cores. They are designed to be non-immunogenic and to be efficiently removed from the circulation by macrophages, which catabolize the complex and render the iron available for apo transferrin. Iron dextrin was the first such preparation to be used clinically, but it has been associated with a wide range of side effects. However, there are now much improved formulations available [50,51]. An advantage of some of these preparations is that doses up to 1 g of iron can be administered in a single infusion [51].
3 Systemic Iron Overload
Iron overload can be present in one of two general forms. Firstly, in situations where erythropoiesis is normal but the plasma iron level exceeds the iron binding capacity of transferrin (e.g., hereditary hemochromatosis); the resulting non-transferrin bound iron (NTBI) is deposited in parenchymal cells of the liver, heart, and endocrine tissues [52]. The second type results from increased catabolism of erythrocytes (e.g., transfusional iron overload). In this situation iron initially accumulates in reticuloendothelial macrophages, but subsequently spills over into the NTBI pool and parenchymal cells. Parenchymal iron deposition leads to organ damage.
3.1 Non-transferrin Bound Iron
When transferrin is fully saturated, any iron entering the blood forms part of the NTBI pool. NTBI, unlike transferrin, lacks an address system and tends to be absorbed by highly vascular tissue, such as the heart and endocrine organs. As iron accumulates in these organs, redox cycling leads to the generation of toxic oxygen radicals. Transferrin also contributes to defence against infection by depriving microorganisms of an iron supply [29]. Conversly, the presence of NTBI presents a weakness, rendering the host more susceptible towards infection.
There are many small molecules in the blood which are capable of binding iron(III), the principle oxidation state of iron in the serum. These include acetate, phosphate, and citrate. Of these, citrate has the highest affinity for iron(III) [53] and there are two dominating complexes under the conditions provided by serum, [Fe(citrate)2] (9) and [Fe3(citrate)3] (10) [53,54]. At an iron concentration of 1 μM, complex (9) dominates at physiological citrate levels (typically 100 μM), whereas at a level of 10 μM iron the oligomeric iron citrate (10) begins to dominate [53]. Another important iron binding component of the serum is albumin, which is present at a relatively high concentration (600 μM) [55,56]. Which of these three forms of NTBI gains facile entry into parenchymal cells has not been established.

3.2 Hereditary Hemochromatosis
The term hereditary hemochromatosis covers a heterogeneous group of iron overload disorders (Table 6). They are linked to gene mutations associated with hepcidin, leading to low levels of hepcidin. This in turn leads to the hyperabsorption of dietary iron, up to 8–10 mg per day. Over a period of time transferrin saturation gradually increases from the normal value of 30% to 100%, which is associated with the appearance of non-transferrin bound iron and the associated inappropriate tissue distribution of iron [57]. In hereditary hemochromatosis, hepatic iron overload can lead to fibrosis, cirrhosis, and carcinoma of the liver [58] and to non-hepatic symptoms including cardiomyopathy, diabetes, hypogonadism, and arthritis [59]. In contrast, the CNS does not develop iron overload.
The above symptoms tend to develop late in life, but the disease can be managed by phlebotomy (each 500 mL of blood contains 250 mg of iron), the earlier the diagnosis the better. Indeed, early diagnosed patients experience a normal life span. In addition to phlebotomy patients should avoid iron supplementation and limit consumption of red meat and alcohol.
3.2.1 HFE Hemochromatosis
HFE hemochromatosis is the most frequent form of hemochromatosis [60] and there is a particularly high prevalence among North European caucasians (1 in 200). The discovery of HFE was the first cloning success to contribute to our understanding of iron metabolism [61]. This important finding was rendered possible by the observation of Simon et al. that the hemochromatosis phenotype is frequently associated with a particular human leukocyte antigen haplotype [62]. The HFE protein is an atypical membrane-bound MHC class 1 molecule which interacts with β2-microglobulin; its link with iron transport remains to be established. Many patients bear a mutant HFE, with a C282Y substitution and HFEC282Y fails to incorporate in the plasma membrane [63]. The frequency of HFEC282Y homozygosity is 1:200, although the clinical penetrance of the disease is lower [58,64]. Additional HFE mutations are also associated with hemochromatosis [65]. Significantly ablation of HFE promotes a hemochromatotic phenotype in mice [66].
3.2.2 Juvenile Hemochromatosis
Juvenile hemochromatosis unlike HFE hemochromatosis is a relatively rare disease mainly located in Southern Europe [67]. These patients rapidly accumulate iron and are more likely to present with cardiomyopathy and endocrinopathy than with liver disease (Table 6). This pathology is linked to the mutation of the gene responsible for hemojuvelin (a bone morphogenetic protein receptor operating upstream of the hepcidin pathway), which leads to the expression of extremely low levels of hepcidin. A small subset of these patients possesses a mutation to the HAMP gene, which encodes for hepcidin. Low hepcidin leads to enhanced iron absorption (Section 1.4.1).
3.2.3 Ferroportin Disease
Ferroportin disease is characterized by moderate to severe iron overload. It is more frequent than Types 2 and 3 hemochromatosis and is caused by mutations to the ferroportin gene. Affected individuals express high hepcidin levels and the disease displays phenotypic heterogeneity [68].
3.2.4 Treatment by Iron Chelation
Phlebotomy is an extremely efficient method of removing iron. Treatment of severely iron-loaded hemochromatosis patients typically involves weekly phlebotomy; which can lead to removal of up to 1g of iron every month. Iron chelation therapy cannot achieve such high excretion rates but does have the advantage of reducing NTBI levels (vide infra). Patients suffering from juvenile hemochromatosis may rapidly develop cardiac complications and such people can benefit from a combination of phlebotomy and chelation therapy. In principle, orally active iron chelators could find application in the treatment of hemochromatosis, and deferasirox (vide infra) has been reported to show good efficacy [69].
3.3 Transfusional Siderosis
The management of anemias associated with ineffective erythropoiesis (for instance thalassemia, sickle cell disease, hemolytic anemia, and myelodysplastic syndromes) requires frequent blood transfusion. Blood contains plenty of iron (0.5 mg mL–1) and this accumulates in the tissues as man does not actively excrete iron (Section 1.4). In addition, ineffective erythropoiesis inhibits hepcidin expression which leads to the stimulation of dietary iron absorption. Overall this pathology leads to transfusional siderosis, the formation of non-transferrin bound iron and the inappropriate iron loading of liver, cardiac, and endocrine tissue [70].
3.3.1 The Hemoglobinopathies
Thalassemia and sickle cell anemia are the most common worldwide monogenic diseases. They occur at their highest frequency in countries of the developing world and it has been estimated that there are 250 million carriers. 300,000 children are born each year with major hemoglobin disorders [71]. The high incidence of these disorders reflects their association with malaria resistance which has developed over thousands of years [72]. More than 3,000 million people live in malarious areas and currently the disease is responsible for at least two million deaths each year. Any protective mechanism against this dominating infection that develops within the native population will be gradually amplified and this has been the situation with thalassemia and sickle cell anemia where a range of mutations have gradually accumulated within the population. The correspondence between the distribution of malaria in the Old World before major control programs were established and the distribution of thalassemia is striking (Figure 17) [73]. The cellular mechanisms related to this type of protection are complex but increasingly well understood [73,74].

(a) Distribution of malaria in the Old World before major control programs were established. (b) Distribution of α- and β-thalassemia [73].
Hemoglobin production in man is complicated. The type of hemoglobin produced in fetal life is altered after birth (Figure 18) as a result of the sequential suppression and activation of individual genes [75]. Clearly, there are a number of possible hemoglobin disorders that can result from this genetic framework and a breakdown of these disorders as monitored by the annual number of births is given in Table 7 [76].

Changes of human hemoglobin with development. There is a switch from hemoglobin-F to adult hemoglobin-A together with a small amount of hemoglobin-A2 in the immediate post natal period [75]. Hemoglobins; F = α2γ2; A = α2β2; A2 = α2δ2. Reproduced with permission from [75]; copyright 2001, Blackwell Science Ltd.
The thalassemias are characterized by a reduced rate of synthesis of one or more globulin chain(s); α°- and β°-thalassemias are associated with no production of α- or β-chains, respectively, α+- and β+-thalassemias are associated with a reduced production of α- or β-chains. These conditions all lead to an imbalanced globin chain production, which is associated with ineffective erythropoiesis and shortened red blood cell survival. Because thalassemias occur in populations in which hemoglobin variants are also common, some children inherit thalassemia from one parent and a hemoglobin variant from the other. These interactions produce disorders of varying severity. Thus, sickle cell/β-thalassemia may be as severe as sickle cell anemia and hemoglobin E/β-thalassemia can produce similar pathology to that of thalassemia major. Hemoglobin E is inefficiently synthesized and hence produces a mild form of β-thalassemia [75].
3.3.1.1 Thalassemia
Although the α-thalassemias are more common than the β-thalassemias, severe forms lead to intrauterine death and so do not pose a major burden on health care. It is the β-thalassemias and their combination with hemoglobin E that produce severe anemia. These diseases are particularly common throughout the Indian subcontinent and South East Asia. Over 180 different mutations have been identified amongst the β globin chains of β-thalassemia patients. The bulk consists of single base changes which lead to different degrees of reduction in the synthesis of α-chains, thereby leading to the clinical diversity β-thalassemia. The reduction of α-chain production leads to an excess of α-chains, which are unstable and tend to precipitate, preventing the normal maturation and survival of erythrocytes [77].
β-Thalassemia major (β°) is associated with the total absence of β-subunits and consequently is fatal in the absence of regular blood transfusion, which in turn is associated with systemic iron overload. With untreated patients, death generally occurs in the second decade of life as a result of infection or heart disease [78]. Iron chelation therapy prevents the development of iron overload and as a result, the life style of thalassemia patients has dramatically improved over the past 50 years. Desferrioxamine (DFO, 7) a natural siderophore was introduced in the clinic by SephtonSmith in 1962 [79]. It scavenges and binds iron(III) extremely tightly, leading to the formation of a stable non-toxic iron complex (Figure 19) [80] which is excreted via the bile. Unfortunately DFO is not orally active and has to be administered parenterally over prolonged time periods, as it is rapidly cleared by the kidney [81]. Never-the-less it has been a remarkably successful pharmaceutical and has extended the lives of thousands of β-thalassemia major patients.

Ferroxamine, the iron complex of desferrioxamine [80].
There is a wide pathological diversity of β-thalassemia patients due to the large number of different mutations, for instance the inheritance of a severe mutation from one parent and a mild mutation from the second parent or the inheritance of β-thalassemia from one parent and an abnormal Hb from the other [82]. As a group, these patients are described as suffering from β-thalassemia intermedia. Management depends on the severity of the disease and may involve transfusion and chelation or in some cases, chelation therapy alone (vide infra) [82].
3.3.1.2 Sickle Cell Disease
Expression of sickle cell disease, like β-thalassemia is highly variable, ranging from mild phenotypes (mostly SC and Sβ+ genotypes) to severe disease (mostly SS and Sβ° genotypes). Transfusion therapy is a key component of patient management and is an effective treatment for chest syndrome, heart failure, and stroke. Monthly transfusions decrease the risk of recurrent stroke. Straight transfusion is used when the Hb level is less than 8 g dL–1 and exchange transfusion is recommended with normal Hb levels [83]. The target percentage of HbS in patients receiving regular transfusions is 30% [84]. Both straight and exchange transfusion lead to iron overload and although the endocrinopathology is less marked in sickle cell anemia patients when compared with β-thalassemia patients [85], iron overload should be treated by chelation (vide infra).
3.3.2 Myelodysplastic Syndrome
Myelodysplastic syndrome (MDS) is a diverse group of hematological disorders that lead to ineffective production of myeloid blood cells with a risk of transformation to leukemia. This bone marrow stem cell disorder is associated with ineffective erythropoiesis and leads to anemia. Diagnosis of MDS is made typically between 60 and 75 years, diagnoses are rare in children. Many MDS patients become dependent on blood transfusion and develop transfusional iron overload [86] which is associated with cardiac problems [87]. Although this iron overload can be treated with desferrioxamine, the demanding continuous parenteral treatment leads to difficulties with compliance, particularly with this typically elderly group of patients [86]. Treatment with orally active iron chelators has good potential (vide infra).
3.4 Hereditary Disorders of Mitochondrial Iron Overload
Several iron loading disorders are characterized by mitochondrial accumulation in specific tissues, without systemic iron overload. They result from mutations in proteins involved in either heme or iron-sulfur cluster biosynthesis. These two metabolic pathways occur in the mitochondria and under normal conditions consume the majority of cellular iron.
3.4.1 Sideroblastic Anemia
Sideroblastic anemias are a group of disorders that are associated with the formation of a large number of ringed sideroblasts in the marrow. Sideroblasts are erythroblasts (nucleated red blood cells) which contain precipitates of non-heme iron aggregates deposited within the cristae of mitochondria [68]. X-linked sideroblastic anemia (XLSA) is the most common form of sideroblastic anemia which results from defects in 5-aminolevulinate synthase, a key enzyme in heme biosynthesis. Management of this disease frequently requires blood transfusion which leads to iron overload [68].
3.4.2 Friedreich’s Ataxia
Friedreich’s ataxia is an autosomal recessive neurodegenerative disorder linked to the functional inactivation of frataxin (FXN) [88,89]; a mitochondrial protein which has a critical role in the assembly of iron-sulfur clusters [90]. An expanded GAA triplet repeat is found in both alleles of the FXN gene. This triplet repeat leads to decreased RNA transcription and lower levels of frataxin. The degree to which transcription is suppressed is proportional to the length of the GAA repeat; with frataxin levels, 5% of normal, being associated with long GAA repeats and the association of higher frataxin levels (30% of normal) with shorter repeats [91]. Thus, patients with shorter GAA repeats generally have a less severe phenotype. Frataxin deficiency is associated with decreased ATP synthesis and elevated mitochondrial iron levels. The tissues most severely influenced are cardiac [92], gastrointestinal and brain [93]. Treatment with orally active iron chelators can be beneficial (vide infra).
3.4.3 Glutaredoxin-5 Deficiency
Glutaredoxin-5 (Grx 5) is a protein cofactor essential for the iron-sulfur cluster assembly pathway [94]. Its absence is causatively linked to microcytic anemia with a sideroblastic-like phenotype [95]. The disease requires regular blood transfusion which leads to systemic iron overload. The pathogenic mechanism involves inhibition of heme synthesis in erythroid precursor cells via accumulation of apo-IRP1 that represses 5-aminolevulinate synthase (ALAS) mRNA translation [96].
3.5 Animal Models of Iron Overload
Over the past two decades genetic analysis of patients with inherited iron homeostasis disorders and the analysis of mutant mice, rats, zebra fish, and fruit flies has greatly facilitated the present understanding of iron metabolism [97]. These animal models are excellent systems for investigating iron homeostasis and for the identification of new therapeutic agents. Although iron metabolism in zebra fish and the fruit fly is not understood in detail, iron metabolism in rodents is similar to that in man. Models have been developed which simulate iron deficiency anemia, sideroblastic anemia, various forms of defective iron transport, hemochromatosis, and Friedreich’s ataxia (Table 8) [21,98–100,104–120].
The first mutant to provide a useful insight into iron transport was the hypotransferrinaemic mouse (hpx) [98,101], although the mk mouse was discovered in 1970 [102] and the Belgrade rat in 1969 [103]. One of the benefits from this work will be the accurate diagnosis of human iron deficiency and iron overload disorders; for instance, the clinical approach to hemochromatosis has been strongly influenced by diagnostic testing [68].
3.6 Genetic Screening for Thalassemia
The improved understanding of the pathophysiology of the thalassemias has led to improved symptomatic treatment by blood transfusion and chelation therapy. However, this therapy is expensive and complicated due to the increasing problem of obtaining blood that is free from HIV and hepatitis.
Because thalassemia and sickle cell heterozygotes are relatively easy to identify, these diseases are suitable for investigation by prenatal diagnosis and termination of pregnancies of homozygote babies. In the 1970’s with the advent of fetal blood sampling, it became possible to undertake such diagnosis by monitoring hemoglobin chain synthesis [121] and later by DNA analysis [122,123]. Screening was soon established in several countries, for instance Sardinia and Cyprus, with remarkable success (Figure 20) [124]. There has now been success in reducing the number of babies born with thalassemia major in Greece, Italy and United Kingdom [121].

4 Iron-Selective Chelators with Therapeutic Potential
4.1 Design Features
Desferrioxamine-B (DFO) (7), the most widely used iron chelator in hematology over the past 40 years, has a major disadvantage of being orally inactive [125]. In order to identify an ideal iron chelator for clinical use, careful design consideration is essential; a range of specifications must be considered such as metal selectivity and affinity, kinetic stability of the complex, bioavailability, and toxicity. This field has recently been reviewed [126,127].
4.1.1 Metal Selectivity
Chelating agents can be designed for either the iron(II) or iron(III) oxidation state. High-spin iron(III) is a tripositive cation of radius 0.65 Å, and forms most stable bonds with charged oxygen atoms, such as those found in DFO (7). In contrast, the iron(II) cation, which has a lower charge density, prefers chelators containing nitrogen such as 1,10-phenanthroline. Ligands that prefer iron(II) retain an appreciable affinity for other biologically important bivalent metals such as copper(II) and zinc(II) ions. In contrast, iron(III)-selective ligands, typically oxyanions and notably hydroxamates and catecholates, are generally more selective for tribasic metal cations over dibasic cations and as most tribasic cations, for instance aluminium(III) and gallium(III), are not essential for life, iron(III) provides the best target for ‘iron chelator’ design under biological conditions. An additional advantage of high-affinity iron(III) chelators is that, under aerobic conditions, they will chelate iron(II) and rapidly autoxidize it to the corresponding iron(III) species [128].
4.1.2 Ligand Selection
Catechol moieties possess a high affinity for iron(III). This extremely strong interaction with tripositive metal cations results from the high electron density of both oxygen atoms. However, this high charge density is also associated with the high affinity for protons (pK a values of 12.1 and 8.4). Thus the binding of cations by catechol has marked pH sensitivity. The hydroxamate moiety possesses a lower affinity for iron than catechol, but the selectivity of hydroxamates, like catechols, favors tribasic cations over dibasic cations. Due to the lower value of the protonation constant (pK a ~9 ), competition with hydrogen ions at physiological pH is less pronounced than for that of catechol ligands.
Hydroxypyridinones (Figure 21) combine the characteristics of both hydroxamate and catechol groups, forming 5-membered chelate rings in which the metal is coordinated by two vicinal oxygen atoms. The hydroxypyridinones are monoprotic acids at pH 7.0 and thus, like hydroxamates, form neutral tris-iron(III) complexes. 3-Hydroxypyridin-4-ones are highly selective for tribasic metal cations over dibasic cations as indicated by the low reduction potential of iron complexes (−620 mV versus NHE). 8-Hydroxyquinoline binds iron(II) more tightly than 3-hydroxypyridin-4-ones as indicated by its higher redox potential of the iron complex (−150 mV versus NHE). Never-the-less it is capable of scavenging iron under biological conditions, forming a 3:1 complex with iron(III) at pH 7.0.

Iron(III) chelating agents.
Although aminocarboxylates and hydroxycarboxylates bind iron(III) they are less selective, frequently possessing appreciable affinities for calcium(II) and magnesium(II), in addition to zinc(II) and copper(II).
4.1.3 Hexadentate, Tridentate, and Bidentate Iron(III) Complexes
The coordination requirements of high spin iron(III) are best satisfied by six donor atoms ligating in an octahedral fashion to the metal center, the affinity for the ligand generally decreasing in the sequence; hexadentate > tridentate > bidentate (Figure 22). The overall stability constant trends for bidentate and hexadentate ligands are typified by the bidentate ligand N,N-dimethyl-2,3-dihydroxybenzamide (DMB) and the hexadentate congener MECAM (Figure 21) where a differential of 6 log units in stability is observed (40.2 versus 46).

Schematic representation of chelate ring formation in metal-ligand complexes.
Although MECAM binds iron(III) more tightly than its bidentate analogue DMB, other hexadentate catechols, for instance, enterobactin (Figure 21), bind iron(III) even more tightly. The smaller the conformational space of the free ligand, the higher the stability of the complex; as the difference between the flexibility of the ligand and its corresponding iron complex decreases, so does the entropy difference. Thus enterobactin (log stability constant = 48) can be considered to possess a degree of preorganisation in contrast to MECAM which does not [129].
Under biological conditions, a comparison standard which is generally more useful than the stability constant is the pFe3+ value [130]. pFe3+ is defined as the negative logarithm of the concentration of the free iron(III) in solution. Typically pFe3+ values are calculated for total [ligand] = 10–5 M, total [iron] = 10–6 M at pH 7.4. The comparison of ligands under these conditions is useful, as the pFe3+ value, unlike the stability constants log K or log β 3, takes into account the effects of ligand protonation and denticity as well as differences in metal-ligand stoichiometries. Comparison of the pFe3+ values for hexadentate and bidentate ligands reveals that hexadentate ligands are far superior to their bidentate counterparts under typical in vivo conditions. The values for DMB, MECAM, and enterobactin being 15, 28, and 31.5, respectively (Table 9) [130].
The formation of a complex will also be dependent on both free metal and free ligand concentration and as such will be sensitive to concentration changes. The degree of dissociation for a tris-bidentate ligand-metal complex is dependent on the cube of [ligand] whilst the hexadentate ligand-metal complex dissociation is only dependent on [ligand]. Hence the dilution sensitivity to complex dissociation for ligands follows the order hexadentate < tridentate < bidentate. It is for this reason that the majority of natural siderophores are hexadentate compounds and can therefore scavenge iron(III) efficiently at low metal concentrations [131]. In general, pFe3+ values follow the trend hexadentate > tridentate >bidentate as exemplified by the examples in Table 9.
4.1.4 Critical Features for Clinical Application: Molecular Size and Hydrophobicity
In order to achieve efficient oral absorption, the chelator should possess appreciable lipid solubility which may facilitate the molecule to penetrate the gastrointestinal tract (partition coefficient between n-octanol and water greater than 0.2) [132]. Molecular size is also a critical factor which influences the rate of drug absorption [133]. Indeed, it has been proposed by Lipinski et al. that the molecular weight should not exceed 500 in order to achieve efficient oral absorption [134]. This molecular-weight limit provides a considerable restriction on the choice of chelator and may effectively exclude hexadentate ligands from consideration; most siderophores, for instance DFO (7) and enterobactin (Figure 21) have molecular weights in the range 500–900. In contrast, bidentate and tridentate ligands, by virtue of their much lower molecular weights, tend to possess higher absorption efficiencies.
4.1.5 Toxicity of Chelators and Their Iron Complexes
The toxicity associated with iron chelators originates from a number of factors; including inhibition of metalloenzymes, lack of metal selectivity, redox cycling of iron complexes between iron(II) and iron(III), thereby generating free radicals, and the kinetic lability of the iron complex leading to iron redistribution.
Enzyme inhibition: In general, iron chelators do not directly inhibit heme-containing enzymes due to the inaccessibility of porphyrin-bound iron to chelating agents. In contrast, many non-heme iron-containing enzymes such as the lipoxygenase and aromatic hydroxylase families and ribonucleotide reductase are susceptible to chelator-induced inhibition [135]. Lipoxygenases are generally inhibited by hydrophobic chelators, therefore, the introduction of hydrophilic characteristics into a chelator tend to minimize such inhibitory potential [136]. Stereochemistry can also limit chelator access to the metal binding center and the introduction of a rigid side chain close to the chelating center of the molecule can reduce inhibitory properties [137]. Thus, careful control of the bulk and shape of iron chelators leads to minimal inhibitory influence of many metalloenzymes.
Metal selectivity: An ideal iron chelator should be highly selective for iron(III) in order to minimize chelation of other biologically essential metal ions which could lead to deficiency with prolonged usage. Many ligands that possess a high affinity for iron(III) also have appreciable affinities for other metals such as zinc(II), this being especially so with carboxylate- and nitrogen-containing ligands. However, this factor is less of a problem with the bidentate catechol, hydroxamate, and hydroxypyridinone ligand groups, which possess a strong preference for tribasic over dibasic cations. In principle, competition with copper(II) could be expected to be a problem, however under most biological conditions this is not so, as copper is extremely tightly bound to chaperone molecules [138].
Iron-complex structure and redox activity: In order to prevent free radical production, iron should be coordinated in such a manner as to avoid direct access of oxygen and hydrogen peroxide, and to possess a redox potential which cannot be reduced under biological conditions. Most hexadentate ligands with oxygen containing ligands such as DFO are kinetically inert and reduce hydroxyl radical production to a minimum by failing to redox cycle.
Chelators that are capable of binding both iron(II) and iron(III) at neutral pH values have potential to redox cycle. This is an undesirable property for iron scavenging molecules, as redox cycling can also lead to the production of reactive oxygen radicals (Figure 23). Significantly, the high selectivity of siderophores for iron(III) over iron(II) renders redox cycling under biological conditions unlikely. Thus the iron complexes of enterobactin and desferrioxamine are extremely low, namely −750 and −468 mV (versus NHE) [130]. In similar fashion, iron-deferiprone has a low redox potential (−620 mV versus NHE) [139]. Iron complexes with redox potentials above −200 mV (versus NHE) are likely to redox cycle under aerobic conditions.

Redox cycling of an iron complex.
Kinetic lability of iron complexes: Hexadentate ligand iron complexes tend to be inert, the rate of dissociation of the complex being vanishingly small at neutral pH values. This renders such molecules ideal scavengers of iron. In contrast, bidentate and tridentate ligands are less kinetically stable and are able to dissociate at appreciable rates, thereby possibly facilitating iron redistribution. Such a property is undesirable for most therapeutic applications, where efficient iron excretion is required. In order to avoid appreciable redistribution of iron in mammalian body tissues, chelators possessing a high iron(III) affinity are required; generally a pFe value ≥20 appears to be sufficient to minimize the redistribution of iron.
4.2 Orally Active Iron Chelators in Current Use
Iron chelation therapy prevents the development of iron overload and as a consequence the life style of thalassemia major patients has been dramatically improved with the application of DFO (7) (Section 3.3.1.1). However, DFO is not an ideal therapeutic chelator due to its oral inactivity and rapid renal clearance (plasma half-life of 5–10 min) [140]. In order to achieve sufficient iron excretion, it has to be administered subcutaneously or intravenously for 8–12 h/day, 5–7 days/week [141].
Patient compliance with this regimen is frequently poor. Furthermore, NTBI (Section 3.1) is present in such patients whenever the plasma DFO level is low, rapidly reappearing on the cessation of DFO perfusion (Figure 24) [142]. As DFO is typically infused for 5 nights, this only provides protection for 40h per week; that is approximately 25% of the time. As transferrin is saturated in most of these patients, NTBI is present for 75% of the time and therefore has the possibility of gradually loading the heart and endocrine tissue with iron, even in well chelated patients. A large proportion of patients treated with DFO suffer from adverse cardiovascular events [143].

The effect of DFO infusion at 50 mg/kg/24h (intravenous) on NTBI is shown in a single patient with thalassemia major both on starting the DFO infusion and on stopping the infusion at 48 hours [142].
4.2.1 Tridentate Chelators
Unlike hexadentate and bidentate molecules, it is difficult to design tridentate ligands which only possess oxygen anion coordination sites [144], the central ligand typically being nitrogen (Figure 22).
Desferrithiocins: Desferrithiocin (DFT) (11) is a siderophore isolated from Streptomyces antibioticus. It forms a 2:1 complex with iron(III) at neutral pH using a phenolate oxygen, a carboxylate oxygen, and a nitrogen atom as ligands [145]. It possesses a high affinity for ferric iron (pFe3+ = 20.4). Long term studies of DFT in normal rodents and dogs at low doses have shown toxic side effects, such as reduced body weight and neurotoxicity [146]. However, a range of synthetic analogues of DFT have been prepared in an attempt to design molecules lacking renal and neurotoxicity [147] and two such molecules have been identified, namely deferitrin (12) and FBS0701 (13). Deferitrin (12), was found to be highly effective when given orally to rats and primates. Histopathological analysis indicated some nephrotoxicity but much less than that arising from DFT [148]. Phase I clinical trials demonstrated

good oral absorption, however the compound was not progressed beyond Phase II clinical trials due to nephrotoxicity. FBS0701 (13) also binds iron(III) with high affinity and in contrast to deferitrin, demonstrated no observable toxicity at a predicted dose level range in preclinical studies [149]. The compound has entered clinical trials sponsored by Ferrokin Biosciences [150], where it has been shown to be well tolerated and to possess favorable pharmacokinetics [151]. FBS0701 is currently in Phase II clinical trials.
Triazoles: Triazoles have been investigated as potential ligands by Novartis [152]. These molecules chelate iron(III) with two phenolate oxygens and one triazolyl nitrogen. The lead compound deferasirox (14) possesses a pFe3+ value of 22.5 and is extremely hydrophobic, with a log P octanol/water value of 3.8. As a result, it can penetrate membranes easily and possesses good oral availability. Indeed, when orally administered to hypertransfused rats, deferasirox promotes the excretion of chelatable iron from hepatocellular iron stores four to five times more effectively than DFO [153].

By virtue of a high proportion of both the free ligand and the 2:1 iron complex binding to albumin (greater than 98%), the ligand possesses low toxicity despite its strong lipophilic character. The extreme hydrophobicity of this chelator necessitates formulation in dispersion tablets, containing the disintegrants, SDS, povidone, and crospovidone. Thus, deferasirox is typically given once daily each morning as a dispersed solution in water, half an hour before breakfast. Deferasirox has been demonstrated to be efficient at removing liver iron from regularly transfused patients [154] but is apparently less effective at removing cardiac iron [155]. Deferasirox (14) forms a 2:1 iron complex which possesses a net charge of 3– and a molecular weight over 800. Should such a complex form intracellularly, it is possible that the iron will remain trapped within the cell. The redox potential of the 2:1 iron complex is −600 mV (versus NHE) confirming that deferasirox is highly selective for iron(III) and that it will not redox cycle under biological conditions. As with all therapeutic iron chelators there are side effects associated with deferasirox [156], kidney toxicity being particularly prevalent [157].
4.2.2 Bidentate Chelators
On the basis of selectivity and affinity, particularly considering pFe3+ values, 3-hydroxypyridin-4-one (Figure 21) is the optimal bidentate ligand for the chelation of iron(III) over the pH range of 6.0–10.0 and to date is the only bidentate class to have been subjected to extensive clinical study.
Dialkylhydroxypyridinones: The 1,2-dimethyl derivative (deferiprone, Ll, CP20) (15) is marketed by Apotex Inc. Toronto, Canada, as FerriproxTm. Deferiprone was first reported as a potential orally active iron chelator in 1984 [158] and demonstrated to be active in man in 1987 [159]. It was licensed for use in India in 1994 and in Europe in 1999, receiving full marketing authorization in 2002. The FDA provided approval for its use in 2011. There are numerous reports indicating the comparative effectiveness of desferrioxamine and deferiprone [160]. A particularly important property of deferiprone is its ability to penetrate cells, coordinate iron, forming a neutral complex, which is also capable of permeating membranes. Thus, iron can be readily removed from iron-loaded cells including those of cardiac tissue (Figure 25) [161]. This ability extends to the clinical situation [162,163], where it has been demonstrated that deferiprone therapy is associated with significantly greater cardiac protection than DFO in patients with thalassemia major [143,164]. Unfortunately, the dose required to keep a previously well chelated patient in negative iron balance with deferiprone is relatively high, in the region of 75–100 mg kg–1 day–1. One of the major reasons for the limited efficacy of deferiprone in clinical use is that it undergoes extensive metabolism in the liver. The use of deferiprone has a range of associated side effects [165], the most important being a low incidence of reversible agranulocytosis [166].

Schematic representation of the penetration of deferiprone [LH]0 through the plasma membrane. The bidentate ligand scavenges loosely bound intracellular iron, forming the 3:1 complex, which also carries zero net charge. Efflux as the iron complex leads to iron excretion.

High iron affinity hydroxypyridinones: In order to further improve chelation efficacy, considerable effort has been applied to the design of novel hydroxypyridinones with enhanced pFe3+ values [167]. Novartis synthesized a range of bidentate hydroxypyridinone ligands, which possess an aromatic substituent at the 2-position. The lead compound (16) was found to be orally active and highly effective at removing iron from both the iron-loaded rat [168] and marmoset [169]. In similar fashion, Hider and coworkers have demonstrated that the introduction of either a l′-hydroxyalkyl group (17) [170] or an amido function (18) [171] at the 2-position of 3-hydroxypyridin-4-ones enhances the affinity for iron(III) over the pH range 5–8. These changes result in an increase in the corresponding pFe3+ values due to the reduced competition with hydrogen ions; thus the 2-amidopyridin-4-one (18) has a pFe value of 21.7 as compared with that of the analogous deferiprone (15) which possesses a pFe value of 20.5. In practical terms this means that at pH 7.4 (18) binds iron over ten times more tightly than deferiprone.
These novel high pFe3+ HPOs show great promise in their ability to remove iron under in vivo conditions. Detailed dose-response studies suggest that chelators with high pFe3+ values scavenge iron more effectively at lower doses when compared with simple dialkyl substituted hydroxypyridinones and so in principle can be used at the lower dose of 20 mg kg–1. A number of related compounds are currently undergoing preclinical evaluation.
Combined therapy with desferrioxamine and hydroxypyridinones: By virtue of its small size and ability to penetrate cells, deferiprone has the capability of efficiently scavenging excess iron. However due to its bidentate nature, the ability of deferiprone for iron(III) at neutral pH values is highly concentration-dependent and at relatively low concentrations (<5 μM) the iron deferiprone complex will donate iron to competing ligands [172]. If deferiprone is used together with a high affinity hexadentate chelator such as desferrioxamine, the deferiprone iron complex will readily donate iron to the kinetically more stable desferrioxamine [173]. Indeed, deferiprone enhances plasma NTBI removal in the presence of desferrioxamine [174]. Early clinical studies indicated that such combination therapy is effective at increasing iron excretion [175]. These observations led to more extensive clinical investigations using deferiprone and desferrioxamine in sequential fashion and resulted with beneficial effects in survival, iron removal and cardiac disease [176,177].
4.2.3 Use of Iron Chelators to Treat Diseases Other than Thalassemia
4.2.3.1 Sickle Cell Anemia
Transfusion has been introduced for the treatment of sickle cell anemia, due to its beneficial effect in the treatment of crises and in the reduction of the incidence of stroke [178] (Section 3.3.1.2). Again an effective orally active iron chelator permits regular transfusion of such patients without the worry of inducing an associated iron overloaded state. There have been a number of trials comparing the efficacy of different chelators and the outcome of these studies reviewed [179]. At the present time there is no clear preference for a particular chelator. In a multicenterd study of iron overload [180], three patient groups have been compared (transfused thalassemia patients, transfused sickle cell patients and non-transfused sickle cell patients). There were more endocrine problems in the transfusion-dependent thalassemia patients (56.3%) than in both transfused sickle cell patients (13.1%) and non-transfused sickle cell patients (7.8%) and it has been suggested that this difference may relate to the different NTBI levels and hence speciation (Section 3.1).
Generally, NTBI levels in thalassemia patients are higher than those of sickle cell patients [181]. This finding could also explain the low incidence of cardiac disease in transfused sickle cell patients [180]. There was a relatively poor compliance observed with deferasirox in the sickle cell anemia group [182].
4.2.3.2 Myelodysplastic Syndromes
Many patients with myelodysplastic syndromes (MDS) become dependent on blood transfusions (Section 3.3.2) and so treatment by an orally active iron chelator is appropriate. The use of deferiprone is not ideal in view of the risk of agranulocytosis which may be higher in this patient group [183]. In contrast, deferasirox has found useful application in removing excess iron from transfused MDS patients [184,185]. As cardiac problems are the most frequent serious complications associated with iron overload in MDS patients [86], procedures designed to maintain NTBI levels at a low level should be adopted. Daily administration of deferasirox is thus likely to provide a useful option.
4.2.3.3 Friedreich’s Ataxia
Friedreich’s ataxia involves the accumulation of iron in mitochondria; cardiac, gastrointestinal and brain tissues being most severely affected (Section 3.4.2). In principle, an iron chelator which can readily cross membranes, gain access to the mitochondrial matrix, scavenge inappropriately deposited iron and efflux from the mitochondrion as a stable iron complex, should find application in the treatment of this disease. Deferiprone is one such compound (Figure 25) as demonstrated by Cabantchik and coworkers [46,186]. Treatment of Friedreich’s ataxia patients with deferiprone for 6 months led to a decrease in iron levels that had specifically accumulated in dentate nuclei of the brain and also improved gait and control of the gastrointestinal tract [93]. This preliminary clinical study has now been extended to a multicenter investigation.
5 Neuropathology and Iron
Neurodegeneration is a complicated multifaceted process which leads to many chronic disease states. A broad classification can be achieved on the basis of neuropathological changes:
-
(i)
Disorders characterized by the accumulation of abnormal proteins leading to a selective loss of neurons; examples include Alzheimer’s disease (Aβ amyloid plaques), Parkinson’s disease (Lewy bodies), Huntington disease (Huntingtin aggregates), and Pick’s disease (Pick bodies).
-
(ii)
Disorders resulting from dysfunction of motor neurons; examples include multiple sclerosis, amyotrophic lateral sclerosis, Friedreich’s ataxia, and progressive supranuclear palsy.
Significantly, all these disease states have been associated with elevated levels of iron in the brain resulting from a loss of control of iron homeostasis in particular areas of the brain [187]. The control of neuronal cytosolic iron(II) levels is achieved by an interplay of fluxes centered on membrane transporters, mitochondria, lysosomes, and ferritin (Figure 8) together with neuromelanin which acts as an additional storage site for iron and other transition metals [188]. The concentration of iron in the cerebrospinal fluid, and hence the interstitial fluid, ranges between 0.2 and 1.1 μM, whereas transferrin levels are low, typically 0.25 μM [189]. Thus, CSF iron levels will frequently exceed the binding capacity of transferrin and in view of the relatively high levels of citrate, form complexes 9 and 10.
The levels of this non-transferrin bound iron will influence cytosolic levels of iron(II). Abnormal iron accumulation induces the oxidation of reduced glutathione in human neuronal cells [190] and this in turn, by virtue of the involvement of glutathione in iron sulfur cluster synthesis [8,191], can lead to a reduction in the respiratory Complex I activity (Complex I contains 8 Fe-S clusters). Reduction of Complex I activity can result in vicious cycles of increased oxidative stress, increased iron accumulation, and decreased GSH content [189]. Significantly, defects in mitochondrial electron transport have been reported for Alzheimer’s disease, Parkinson’s disease, Huntington disease, and Friedreich’s ataxia [192].
5.1 Alzheimer’s Disease
Alzheimer’s disease is the most common form of dementia, there being over 24 million sufferers at the present time and by 2040 it has been estimated that there will be 80 million worldwide. Central to the disease is the formation of amyloid plaques which predominately consist of Aβ amyloid peptide, a 42 membered peptide resulting from the breakdown of a membrane-bound protein (APP) (Figure 26) [193].

Amyloidogenic processing of amyloid precursor protein (APP). APP is cleaved to produce membrane bound Aβ42 peptide, which is in equilibrium with water soluble oligomers of Aβ42 peptide. The oligomers aggregate to form amyloid plaques.
There is continual production of Aβ42 in normal individuals, but in Alzheimer’s disease patients, proteosome processing becomes less efficient and the peptide accumulates both in the membrane and as soluble oligomers. The latter aggregate to form amyloid plaques (Figure 26). The oligomers and the membrane bound Aβ42 molecules possess an iron binding site [194,195] which endows the peptide with pro-oxidant activity [196,197]. It has recently been proposed that APP possesses ferroxidase activity, which is inhibited in Alzheimer’s disease, thereby facilitating neuronal iron accumulation [198,199]. Brain iron accumulation has been demonstrated in Alzheimer’s disease patients by both MRI [200] and ICP-MS [201]. This sequence of events could lead to mitochondrial toxicity as outlined in Section 5.1. Clearly there is potential for iron chelation therapy, both in prophylactic and therapeutic treatments of Alzheimer’s disease (Section 5.5).
5.2 Parkinson’s Disease
Parkinson’s disease, like Alzheimer’s disease, tends to be more common in the elderly, about 4% of those over 80 years experience symptoms. Although stiffness and tremor are common symptoms, there are many features of the disease [202], for instance the nigrostriatal and caudate nucleus dopaminergic neurons are adversely effected leading to cell death and neuronal loss. Genetic factors, environmental factors and aging can all influence the onset of the disease [203]. Lewy bodies appear and there is multiple evidence of both damage induced by free radicals [204,205] and for an increased nigral iron content [206,207]. In particular, there are reduced glutathione levels in the nigral brain region and the mitochondrial Complex I is inhibited [205].
As with Alzheimer’s disease there is clear potential for iron chelation therapy (Section 5.5).
5.3 Pantothenate Kinase-2 Deficiency
Pantothenate kinase-2 (PANK 2) deficiency is a relatively uncommon autosomal recessive trait which is characterized by progressive Parkinson-like rigidity, together with mental and emotional retardation (previously termed Hallervorden-Spatz syndrome) [208]. Onset is in late childhood, death typically occurring within 10 years.
The globus pallidus and substantia nigra possess elevated levels of iron, mostly located in microglia and macrophages giving rise to the ‘the eye of the tiger’ MRI brain image [209,210]. PANK 2 is the first enzyme involved in the biosynthesis of coenzyme A and is located in the mitochondria. Patients with PANK 2 deficiency suffer from a coenzyme A deficiency and a related elevation of cysteine [211] which has been suggested to increase iron levels by chelation [212].
5.4 Macular Degeneration
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in the developed nations, in people aged 65 and older [213]. Oxidative stress and free radical damage have been implicated in the pathogenesis of AMD [214] and iron is a likely oxidant, as AMD-affected maculars possess a higher iron content than healthy age-matched maculars, as demonstrated by Perl’s stain [215]. Whereas iron accumulation is observed in AMD retinas, it is unclear whether iron is directly involved in AMD pathogenesis or is simply a byproduct of AMD pathology. However, there are several lines of evidence that link iron with AMD pathogenesis; for instance, the levels of bone morphogenetic protein 6 (BMP 6), a major regulator of systemic iron (Section 1.4.1), is increased in advanced AMD eyes [216] and hepcidin-knockout mice experience age-dependent increases in retinal iron followed by retinal degeneration [217]. It is significant that retinal iron accumulation resulting from Friedreich’s ataxia and PANK 2 disease is associated with retinal degeneration [218].
5.5 Potential of Iron Chelators for the Treatment of Neurodegeneration
A critical prerequisite for a chelator capable of treating various forms of neurodegeneration is to be orally active and to readily permeate the blood brain barrier or in the case of the eye, the blood-retinal barrier. Thus, the chelator should have a molecular weight <500, be sufficiently hydrophobic to partition in membranes and yet be sufficiently hydrophilic to achieve water solubility at pharmacological levels [219]. Of the three chelators currently used in the clinic, only deferiprone possesses these properties (Figure 25), which accounts for why it has been so extensively investigated for the treatment of neurodegenerative diseases [93,220–223]. A range of 8-hydroxyquinoline derivatives, including clioquinol (19) [224], VK-28 (20) [225], and an iron-binding peptide (21) [226], have also been investigated.

Deferiprone (15), VK-28 (20), and clioquinol (19) have each demonstrated neuroprotective action in various models of Alzheimer’s disease [227–229] and clioquinol has been investigated in a related clinical trial [230]. A similar situation exists with Parkinson’s disease, where a number of successful studies have been achieved in various animal models with both 8-hydroxyquinolines [225,231] and deferiprone [220]. Deferiprone is currently under clinical investigation for efficacy and safety in Parkinson’s disease [232,233]. Deferiprone has also been demonstrated to have a beneficial effect in the clinical treatment of age-dependent retinal degeneration [221,234] and PANK 4 disease [222,223].
Various strategies have been investigated in an attempt to enhance the transfer of chelators across the BBB. In principle pendant glucose molecules can be attached to molecules in order to enhance their ability to permeate the BBB. Due to the critical requirement of the brain for glucose, the BBB is endowed with a high density of GLUT 1 hexose transporter protein. With this strategy in mind, glucose conjugates (22) and (23) have been synthesized [235,236], but to date have not been demonstrated to cross the BBB [237]. Another approach involves the attachment of chelators to nanoparticles [238]. Chelator hybrid molecules are also under current investigation in a range of neurodegenerative models, for instance, radical scavengers (24) [239] and monoamine oxidase inhibitors (25) [240]. This field has recently been reviewed [241].

6 The Role of Iron Chelation in Cancer Therapy
The importance of iron in tumor cell growth has been discussed over the past 50 years [242], high levels of dietary iron having been linked to increased tumor development [243,244]. More recently an iron regulatory gene signature has been linked to the incidence of breast cancer [245], and both gastrointestinal and liver cancer have been specifically associated with dietary iron [246]. Cell proliferation is dependent on a plentiful supply of iron and iron chelators can interfere with the cell cycle, causing G1/S arrest by removing iron from ribonucleotide reductase [247] and deoxyhypusine hydroxylase [248]. Many chelating agents have been investigated for their potential to selectively inhibit tumor cell growth, in particular desferrioxamine [249], spermidine catecholamides [250], and PIH analogues [247]. At the present time it has not been possible to achieve an acceptable selective toxicity against tumor cells in the clinic.

Pyridoxal isonicotinoyl hydrazine (PIH) derivatives (26) show potential in this regard and Richardson and his coworkers have thoroughly investigated this group of chelators. The β-napthol derivative (27) has been reported to be a particularly effective antiproliferative agent [251], as have the closely related thiosemicarbazones (28) [252]. The precise role of iron in the cell-cycle control remains unclear, but the posttranscriptional regulation of a cyclin-dependent kinase inhibitor may also feature in this network of iron-dependent reactions [253].
7 Iron and Infection
Iron is essential for the growth of almost all microorganisms and thus bacteria and fungi have evolved strategies to scavenge iron from the soil, fresh and marine water, and living organisms. One of the most common strategies is siderophore production. Siderophores are low molecular weight compounds (500–1500 daltons) which possess a high affinity and selectivity for iron. There are over 500 different siderophores 270 of which have been structurally characterized [131]; desferrioxamine (7), desferrithiocin (11), and enterobactin (Figure 21) are typical examples.
Pathogenic bacteria and fungi have developed the means of survival in animal tissue. They may invade the gastrointestinal tract (Escherichia, Shigella, and Salmonella), the lung (Pseudomonas, Bordatella, Streptococcus, and Corynebacterium), skin (Staphylococcus) or the urinary tract (Escherichia and Pseudomonas). Such bacteria may colonize wounds (Vibrio and Staphylococcus) and be responsible for septicaemia (Yersinia and Bacillus). Some bacteria survive for long periods of time in intracellular organelles, for instance Mycobacterium. Because of this continual risk of bacterial and fungal invasion, animals have developed a number of lines of defence based on immunological strategies, the complement system, the production of iron-siderophore binding proteins and the general “withdrawal” of iron [254].
There are two major types of iron-binding proteins present in most animals that provide protection against microbial invasion – extracellular protection is achieved by the transferrin family of proteins and intracellular protection is achieved by ferritin (Section 1.2.4). Under normal conditions transferrin is about 25–40% saturated, which means that any freely available iron in the serum will be immediately scavenged – thus preventing microbial growth. Most siderophores are unable to remove iron from transferrin, although some, for instance aerobactin, can compete [255,256]. Mammals also produce lactoferrin, which is similar to serum transferrin but possesses an even higher affinity for iron [257]. Lactoferrin is present in secretory fluids, such as sweat, tears and milk, thereby minimizing bacterial infection. Ferritin is present in the cytoplasm of cells and limits the intracellular iron level to approximately 1 μM. Siderophores are unable to mobilize iron from ferritin. In addition to these two classes of iron binding proteins, hepcidin is involved in controlling the release of iron from absorptive enterocytes, iron-storing hepatocytes, and macrophages (Section 1.4.1). Infection leads to inflammation and the release of interleukin-6 (IL-6) which stimulates hepcidin expression. In humans, IL-6 production results in low serum iron, making it difficult for invading pathogens to infect.
In addition to these “iron withdrawal” tactics, mammals produce an iron-siderophore binding protein, siderochelin [258]. Siderocalin is a potent bacteriostatic agent against E. coli. As a result of infection, it is secreted by both macrophages and hepatocytes, enterobactin being scavenged from the extracellular space. Indeed, mice are highly susceptible to E. coli infections when the siderocalin gene is “knocked-out” [259]. Siderocalin binds a wide range of tris-catecholate siderophores.
7.1 Tuberculosis
Tuberculosis (TB) caused by the pathogen Mycobacterium tuberculosis infects one-third of the world population. Despite the development of new drugs, the incidence of TB continues to increase. Increased iron status enhances tuberculosis infection [260]. Siderophore molecules used by Mycobacterium for iron acquisition are potential therapeutic targets, as are synthetic iron chelators which are capable of outcompeting siderophores for iron.
7.2 Malaria
Many metabolic components of the erythrocytic malaria parasite are dependent on iron, including ribonucleotide reductase, and mitochondrial function. Several studies have indicated that the erythrocyte labile iron pool is the iron source for malaria parasites (Section 1.3) [246]; a finding somewhat confirmed by the observation that iron chelators suppress the growth of P. falciparum in human erythrocytes. A wide range of chelators have been demonstrated to inhibit the growth of erythrocytic malaria parasites under in vitro conditions [261,262], however with the exception of desferrioxamine, extension to animal studies has proved to be less impressive [262].
There have been a number of clinical investigations centered on the use of desferrioxamine, where it has displayed an appreciable antimalarial activity but failed to effect a cure [246].
8 Overview and Future Developments
The development of iron biochemistry has been dramatic over the past 50 years. A large range of iron-dependent enzymes have been characterized, the chemistry of electron transfer proteins, containing iron-sulfur clusters, is beginning to be understood and oxygen-binding iron centers are very well characterized. The highly controlled transport of iron throughout multicellular organisms and the mechanism of intracellular distribution is now understood, although there is one tissue where iron distribution is far from clear: the brain. Transferrin levels are much lower in CNS than in blood and there are a number of brain proteins which appear to be influenced by iron levels and yet currently have no known function, for instance amyloid precursor protein and α-synuclein. Clearly, this is an area that deserves intensive investigation, particularly as the progression of many forms of neurodegeneration appears to be enhanced by elevated iron levels.
Over the same period iron chelators have emerged as an important therapeutic class. For many years the orally inactive desferrioxamine was the only iron chelator available for clinical use, but during the past twenty years two other chelators have been introduced, deferiprone and deferasirox. Both are orally active and this has rendered the treatment of iron overload to be more “patient friendly” thereby enabling clinicians to investigate the use of iron chelation for diseases other than systemic iron overload, for instance Parkinson’s disease and macular degeneration.
There are more iron chelators under development and it is likely that in years to come there will be a selection of iron chelators available for clinical use that will cover ranges of both iron affinity and membrane penetrative ability. Studies are in progress to design lysosomotrophic and mitochondrotrophic chelators.
Improved therapeutic control of a wide range of anemias has been accomplished over the past 50 years and in particular the development of a range of relatively nontoxic parential iron preparations has proved to be highly beneficial. Surprisingly, the most common oral supplement for the treatment of iron deficiency anemia is ferrous sulfate, which was first introduced to medicine in 1832. There is a very clear requirement for the introduction of a replacement oral therapy due to the various toxic side effects of ferrous sulfate. Hopefully one or more such replacements will be developed in the near future.
- ALAS:
-
5-amino levulinate synthase
- AMD:
-
age-related macular degeneration
- APP:
-
amyloid precursor protein (Fig. 26)
- AscH– :
-
ascorbic acid
- ATP:
-
adenosine 5′-triphosphate
- BBB:
-
blood brain barrier
- BMP:
-
bone morphogenetic protein
- CD91:
-
lipoprotein receptor
- CNS:
-
central nervous system
- CSF:
-
cerebrospinal fluid
- DCYTB:
-
duodenal cytochrome b
- DF:
-
desferrithiocin
- DFO:
-
desferrioxamine
- DMT1:
-
divalent metal-iron transporter 1
- EDTA:
-
ethylenediamine-N,N,N′,N′-tetraacetic acid
- FBXL5:
-
F-box and leucine-rich repeat protein 5
- FDA:
-
Food and Drug Administration
- FPN1:
-
ferroportin
- FXN:
-
frataxin
- GDF15:
-
growth differentiation factor 15
- GLUT:
-
1 glucose transporter 1
- Grx:
-
5 glutaredoxin-5
- HAMP:
-
hepcidin synthesis gene
- Hb:
-
hemoglobin (Fig. 16)
- HCP1:
-
heme carrier protein 1
- HEIDI:
-
2,2′-(2-hydroxyethylazanediyl)diacetic acid
- HFE:
-
hemochromatosis protein
- HIV:
-
human immunodeficiency virus
- HPO:
-
hydroxypyridinone
- ICP-MS:
-
inductively coupled plasma mass spectrometry
- IL-6:
-
interleukin 6
- IRE:
-
iron responsive elements
- IRP:
-
iron responsive proteins
- JAK/STAT:
-
janus kinase/signal transducer and activator of transcription
- MDS:
-
myelodysplastic syndrome
- MECAM:
-
N,N′-(5-((2,3-dihydroxycyclohexa-1,3-dienecarboxamido)methyl)-1,3-phenylene)bis(methylene)bis(2,3-dihydroxybenzamide)
- MHC:
-
major histocompatibility complex
- MRI:
-
magnetic resonance imaging
- MTP:
-
metal transporter protein
- NHE:
-
normal hydrogen electrode
- NTBI:
-
non-transferrin bound iron
- PANK:
-
2 pantothenate kinase-2
- PIH:
-
pyridoxal isonicotinoyl hydrazine
- SDS:
-
sodium dodecyl sulfate
- SMAD:
-
4 tumor suppressor gene
- SS:
-
sickle cell disease
- TB:
-
tuberculosis
- TfR1:
-
transferrin receptor 1
- TfR2:
-
transferrin receptor 2
- WHO:
-
World Health organization
- XLSA:
-
X-linked sideroblastic anemia
References
W. Schneider, Chimia 1988 , 42, 9–20.
R. R. Crichton, Iron Metabolism, 3rd edn., Wiley, Chichester, 2009.
A. K. Powell, S. L. Heath, D. Gatteschi, L. Pardi, R. Sessoli, G. Spina, F. Del Giallo, F. Pieralli, J. Am. Chem. Soc. 1995, 117, 2491–2502.
W. R. Harris, in “Molecular and Cellular Iron Transport”, Ed D. M. Templeton, Marcel Dekker, Inc., New York, 2002, pp. 1–40.
B. Halliwell, J. M. C. Gutteridge, Arch. Biochem. Biophys. 1986, 246, 501–514.
A. Ozer, R. K. Bruick, Nature Chem. Biol. 2007, 3, 144–153.
C. Andreini, I. Bertini, G. Cavallaro, R. J. Najmanovich, J. M. Thornton, J. Mol. Biol. 2009, 388, 256–380
R. C. Hider, X. Kong, Dalton Trans. 2013, 42, 3220–3229.
K. Mizutani, M. Toyoda, B. Mikami, Biochim. Biophys. Acta 2012, 1820, 203–211.
M. K. Mayle, A. M. Le, D. T. Kamei, Biochim. Biophys. Acta 2012, 1820, 264–281.
W. R. Harris, Biochim. Biophys. Acta 2012, 1820, 348–361.
P. M. Harrison, P. Arosio, Biochim. Biophys. Acta 1996, 1275, 161–203.
G. A. Clegg, J. E. Fitton, P. M. Harrison, A. Treffry, Progr. Biophys. Mol. Biol. 1981, 36, 53–86.
S. Roberts, A. Bomford, J. Biol. Chem. 1988, 241, 3630–3637.
R. C. Hider, X. Kong, BioMetals 2011, 24, 1179–1187.
H. Gonshin, B. Mackenzie, U. V. Berger, Y. Gunshin1, M. F. Romero, W. F. Boron, S. Nussberger, J. L. Gollan, M. A. Hediger, Nature 1997, 388, 482–488.
M. Shayeghi, G. O. Latunde-Dada, J. S. Oakhill, A. H. Laftah, K. Takeuchi, N. Halliday, Y. Khan, A. Warley, F. E. McCann, R. C. Hider, D. M. Frazer, G. J. Anderson, C. D. Vulpe, R. J. Simpson, A. T. McKie, Cell 2005, 122, 789–801.
P. N. Paradkar, K. B. Zumbrennen, B. H. Paw, D. M. Ward, J. Kaplan, Mol. Cell. Biol. 2009, 29, 1007–1016.
W. Chen, H. A. Dailey, B. H. Paw, Blood 2010, 116, 628–630.
A.T. McKie, P. Marciani, A. Rolfs, K. Brennan, K. Wehr, D. Barrow, S. Miret, A. Bomford, T. J. Peters, F. Farzaneh, M. A. Hediger, M. W. Hentze, R. J. Simpson, Mol. Cell 2000, 5, 299–309.
A. Donovan, A. Brownlie, Y. Zhou, J. Shepard, S. J. Pratt, J. Moynihan, B. H. Paw, Anna Drejer, B. Barut, A. Zapata, T. C. Law, C. Brugnara, S. E. Lux, G. S. Pinkus, J. L. Pinkus, P. D. Kingsley, J. Palis, M. D. Fleming, N. C. Andrews, L. I. Zon, Nature 2000, 403, 776–781.
S. Abboud, D. J. Haile, J. Biol. Chem. 2000, 275, 19906–19912.
M. U. Muckenthaler, R. Hill in Iron Physiology and Pathophysiology in Humans, Eds J. G. Anderson, G. D. McLaren, Humana Press, New York, 2012, pp. 27–50.
E. W. Mullner, B. Neupert, L. C. Kuhn, Cell 1989, 58, 373–382.
K. J. Addess, J. P. Basilion, R. D. Klausner, T. A. Rouault, A. Pardi, J. Mol. Biol. 1997, 274, 72–83.
D. J. Haile, M. W. Hentze, T. A. Rouault, J. B. Harford, R. D. Klausner, Mol. Cell. Biol. 1989, 9, 5055–5061.
T. Ganz, S.Vaulont, in Iron Physiology and Pathophysiology in Humans, Eds G. J. Anderson, G. D. McLaren, Humana Press, New York, 2011, pp. 173–190.
M. W. Hentze, M. U. Muckenthaler, B. Galy, C. Camaschella, Cell 2010, 142, 24–38.
J. J. Bullen, E. Griffiths, Iron and Infection, 2nd edn., Wiley Press, New York, 1999.
P. R. Dallman, M. A. Siimes, A. Stekel, Am. J. Clin. Nutr. 1980, 33, 86–118.
W. I. Leong, B. Lönnerdal in Iron Physiology and Pathophysiology in Humans, Eds G. J. Anderson, G. D. McLaren, Humana Press, New York, 2011, pp. 81–99.
World Health Organisation, Iron Deficiency Anaemia: Assessment, Prevention and Control. A Guide to Programme Managers, Geneva, WHO, 2001.
R. D. Baynes, T. H. Bothwell, Ann. Rev. Nutr. 1990, 10, 133–148.
F. E. Viteri, B. Torun, in Clinics in Haematology, Vol 3, Ed L. Garby, Saunders, London, 1974, pp 609–626.
G. W. Gardner, V. R. Edgerton, B. Senewiratne, R. J. Barnard, Y. Ohira, Am. J. Clin. Nutr. 1977, 30, 910–917.
The British Nutrition Foundation, Iron, Nutritional and Physiological Significance, Chapman and Hall, London, 1995, pp. 65–78.
J. D. Cook, M. B. Reddy, J. Burri, M. A. Juillerat, R. F. Hurrell, Am. J. Clin. Nutr. 1997, 65, 964–969.
P. Blaud, Rev. Med. Franç. Etranger, 1832, 45, 341–367.
I. H. Stockley, Stockley’s Drug Interactions, 6th edn, Pharmaceutical Press, London, 2002.
D. M. Reffitt, T. J. Burden, P. T. Seed, J. Wood, R. P. H. Thompson, J. Powell, Ann. Clin. Biochem. 2000, 37, 457– 466.
P. Jacobs, G. Johnson, L. Wood, J. Medicine 1984, 15, 367–377.
W. Schneider, W. Forth, Arzneim. Forsch. Drug Res. 1987, 37, 89–95.
C.N. Roy, in Iron Physiology and Pathophysiology in Humans, Eds G. J. Anderson, G. D. McLaren, Humana Press, New York, 2011, pp. 303–320.
C. N. Roy, D. A. Weinstein, N. C. Andrews, Pediatr. Res. 2005, 53, 507–512.
E. Nemeth, S. Rivera, V. Gabayan, C. Keller, S. Taudorf, B. K. Pedersen, T, Ganz, Blood 2003, 101, 2461–2463.
Y. S. Sohn, W. Brever, A. Munnich, Z. I. Cabantchik, Blood 2008, 111, 1690–1699.
R. W. Evans, X. Kong, R. C. Hider, Biochim. Biophys. Acta 2012, 1820, 282–290.
J. P. Kaltwasser, U. Kessler, R. Gottschalk, G. Stucki, B. Moller, J. Rheumatol. 2001, 28, 2430–2436.
L. T. Goodnough, B. Skikne, C. Brugnara, Blood 2000, 96, 823–833.
W. Y. Qunibi, Arzneimittelforschung 2010, 60, 399–412.
P. Geisser, S. Burckhurdt, Pharmaceutics 2011, 3, 12–33.
L. J. Noetzli, S. M. Carson, A. S. Nord, T.D. Coates, J. C. Wood, Blood 2009, 114, 4021–4026.
M. N. Silva, X. Kong, M. C. Parkin, R. Cammack, R. C Hider, Dalton Trans. 2009, 40, 8616–8625.
J.L. Pierre, I. Gautier-Luneau, BioMetals 2000, 13, 91–96.
R. W. Evans, R. Rafique, A. Zarea, C. Rapisarda, R. Cammack, P. J. Evans, J. B. Porter, R. C. Hider, J. Biol. Inorg. Chem. 2008, 13, 57–74.
M. N. Silva, R. C. Hider, Biochim. Biophys. Acta 2009, 1794, 1449–1458.
P. Brissot, M. Ropert, C. Le Lan, O. Loreal, Biochim. Biophys. Acta 2012, 1820, 403–410.
P. C. Adams, J. C. Barton, Lancet 2007, 370, 1855–1860.
N. C. Andrews, New Engl. J. Med. 1999, 341, 1986–1995.
A. Pietrangelo, New Engl. J. Med. 2004, 350, 2383–2397.
J. N. Feder, A. Gnirke, W. Thomas, Z. Tsuchihashi, D. A. Ruddy, A. Basava, F. Dormishian, R. Jr Domingo, M. C. Ellis, A. Fullan, L. M. Hinton, N. L. Jones, B. E. Kimmel, G. S. Kronmal, P. Lauer, V. K. Lee, D. B. Loeb, F. A. Mapa, E. McClelland, N. C. Meyer, G. A. Mintier, N. Moeller, T. Moore, E. Morikang, C. E. Prass, L. Quintana, S. M. Starnes, R. C. Schatzman, K. J. Brunke, D. T. Drayna, N. J. Risch, B. R. Bacon, R. K. Wolff, Nature 1996, 13, 399–408.
M. Simon, M. Bourel, R. Fauchet, B. Genetet, Gut 1976, 17, 332–334
J. N. Feder, Z. Tsuchihashi, A. Irrinki, V. K. Lee, F. A. Mapa, E. Marikang, C. E. Prass, S. M. Starnes, R. K. Wolff, S. Parkkilay, W. S. Sly, R. C. Schatzman, J. Biol. Chem. 1997, 272, 14025–14028.
K. J. Allen, L. C. Gurrin, C. C. Constantine, N. J. Osborne, M. B. Delatycki, A. J. Nicol, C. E. Mcharen, M. Bahlo, A. E. Nisselle, C. D. Vulpe, G. J. Anderson, M. C. Southey, G. G. Giles, D. R. English, J. L. Hopper, J. K. Olynk, L. W. Powell, D. M. Gertig, New Engl. J. Med. 2008, 358, 221–230.
P. L. Lee, E. Beutler, Annu. Rev. Pathol. Mech. Dis. 2009, 4, 489–515.
T. J. Sproule, E. C. Jazwinska, R. S. Britton, B. R. Bacon, R. E. Fleming, W. S. Sly, D. C. Roopeniom, Proc. Natl. Acad. Sci. USA 2001, 98, 5170–5174.
C. Camaschella, A. Roetto, M. Cicilano, P. Pasquero, S. Bosio, L. Gubetta, F. Di Vito, D. Girelli, A. Totaro, M. Carella, A. Grifa, P. Gasparini, Eur. J. Hum. Genet. 1997, 5, 371–375.
G. Sebastiani, K. Pantopoulos, Metallomics 2011, 3, 971–986.
L. P. Yang, S. J. Keam, G. M. Keating, Drugs 2007, 67, 2211–2230.
S. Rivella, in Iron Physiology and Pathophysiology in Humans, Eds G. J. Anderson, G. D. McLaren, Humana Press, New York, 2012, pp. 321–341.
D. J. Weatherall, Ann. New York Acad. Sci. 2010, 1202, 17–23.
J. B. S. Haldane, Hereditas 1949, 35, 267–273.
D. J. Weatherall, Brit. J. Haematol. 2008, 141, 276–286.
H. F. Bunn, Blood 2013, 121, 20–25.
D. J. Weatherall, J. B. Clegg, Thalassaemia Syndromes, 3rd edn, Blackwell, London, 1981.
B. Model, M. Darlison, Bull. World Health Organ. 2008, 86, 480–487.
D. J. Weatherall, Brit. J. Heamatol. 2000, 321, 1117–1120.
G. M. Brittenham, in Hematology: Basic Principles and Practice, Eds R. Hoffman, E. Benz, S. Shattil, B. Furie, H. Cohen, Churchill Livingstone, New York, 1991, pp. 327–342.
R. Sephton Smith, Brit. Med. J. 1962, 1577–1580.
S. Dhungana, P. S. White, A. L. Crumbliss, J. Biol. Inorg. Chem. 2001, 6, 810–818.
R. D. Propper, S. B. Schurin, D. G. Nathan, New Engl. J. Med. 1976, 294, 1421–1423.
A. T. Taher, K. M. Musallam, M. D. Cappellini, D. J. Weatherall, Brit. J. Haematol. 2010, 152, 512–523.
S. Claster, E. P. Vichinsky, Brit. Med. J. 2003, 327, 1151–1155.
M. J. Stuart, R. L. Nagel, Lancet 2004, 364, 1343–1360.
E. B. Fung, P. R. Harmatz, P. D. K. Lee, M. Milet, R. Bellevue, M. R. Jeng, K. A. Kalinyak, M. Hudes, S. Bhatia, E. P. Vinchinsky, Brit. J. Haematol. 2006, 135, 574–582.
N. Galtermann, E. A. Rachmilewitz, Ann. Hematol. 2011, 90, 1–10.
N. B. A. Roy, S. Myerson, A. H. Schuh, P. Bignell, R. Patel, J. S. Wainscoat, S. McGowan, E. Marchi, W. Atoyebi, T. Littlewood, J. Chacko, P. Vyas, S. B. Killick, Brit. J. Haematol. 2011, 154, 521–524.
V. Campuzano, L. Montermini, M.D. Molto, L. Pianese, M. Cossee, F. Cavalcanti, E. Monros, F. Rodius, F. Duclos, A. Monticelli, F. Zara, J. Cañizares, H. Koutnikova, S. Bidichandani, C. Gellera, A. Brice, P.Trouillas, G. De Michele, A. Filla, R. De Frutos, F. Palau, P. I. Patel, S. D. Donato, J. L. Mandel, S. Cocozza, M. Koenig, M. Pandolfo, Science 1996, 271, 1423–1427.
M. Babcock, D. deSilva, R. Oaks, S. Davis-Kaplan, S. Jiralerspong, L. Montermini, M. Pandolfo, J. Kaplan, Science 1997, 276, 1709–1712.
T. L. Stemmler, E. Lesuisse, D. Pain, A. Dancis, J. Biol. Chem. 2010, 285, 26737–26743.
V. Campuzano, L. Montermini, M. D. Moltò, L. Pianese, M. Cossée, F. Cavalcanti, Hum. Mol. Genet. 1997, 6, 1771–1780.
D. R. Lynch, S. R. Regner, K. A. Schadt, L. S. Friedman, K. Y. Lin, M. G. St John Sutton, Expert Rev. Cardiovasc. Ther. 2012, 10, 767–777.
N. Boddaert, K. H. Le Quan Sang, A. Rötig, A. Leroy-Willig, S. Gallet, F. Brunelle, D. Sidi, J. Thalabard, A. Munnich, Z. I. Cabantchik, Blood 2007, 100, 401–408.
M. T. Rodríguez-Manzaneque, J. Tamarit, G. Bellí, J. Ros, E. Herrero, Mol. Biol. Cell. 2002, 13, 1109–1121.
C. Camaschella, A. Campanella, L. D. Falco, L. Boschetto, R. Merlini, L. Silvestri, S. Levi, A. Iolascon, Blood 2007, 110, 1353–1358.
H. Ye, Jeong, M. C. Ghosh, G. Kovtunovych, L. Silvestri, D. Ortillo, N. Uchida, J. Tisdale, C. Camaschella, T. A. Rouault, J. Clin. Invest. 2010, 1120, 1749–1761.
N. C. Andrews, Nat. Rev. Genetics 2000, 1, 208–217.
C. C. Trenor, D. R. Campagna, V. M. Sellers, N. C. Andrews, M. D. Fleming, Blood 2000, 96, 1113–1118.
J. E. Levy, O. Jin, Y. Fujiwara, F. Kuo, N. C. Andrews, Nat. Genetics, 1999, 21, 396–399.
M. D. Fleming, M. A. Romano, M. A. Su, L. M. Garrick, M. D. Garrick, N. C. Andrews, Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153.
S. E. Bernstein, J. Lab. Clin. Med. 1987, 110, 690–705.
E. S. Russell, E. C. McFarland, E. L. Kent, Transpl. Proc. 1970, 2, 144–151.
D. Sladic-Simic, P. N. Martinovich, N. Zivkovic, D. Pavic, J. Martinovic, M. Kahn, H. M. Ranney, Ann. N. Y. Acad. Sci. 1969, 165, 93–99.
C. D. Vulpe, Y. M. Kuo, T. L. Murphy, L. Cowley, C. Askwith, N. Libina, J. Gitschier, G. J. Anderson, Nat. Genet. 1999, 21, 195–199.
X. Y. Zhou, S. Tomatsu, R. E. Fleming, S. Parkkila, A. Waheed, J. Jiang, Y. Fei, E. M. Brunt, D. A. Ruddy, C. E. Prass, R. C. Schatzman, R. O’Neill, R. S. Britton, B. R. Bacon, W. S. Sly, Proc. Natl. Acad. Sci. USA 1998, 95, 2492–2497.
V. Niederkofler, R. Salie, S. Arber, J. Clin. Invest. 2005, 115, 2180–2186.
F. W. Huang, J. L. Pinkus, G. S. Pinkus, M. D. Fleming, N. C. Andrews, J. Clin. Invest. 2005, 115, 2187–2191.
G. Nicolas, M. Bennoun, I. Devaux, C. Beaumont, B. Grandchamp, A. Kahn, Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785.
R. E. Fleming, J. R. Ahmann, M. C. Migas, A. Waheed, H. P. Koeffler, H. Kawabata, R. S. Britton, B. R. Bacon, and W. S. Sly, Proc. Natl. Acad. Sci. USA 2002, 99, 10653–10658.
I. E. Zohn, I. D. Domenico, A. Pollock, D. M. Ward, J. F. Goodman, X. Liang, A. J. Sanchez, L. Niswander, J. Kaplan, Blood 2007, 109, 4174–4180.
R. H. Wang, C. Li, X. Xu, Y. Zheng, C. Xiao, P. Zerfas, S. Cooperman, M. Eckhaus, T. Rouault , L. Mishra, C. X. Deng, Cell Metab. 2005, 2, 399–409.
R. S. Ohgami, D. R. Campagna, E. L. Greer, B. Antiochos, A. McDonald, J. Chen, J. J. Sharp, Y. Fujiwara, J. E Barker, M. D. Fleming, Nature Genet. 2005, 37, 1264–1269.
M. Yamamoto, O. Nakajima, Int. J. Hematol. 2000, 72, 157–164.
S. Lyoumi, M. Abitbol, V. Andrieu, D. Henin, E. Robert, C. Schmitt, L. Gouya, H. D. Verneuil, J. C. Deybach, X. Montagutelli, C. Beaumont, H. Puy, Blood 2007, 109, 811–818.
S. Al-Mahdawi, R. M. Pinto, D. Varshney, L. Lawrence, M. B. Lowrie, S. Hughes, Z. Webster, J. Blake, J. M. Cooper, R. King, M. A. Pook, Genomics 2006, 88, 580–590.
W. Chen, P. N. Paradkar, L. Li, E. L. Pierce, N. B. Langer, N. Takahashi-Makise, B. B. Hyde, O. S. Shirihai, D. M. Ward, J. Kaplan, B. H. Paw, Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268.
A. Brownlie, A. Donovan, S. J. Pratt, B. H. Paw, A. C. Oates, C. Brugnara, H. E. Witkowska, S. Sassa, L. I. Zon, Nature Genet. 1998, 20, 244–250.
R. A. Wingert, J. L. Galloway, B. Barut, H. Foott, P. Fraenkel, J. L. Axe, G. J Weber, K. Dooley, A. J. Davidson, B. Schmidt, B. H. Paw, G. C. Shaw, P. Kingsley, J. Palis, H. Schubert, O. Chen, J. Kaplan, L. I. Zon, Nature 2005, 436, 1035–1039.
A. Donovan, A. Brownlie, M. Dorschner, Y. Zhou, S. J. Pratt, B. H. Paw, C. B. Thisse, B. Thisse, L. I. Zon, Blood 2002, 100, 4655–4659.
G. C. Shaw, J. J. Cope, L. Li, K. Corson, C. Hersey, G. E. Ackermann, B. Gwynn, A. J. Lambert, R. A. Wingert, D. Traver, N. S. Trede, B. A. Barut, Y. Zhou, E. Minet, A. Donovan, A. Brownlie, R. Balzan, M. J. Weiss, L. L. Peters, J. Kaplan, L. I. Zon, B. H. Paw, Nature 2006, 440, 96–100.
M. Petrou, B. Modell, Prenatal Diagnosis 1995, 15, 1275–1295.
Y. W. Kan, M. S. Golbus, A. M. Dozy, N. Eng. J. Med. 1976, 295, 1165–1167.
Y. W. Kan, A. M. Dozy, Lancet(II) 1978, 910–913.
A. Cao, R. Galanello, M. C. Rosatelli, Clin. Haematol. 1993, 6, 263–286.
C. Hershko, A. M. Konijn, G. Link, Br. J. Haematol. 1998, 101, 399–406.
P. C. Sharpe, D. R. Richardson, D. S. Kalinowski, P. V. Bernhardt, Curr. Top. Med. Chem. 2011, 11, 591–607.
Y. Ma, T. Zhou, X. Kong, R. C. Hider, Curr. Med. Chem. 2012, 19, 2816–2927.
D. C. Harris, P. Aisen, Biochim. Biophys. Acta 1973, 329, 156–158.
S. J. Rodgers, C. W. Lee, C. Y. Ng, K. N. Raymond, Inorg. Chem. 1987, 26, 1622–1625.
K. N. Raymond, C. J. Carrano, Acc. Chem. Res. 1979, 12, 183–190.
R. C. Hider, X. Kong, Nat. Prod. Reports 2010, 27, 637–657.
G. S. Tilbrook, R. C. Hider, in Iron Transport and Storage in Microorganisms, Plants and Animals, Vol. 35 of Metal Ions in Biological Systems, Marcel Dekker, New York, 1998, 691–730.
U. Fagerholm, D. Nilsson, L. Knutson, H. Lennernas, Acta Physiol. Scand. 1999, 165, 315–324.
C. A. Lipinski, F. Lombardo, B. W. Dominy, P. J. Feeney, Adv. Drug Delivery Rev. 1997, 23, 3–25.
R. C. Hider, Toxicol. Lett. 1995, 82–3, 961–967.
R. D. Abeysinghe, P. J. Robert, C. E. Cooper, K. H. MacLean, R. C. Hider, J. B. Porter, J. Biol. Chem. 1996, 271, 7965–7972.
Z. D. Liu, R. S. Kayyali, R. C. Hider, J. B. Porter, A. E. Theobald, J. Med. Chem. 2002, 45, 631–639.
L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo, P. Palumaa, Nature 2010, 465, 645–648.
M. Merkofer, R. Kissner, R.C. Hider, W. H. Koppenol, Helv. Chim. Acta 2004, 87, 3021–3034.
M. R. Summers, A. Jacobs, D. Tudway, P. Perera, C. Ricketts, Br. J. Haematol. 1979, 42, 547–555.
M. J. Pippard, S. T. Callender, D. J. Weatherall, Clin. Sci. Mol. Med. 1978, 54, 99–106.
J. B. Porter, R. D. Abeysinghe, S. Singh, R. C. Hider, Blood, 1996, 88, 705–713.
C. Borgna-Pignatti, M. D. Cappellini, P. De Stefano, G. C. Del Vecchio, G. L. Forni, M. R. Gamberini, R. Ghilardi, A. Piga, M. A. Romeo, H. Zhao, A. Cnaan, Blood, 2006, 107,3733–3737.
M. Y. Moridani, G. S. Tilbrook, H. H. Khodr, R. C. Hider, J. Pharm. Pharmacol. 2002, 54, 349–364.
F. N. Hahn, T. J. McMurry, A. Hugi, K. N. Raymond, J. Am. Chem. Soc. 1990, 112, 1854–1860.
L. C. Wolfe, Sem. Hematol. 1990, 27, 117–120.
R. J. Bergeron, J. Wiegand, W. R. Weimar, J. R. T. Vinson, J. Bussenius, G. W. Yao, J. S. McManis, J. Med. Chem. 1999, 42, 95–108.
R. J. Bergeron, J. Wiegand, J. S. McManis, B. H. McCosar, W. R. Weimar, G. M. Brittenham, R. E. Smith, J. Med. Chem. 1999, 42, 2432–2440.
J. C. Barton, Drugs 2007, 10, 480–490.
R. J. Bergeron, J. Wiegand, N. Bharti, J. S. McManis, S. Singh, BioMetals 2011, 24, 239–258.
H. Y. Rienhott, V. Viprakasit, L. Tay, P. Harmatz, E. Vichinsky, D. Chirnomas, J. L. Kwiatkowski, A. Tapper, W. Kramer, J. B. Porter, E. Neufeld, J. Haematol. 2011, 96, 521–525.
U. Heinz, K. Hegetschweiler, P. Acklin, B. Faller, R. Lattmann, H. P. Schnebli, Angew. Chem. Int. Ed. 1999, 38, 2568–2570.
C. Hershko, A. M. Konijn, H. P. Nick, W. Breuer, Z. I. Cabantchik, G. Link, Blood, 2001, 97, 1115–1122.
M. D. Cappellini, J. Porter, A. El-Beshlawy, C. K. Li, J. F. Seymour, M. Elalfy, N. Gattermann, S. Giraudier, J. W. Lee, L. L. Chan, K. H. Lin, C. Rose, A. Taber, S. W. Thein, V. Viprakasit, D. Habr, G. Domokos, B. Roubert, A. Kattamis, Haematologica, 2010, 95, 557–566.
J. C. Wood, B. P. Kang, A. Thompson, P. Giardina, P. Harmatz, T. Glynos, C. Paley, T. D. Coates, Blood 2010, 116, 537–543.
R. Galanello, A. Piga, D. Alberti, M. C. Rouan, H. Bigler, R. Sechaud, J. Clin. Pharmacol. 2003, 43, 565–572.
C. Rafat, F. Fakhouri, J. A. Ribeil, R. Delarue, M. LeQuintrec, Amer. J. Kidney Dis. 2009, 54, 931–934.
R. C. Hider, G. Kontoghiorghes, J. Silver, M. A. Stockham, G. B. Patent 1984, 2118176.
G. J. Kontoghiorghes, M. A. Aldouri, A. V. Hoffbrand, J. Barr, B. Wonke, T. Kourouclaris, L. Sheppard, Br. Med. J. 1987, 295, 1509–1512.
A. Maggio, G. D. Amico, A. Morabito, M. Capra, C. Ciaccio, P. Cianciulli, F. DiGregorio, G. Garozzo, R. Malizia, C. Magnano, A. Mangiagli, G. Quarta, M. Rizzo, D. G. D`Ascola, A. Rizzo, M. Midiri, Blood Cells, Mol. Dis. 2002, 28, 196–208.
H. Glickstein, R. BenEl, G. Link, W. Breuer, A. M. Konijn, C. Hershko, H. Nick, Z. I. Cabantchik, Blood 2006, 108, 3195 –3203.
D. J. Pennell, V. Berdoukas, M. Karagiorga, V. Ladis, A. Piga, A. Aessopos, E. D. Gotsis, M. A. Tanner, G. C. Smith, M. A. Westwood, B. Wonke, Blood, 2006, 107, 3738 –3744.
C. Hershko, M. D. Cappellini, R. Galanello, A. Piga, G. Tognoni, G. Masera, Br. J. Haematol. 2004, 125, 545–551.
A. Pepe, A. Meloni, M. Capra, P. Cianciulli, L. Prossomariti, C. Malaventura, M. C. Putti, A. Lippi, M. A. Romeo, M. G. Bisconte, A. Filosa, V. Caruso, A. Quarta, L. Pitrolo, M. Missere, M. Midiri, G. Rossi, V. Positano, M. Lombardi, A. Maggio, Haematologica 2011, 96, 41–47.
A. Cohen, R. Galanello, A. Piga, V. Sanctis, F. Tricta, Blood 2003, 102,1583–1587.
A. Ceci, P. Baiardi, M. Felisi, M. D. Cappellini, V. Carnelli, V. De Sanctis, R. Galanello, A. Maggio, G. Mosera, A. Piga, F. Schettini, I. Stefano, F. Tricta, Br. J. Haematol. 2002, 118, 330–336.
R.C. Hider, G. S. Tilbrook, Z. D. Liu, International Patent WO98/54138 (1998).
N. Lowther, P. Fox, B. Faller, H. Nick, Y. Jin, T. Sergejew, Y. Hirschberg, R. Oberle, H. Donnelly, Pharm. Res. 1999, 16, 434 –440.
T. Sergejew, P. Forgiarini, H. P. Schnebli, Br. J. Haematol. 2000, 110, 985–92.
Z. D. Liu, H. H. Khodr, D. Y. Liu, S. L. Lu, R. C. Hider, J. Med.Chem. 1999, 42, 4814–4823.
S. Piyamongkol, Y. M. Ma, X. L. Kong, Z. D. Liu, M. D. Aytemir, D. van der Helm, R. C. Hider, Chem. Eur. J. 2010, 16, 6374–6381.
L. D. Devanur, H. Neubert, R. C. Hider, J. Pharm. Sci. 2008, 97, 1454–1467.
L. D. Devanur, R. W. Evans, P. J. Evans, R. C. Hider, Biochem. J. 2008, 409, 439–441.
P. Evans, R. Kayyali, R. C. Hider, J. Eccleston, J. B. Porter, Transl. Res. 2010, 156, 55–61.
P. J. Giardina, R. W. Grady, Semin. Hematol. 2001, 38, 360–366.
M. E. Lai, R. W. Grady, S. Vacquer, A. Pepe, M. P. Carta, P. Bina, F. Saw, P. Cianciulli, A. Maggio, R. Galanello, P. Farci, Blood Cells Mol. Dis. 2010, 45, 136–143.
V. Berdoukas, G. Chouliaras, P. Moraitis, K. Zannikos, E. Berdoussi, V. Ladis, J. Cardiovasc. Magn. Reson. 2009, 28, 11–20.
W. Hagar, E. Vichinsky, Brit. J. Haematol. 2008, 141, 346–356.
G. Lucania, A. Vitrano, A. Filosa, A. Maggio, Brit. J. Haematol. 2011, 154, 545–555.
E. B. Fung, P. Harmatz, M. Milet, S. K. Ballas, L. DeCastro, W. Hagar, L. Hilliard, W. Owen, N. Olivieri, K. Smith-Whitley, D. Darbari, W. Wang, E. Vichinsky, Amer. J. Hematol. 2007, 82, 255–265.
W. C. Wang, N. Ahmed, M. Hanna, J. Pediat. 1986, 108, 552–557.
E. Vichinsky, F. Bernaudin, G. L. Forni, R. Gardner, K. Hassell, M. M. Heeney, B. Inusa, A. Kutlar, P. Lane, L. Mathias, J. Porter, C. Tebbi, F. Wilson, L. Griffel, W. Deng, V. Giannone, T. Coates, Br. J. Haematol. 2011, 154, 387–397.
M. Jaeger, C. Anul, D. Sohngen, U. Germing, W. Scheier, Drugs Today 1992, 28, 143–147.
G. Metzgeroth, D. Dinter, B. Schultheis, A. Dorn-Beineke, K. Lutz, O. Leismann, R. Hehlmann, J. Hastka, Ann. Hematol. 2009, 88, 301–310.
N. Galtermann, C. Finelli, M. D. Porta, P. Fenaux, A. Ganser, A. Guerci-Bresler, M. Schmid, K. Taylor, D. Vassilieff, D. Habr, G. Domokos, B. Roubert, C. Rose, Leuk. Res. 2010, 34, 1143–1150.
O. Kakhlon, H. Manning, W. Breuer, N. Melamed-Book, C. Lu, G. Cortopassi, A. Munnich, Z. I. Cabantchik, Blood, 2008, 112, 5219–5227.
A. Gaeta, R. C. Hider, Brit. J. Pharmacol. 2005, 146, 1041–1059.
K. L. Duble, M. Gerlach, V. Schunemann, A. X. Trautwein, L. Zecca, M. Gallorini, M. B. Youdim, P. Riederer and D. Ben-Shachar, Biochem. Pharmacol. 2003, 66, 489–494.
M. T. Núñez, P. Urrutia, N. Mena, P. Aguirre, V. Tapia, J. Salazar, BioMetals 2012, 25, 761–776.
M. T. Núñez, V. Gallardo, P. Muñoz, V. Tapia, A. Esparza, J. Salazar, H. Speisky, Free Radic. Biol. Med. 2004, 37, 953–960.
R. Lill, U. Mühlenhoff, Annu. Rev. Biochem. 2008, 77, 669–700.
A. H. Schapira, Lancet 2006, 368, 70–82.
K. Blennow, M. J. de Leon, H. Zetterberg, Lancet 2006, 368, 387–403.
R. C. Hider, Y. M. Ma, F. Molina-Holgado, A. Gaeta, S. Roy, Biochem. Soc. Trans. 2008, 36, 1304–1308.
F. Bousejra-ElGarah, C. Bijani, Y. Coppel, P. Faller and C. Hureau, Inorg. Chem. 2011, 50, 9024–9030.
M. A. Smith, P. L. R. Harris, L. M. Sayre, G. Perry, Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868.
A. Kontush, Free Radic. Biol. Med. 2001, 31, 1120–1131.
J. A. Duce, A. Tsatsanis, M. A. Cater, S. A. James, E. Robb, K. Wikhe, S. L. Leong, K. Perez, T. Johanssen, M. A. Greenough, H. Cho, D. Galatis, R. D. Moir, C. L. Masters, C. McLean, R. E. Tanzi, R. Cappai, K. J. Barnham, G. D. Ciccotosto, J. T. Rogers, A. I. Bush, Cell 2010, 142, 857–867.
P. Lei, S. Ayton, D. I. Finkelstein, L. Spoerri, G. D. Ciccotosto, D. K. Wright, B. X. W. Wong, P. A. Adlard, R. A. Cherny, L. Q. Lam, B. R. Roberts, I. Volitakis, G. F. Egan, C. A. McLean, R. Cappai, J. A. Duce, A. I. Bush, Nature Medicine 2012, 18, 291–296.
J. F. Schenck, E. A. Zimmerman, Z. Li, S. Adak, A. Saha, R. Tandon, K. M. Fish, C. Belden, R. W. Gillen, A. Barba, D. L. Henderson, W. Neil, T. O’Keefe, Top. Magn. Reson. Imaging 2006, 17, 41–50.
H. Akatsu, A. Hori, T. Yamamoto, M. Yoshida, M. Mimuro, Y. Hashizume, I. Tooyama, E. M. Yezdimer, BioMetals 2012, 25, 337–350.
C. E. Clarke, Brit. Med. J. 2007, 335, 441–445.
Y. Agid, Lancet 1991, 337, 1321–1324.
P. Jenner, Acta Neurol. Scond. 1991, 84 Suppl. 136, 6–15.
P. Jenner, Lancet, 1994, 344, 796–798.
D. T. Dexter, F. R. Wells, F. Agid, Y. Agid, A. J. Lees, P. Jenner, C. D. Marsden, Lancet 1987, 330, 1219–1220.
E. Sofic, P. Riederer, H. Heinsen, H. Beckmann, G. P. Reynolds, G. Hebenstreit, M. B. Youdim, J. Neural. Transm. 1988, 74, 199–205.
J. Hallervorden, H. Spatz, Z. Ges. Neurol. Psychiat. 1922, 79, 254–302.
S. J. Hayflick, J. M. Penzien, W. Michl, U. M. Sharif, N. P. Rosman, P. G. Wheeler, Pediatric Neurology 2001, 25, 166–169.
A. Gregory, S. J. Hayflick, Folia Neuropathologica 2005, 43, 286–296.
N. Gordon, Eur. J. Pediatr. Neurol. 2002, 6, 243–247.
T. L. Perry, M. G. Norman, V. W. Yong, S. Whiting, J. C. Crichton, S. Hansen, S. J. Kish, Ann. Neurol. 1985, 482–489.
R. Klein, Q. Wang, B. E. K. Klein, S. E. Moss, S. M. Meuer, Invest. Ophthalmol. Vis. Sci. 1995, 36, 182–191.
M. A. Zarbin, Arch. Ophthalmol. 2004, 122, 598–614.
P. Haly, A. H. Milam, J. L. Dunaief, Arch. Ophthalmol. 2003, 121, 1099–1105.
M. Hadziahmetovic, Y. Song, N. Wolkow, J. Iacovelli, L. Kautz, M. P. Roth, J. L. Dunaief, Amer. J. Pathol. 2011, 179, 335–348.
M. Hadziahmetovic, Y. Song, P. Ponnuru, J. Iacovelli, A. Hunter, N. Haddad, J. Beard, J. R. Connor, S. Vaulont, J. L. Dunaief, Invest. Ophthalmol. Vis. Sci. 2011, 52, 109–118.
X. He, P. Hahn, J. Iacovelli, R. Wong, C. King, R. Bhisitkul, M. Massaro-Giordano, J. L. Dunaief, Prog. Retin. Eye Res. 2007, 26, 649–673.
T. Zhou, Y. Ma, X. Kong, R. C. Hider, Dalton. Trans. 2012, 41, 6371–6389.
D. T. Dexter, S. A. Statton, C. Whitmore, W. Freinbichler, P. Weinberger, K. F. Tipton, L. Della Corte, R. J. Ward, R. R. Crichton, J. Neural. Transm. 2011, 118, 223–231.
M. Hadziahmetovic, Y. Song, N. Wolkow, J. Iacovelli, S. Grieco, J. Lee, A. Lyubarsky, D. Pratico, J. Connelly, M. Spino, Z. L. Harris, J. L. Dunaief, Invest. Ophthalmol. Vis. Sci. 2011, 52, 959–968.
G. L. Forni, M. Balocco, L. Cremonesi, G. Abbruzzese, R. C. Parodi, R. Marchese, Movement Disorders 2008, 23, 904–907.
G. Abbruzzese, G. Cossu, M. Balocco, R. Marchese, D. Murgia, M. Melis, R. Galanello, S. Barella, G. Matta, U. Ruffinengo, U. Bonuccelli, G. L. Forni, Haematologica 2011, 96, 1708–1711.
A. I. Bush, Neurobiology of Aging 2002, 23, 1031–1038.
H. Zheng, L. M. Weiner, O. Bar-Am, S. Epsztejn, Z. I. Cabantchik, A. Warshawsky, M. B. H. Youdim, M. Fridkin, Bioorg. Med. Chem. 2005, 13, 773–783.
H. Zheng, D. Blat, M. Fridkin, J. Neural. Trans. 2006, 71, 163–172.
F. Molina-Holgado, A. Gaeta, P. T. Francis, R. J. Williams, R. C. Hider, J. Neurochem. 2008, 105, 2466–2476.
Y. Avramovich-Tirosh, T. Amit, O. Bar-Am, H. Zheng, M. Fridkin, M. B. Youdim, J. Neurochem. 2007, 100, 490–502.
R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters, A. I. Bush, Neuron 2001, 30, 665–676.
C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q. X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi, C. L. Masters, Arch. Neurol. 2005, 60, 1685–1691
D. Kaur, F. Yantiri, S. Rajagopalan, J. Kumar, J.Q. Mo, R. Boonplueang, V. Viswanath, R. Jacobs, L. Yang, M. F. Beal, D. DiMonte, I. Volitaskis, L. Ellerby, R. A. Cherny, A. I. Bush, J. K. Andersen, Neuron 2003, 37, 899–909.
University Hospital Lille Trial (FAIR-PARK1), http://clinicaltrials.gov/ct2/show/NCT00943748?term=FAIR-PARK-I&rank=1
Imperial College London Trial (DeferipronPD), http://clinicaltrials.gov/ct2/show/NCT01539837?term=deferiprone&rank=4
D. Song, Y. Song, M. Hadziahmetovic, Y. Zhong, J. L. Dunaief, Free Rad. Biol. Med. 2012, 53, 64–71.
T.P. Kruck, J.G. Cui, M.E. Percy, W.J. Lukiw, Cell Mol. Neurobiol. 2004, 24, 443–459.
H. Schugar, D. E. Green, M. L. Bowen, L. E. Scott, T. Storr, K. Bohmerle, F. Thomas, D. D. Allen, P. R. Lockman, M. Merkel, K. H. Thompson, C. Orvig, Angew. Chem. Int. Ed. 2007, 46, 1716–1718.
R. C. Hider, S. Roy, Y. M. Ma, X. L. Kong, J. Preston, Metallomics 2011, 3, 239–249.
G. Liu, P. Men, W. Kudo, G. Perry, M. A. Smith, Neurosci. Lett. 2009, 455, 187–190.
D. Bebbington, C. E. Dawson, S. Gaur, J. Spencer, Bioorg. Med. Chem. Lett. 2002, 12, 3297–3300.
H. Zheng, M. B. H. Youdim, M. Fridkin, J. Med. Chem. 2009, 52, 4095–4098.
L. R. Perez, K. J. Franz, Dalton Trans. 2010, 39, 2177–2187.
S. V. Torti, F. M. Torti, Cancer Res. 2011, 71, 1511–1514.
R. G. Stevens, B. I. Graubard, M. S. Micozzi, K. Neriishi, B. S. Blumberg, Int. J. Cancer 1994, 56, 364–369.
A. G. Smith, J. E. Francis, P. Carthew, Carcinogenesis 1990, 11, 437–444.
L. D. Miller, L. G. Coffman, J. W. Chou, M. A. Black, J. Bergh, R. D’Agostino, Jr., S. V. Torti, F. M. Torti, Cancer Res. 2011, 71, 6728–6737.
S. Nekhai, V. R. Gardeuk, in Iron Physiology and Pathophysiology in Humans, Eds G. J. Anderson, G. D. McLaren, Humana Press, New York, 2012, 477–495.
D. B. Lovejoy, D. R. Richardson, Curr. Med. Chem. 2003, 10, 1035–1049.
H.M. Hanauske-Abel, M.-H. Park, A.-R. Hanauske, A.M. Popowicz, M. Lalande, J.E. Folk, Biochim. Biophys. Acta, 1994, 1221, 115–124.
C. Hershko, Mol. Asp. Med. 1992, 13, 113–165.
R. J. Bergeron, Trends Biol. Sci. 1986, 11, 133–136.
J. Gao, D. R. Richardson, Blood 2001, 98, 842–850.
J. Yuan, D. B. Lovejoy, D. R. Richardson, Blood 2004, 104, 1450–1458.
D. Fu, D. R. Richardson, Blood 2007, 110, 752–761.
E. D. Weinberg, Biochim. Biophys. Acta 2009, 1790, 600–605.
N. H. Carbonetti, S. Boonchai, S. H. Parry, V. Vaisanenrhen, T. K. Korhonen, P. H. William, Infection and Immunity 1986, 51, 966–968.
S. Ford, R. A. Cooper, R. W. Evans, R. C. Hider, P. H. Williams, Eur. J. Biochem. 1988, 178, 477–481.
I. A. García-Montoya, T. S. Cendón, S. Arévalo-Gallegos, Q. Rascón-Cruz, Biochim. Biophys. Acta 2012, 1820, 226–236.
K. N. Raymond, E. A. Dertz, S. S. Kim, Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588.
M. A. Holmes, W. Paulsene, X. Jide, C. Ratledge, R. K. Strong, Structure 2005, 13, 29–41.
J. R. Boelaert, S. J. Vandecasteele, R. Appelberg, V. R. Gordeuk, J. Infect. Dis. 2007, 195, 1745–1753.
R. C. Hider, Z. Liu, J. Pharm. Pharmacol. 1997, 49, 59–64.
G. F. Mabeza, Pharmacol. Ther. 1999, 81, 53–75.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Hider, R.C., Kong, X. (2013). Iron: Effect of Overload and Deficiency. In: Sigel, A., Sigel, H., Sigel, R. (eds) Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences, vol 13. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7500-8_8
Download citation
DOI: https://doi.org/10.1007/978-94-007-7500-8_8
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7499-5
Online ISBN: 978-94-007-7500-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)