
Abstract
All currently approved antidepressant medications for major depressive disorder (MDD) and bipolar disorder act primarily on the monoaminergic system and have varying affinities for serotonergic, norepinephrine-ergic, and/or dopaminergic receptors. Unfortunately, these drugs are only effective in approximately two thirds of patients. Glutamate is the major excitatory neurotransmitter in the central nervous system, and the glutamatergic system has been implicated in the pathophysiology of MDD. Here, we review the putative involvement of the glutamate receptor subtypes—N-methyl-d-aspartate (NMDA), α-amino-3-hydroxyl-5-methyl-4-isoxazoleproprionic acid (AMPA), kainate, and the group I, II, and III metabotropic glutamate receptors (mGluRs)—in the development of novel and more effective treatments for mood disorders as well as preclinical and clinical trials of drugs targeting these receptors. The rapid and robust antidepressant effects of ketamine—an NMDA receptor antagonist—have been consistently replicated in multiple trials. Other glutamatergic drugs have been investigated with varying success. Here, we highlight some of the most interesting results, including the following: (1) repeated oral, intramuscular, and sublingual ketamine appears to be less robustly effective than intravenous ketamine, but also causes fewer significant adverse effects; (2) the glycine partial agonist GLYX-13 appears to be effective both as monotherapy and adjunctive treatment in the treatment of MDD. An oral analogue, NRX-1074, is currently under investigation; and (3) mGluR modulators targeting mGluR5 have demonstrated convincing preclinical results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is the most common psychiatric disorder in developed countries, with an estimated prevalence of nearly 17 % [1, 2]. In 2004, the World Health Organization (WHO) ranked it third among the leading causes of global disease burden [3]. Currently available conventional antidepressants, which target monoamines (dopamine, norepinephrine, and/or serotonin), unfortunately have low rates of treatment response. While one third of MDD patients respond to these agents, approximately two thirds will respond only after trying several classes of antidepressants. Such patients display treatment-resistant depression (TRD); its definition has evolved to encompass failure to respond to at least two antidepressants from different pharmacological classes and of adequate dose and duration [4].
In the early 1990s, a seminal study using a rodent behavioral despair model of depression first demonstrated that the N-methyl-d-aspartate (NMDA) receptor complex played a major role in antidepressant action [5]. Nearly a decade later, the first report of rapid antidepressant effects associated with the NMDA receptor antagonist ketamine in patients with MDD (n = 7) was published [6]. Since 2006, when ketamine began to be systematically studied [7]—particularly in TRD—the number of studies investigating ketamine as well as other glutamatergic modulators in MDD has multiplied exponentially. Here, we review the classification and characteristics of central glutamate receptors and current hypotheses regarding how NMDA receptor antagonism results in rapid and robust antidepressant response. This paper also briefly reviews novel developments with regard to ketamine for the treatment of mood disorders, and describes recent preclinical and clinical studies of various glutamate receptor modulators in the treatment of TRD.
Classification and Characteristics of Glutamate Receptors
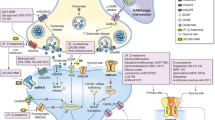
Glutamate is the most abundant excitatory amino acid neurotransmitter in the central nervous system. Glutamate receptors have historically been divided into two classes—ionotropic and metabotropic—based on their biochemical properties and functions (see Fig. 1) [8]. Ionotropic receptors include NMDA, α-amino-3-hydroxyl-5-methyl-4-isoxazoleproprionic acid (AMPA), and kainate receptors. Metabotropic receptors (mGluRs) are categorized into three groups (group I, group II, and group III). All ionotropic glutamate receptors are located postsynaptically, so agonist binding at these receptors results in local excitation and the generation of action potential. The NMDA receptor primarily fluxes Ca2+ when activated by the binding of the obligatory co-agonists glutamate and glycine. On the other hand, AMPA and kainate receptors are primarily Na+ flux channels activated by the binding of glutamate only. mGluRs differ from ionotropic receptors in that they are also located presynaptically. The group I mGluRs are mostly postsynaptic, while groups II and III have predominantly presynaptic localization. Group I mGluRs include mGluR1 and mGluR5, group II mGluRs include mGluR2 and mGluR3, and group III mGluRs include mGluR4, mGluR6, mGluR7, and mGluR8. All are G-protein coupled receptors that produce intracellular action through downstream signal transduction/secondary messenger cascades and, hence, often result in slower and longer lasting modulatory tone. In contrast to ionotropic receptors, the net effect of mGluR activation can be either excitatory or inhibitory. Group I receptor stimulation generally enhances glutamatergic neurotransmission while group II and II receptors dampen excitatory tone.

Glutamate receptor subtypes and novel drugs
In theory, glutamatergic signaling can be modulated by regulating postsynaptic signaling, presynaptic glutamate release, or synaptic glutamate concentrations [9•]. These actions can be accomplished by designing drugs that (1) directly activate the receptors; (2) change the available synaptic concentrations of natural ligands such as glutamate, glycine (or D-serine), or other ions; or (3) change the affinity and efficacy of the receptors to their natural ligands. Drugs that elicit these actions include NMDA receptor antagonists, NMDA receptor subunit (especially NR2B) antagonists, NMDA receptor glycine-site partial agonists, group I and II mGluR antagonists, and negative allosteric modulators (NAMs); all have been studied in clinical trials for MDD.
Two main hypotheses—largely drawn from the preclinical literature—are thought to explain why NMDA receptor antagonism has rapid and robust antidepressant effects. The first hypothesis proposes that the primary event is NMDA receptor blockade on cortical gamma aminobutyric acid (GABA)ergic interneurons that normally provide tonic inhibitory tone on cortical excitatory pyramidal output neurons. The antagonism at these receptors, therefore, would decrease inhibitory tone [10] with the net result of increased excitatory glutamate release, AMPA receptor activation, and downstream synaptogenesis and dendritic remodeling [11]. These changes have been associated with antidepressant effects in pre-clinical behavioral models [12, 13]. The second hypothesis suggests that the downstream action of direct NMDA antagonism is the inactivation of eukaryotic elongation factor (eEF2) kinase. This results in dephosphorylation and activation of eEF2, which then stimulates local protein translation and release in dendrites, including brain-derived neurotrophic factor (BDNF). This increase in BDNF is then associated with synaptogenesis and neurogenesis [10, 14–16] (see Fig. 2).

Hypothesized mechanisms of action of glutamate in treatment-resistant depression (TRD)
Drugs Targeting Ionotropic Glutamate Receptors
NMDA Receptor Non-competitive Antagonists
Ketamine
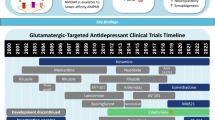
Ketamine has been used safely for decades as a dissociative anesthetic. Since 2000, several clinical trials have demonstrated ketamine’s rapid and robust antidepressant effects in MDD. One study found that a single subanesthetic ketamine infusion (0.5 mg/kg) for 40 min resulted in a 14-point decrease in Hamilton Depression Rating Scale (HAM-D) score 72 h post-infusion [6]. Another study noted that the same dose of ketamine was associated with a 71 % response rate and a 29 % remission rate within 24 h [7]. The initial pioneering studies were associated with several concerns, including the integrity of a saline solution placebo, the abuse potential of ketamine, and difficulties in translation to real-world use. However, subsequent trials addressed some of these concerns (see below).
Murrough and colleagues addressed the issue of integrity of the blind by substituting the inert saline with a psychoactive placebo [17•]. Midazolam was used because of its rapid sedative and anxiolytic effects. It should be noted, however, that the blinding was not perfect because midazolam does not induce the same dissociative, psychotomimetic, and hemodynamic effects as ketamine. Patients had a greater antidepressant response to ketamine than midazolam (64 vs. 28 %, respectively) 24 h post-infusion. The time response curve over the course of the week was nearly identical to that observed with earlier ketamine studies [17•].
Initial studies noted that antidepressant response to a single ketamine infusion—though robust—was transient, lasting approximately up to 1 week in responders. Furthermore, the neurotoxic effects of chronic exposure to ketamine and other NMDA antagonists (e.g., phencyclidine (PCP)) raised concerns regarding the safety of repeated ketamine infusions and/or its long-term use. One study investigated 24 unmedicated patients with TRD who received six repeated ketamine infusions (0.5 mg/kg over 40 min) over 12 days. At the end of 2 weeks, the response rate among patients was 70.8 % [18•], and only mild and transient side effects were associated with the repeated infusions. Mean time to relapse after the last infusion was 18 days but, in about 30 % of responders, antidepressant response lasted until the end of the naturalistic observation phase (83 days after the last infusion). Another open-label study of 13 MDD patients receiving six ketamine infusions noted similar results [19]. Eleven subjects (92 %) responded to ketamine and 60 % remitted. Of the responders, six subjects relapsed, on average 16 days after the last infusion (range 7 to 28 days). Four weeks after the last infusion, five patients had still not relapsed. In addition, visual and working memory performance was also improved [19]. When the same amount of total ketamine was infused at a lower concentration for a longer period of time (0.3 mg/kg over 100 min), the side effect profile did not worsen significantly (although there was no side-by-side comparison with the typical ketamine dose to confirm this) [20]. Another study looked at weekly vs. biweekly ketamine infusions (0.5 mg/kg × 40 min) over 6 weeks (six infusions vs. three infusions, total) [21]. In 28 medicated patients with MDD and bipolar (BD) depression, remission rates of 29 and 14 %, respectively, were observed with no added cognitive difficulties [21]. During the 6-month naturalistic follow-up, a broad range in time-to-relapse was observed (approximately 70 days). Finally, in a dose-finding, placebo-controlled, pilot study of four TRD subjects, two patients experienced an antidepressant response to 0.1 mg/kg of ketamine infused over 2 to 5 min; both relapsed within 1 week [22]. The higher ketamine dose was associated with greater antidepressant response, and infusion over 2 min was associated with elevated psychotomimetic side effects compared to the longer infusion.
In terms of clinical practice, ketamine infusions are labor- and resource-intensive, which may render them impractical in many clinical psychiatric settings, particularly office-based private practice. Less intensive modes of administration are therefore an attractive alternative. Intranasal [23•], intramuscular [24], sublingual [25•], and oral [26–29] alternatives have all been explored. In a randomized, double-blind, crossover study of intranasal ketamine (50 mg) vs. saline, 20 MDD patients showed a seven-point reduction in mean Montgomery Åsberg Depression Rating Scale (MADRS) score at 24 h post-inhalation [23•]. Intranasal ketamine produced only minor hemodynamic, dissociative, and psychotomimetic effects. However, no antidepressant effects were noted compared to placebo 72 h post-administration.
Intramuscular ketamine has 93 % bioavailability compared to intravenous ketamine, and a small study of two female patients found that it had dose-dependent antidepressant effects [30]. In another case series, two patients with BD-II depression achieved symptom reduction for 4 to 6 months after intravenous ketamine followed by repeated intramuscular injections (from 32 to 100 mg/kg every 3 to 4 days over several months) [24]. Side effects included irritability, headaches, nightmares, and dissociation. Complications associated with chronic ketamine abuse (e.g., increased intracranial pressure, muscle rigidity, or cystitis) were not observed [24].
Sublingual ketamine has 30 % bioavailability compared to intravenous ketamine. In 27 depressed MDD and BD outpatients, variable administration (every 2 to 7 days) of add-on, escalating dose sublingual ketamine had antidepressant effects in 20 patients (77 %) [25•]. Sublingual ketamine was well tolerated, with no reported psychotomimetic effects; the most common side effect was transient lightheadedness.
Only two small cases studies of oral ketamine—whose bioavailability is approximately 20 % of intravenous ketamine’s—have been conducted [27]. Eight depressed hospice patients received oral ketamine (0.5 mg/kg) for 28 days and reported significant antidepressant and anxiolytic effects [28]. A major limitation of the aforementioned studies with alternative modes of administration is that ketamine levels, as well as levels of bioactive metabolites such as hydroxynorketamine, were not measured and/or reported. Such indicators are critical for contrasting the results with those from intravenous infusions.
Esketamine
Esketamine (S-enantiomer of ketamine) has two- to threefold higher potency than racemic (R- and S-) ketamine [31]. At the 2014 International College of Neuropharmacology meeting, Janssen presented results from a double-blind, randomized, placebo-controlled trial of intravenous esketamine (0.2 vs. 0.4 mg/kg vs. placebo) in 30 TRD patients. Both doses of esketamine had similar antidepressant effects (0.2 mg/kg, −16.8 MADRS points over 2 days vs. 0.4 mg/kg, −16.9 MADRS points) in contrast to a mean 3.8-point reduction on the MADRS for individuals receiving placebo. Although not compared head-to-head, the results with esketamine were equivalent to those obtained with the racemic mixture. Common side effects included headache, nausea, and dissociation. The prevalence and severity of these side effects were not reported. As with the racemic mixture, oral esketamine’s clinical utility may be hampered by its poor oral bioavailability. Intranasal esketamine is undergoing current clinical trials for safety and efficacy in TRD (clinicaltrials.gov identifier: NCT01998958).
Nitrous Oxide
Nitrous oxide (N2O) is an inhaled general anesthetic commonly used in dentistry and obstetrics due to its ability to non-competitively inhibit NMDA receptors. In a double-blind, placebo-controlled, crossover study, 20 patients with TRD received a 1-h inhalation of 50 % N2O or 50 % nitrogen as placebo [32•]. Compared to placebo, N2O at both 2 h (−4.8 points, p = 0.002) and 24 h (− 5.5 points, p < 0.001) post-inhalation significantly improved outcome as assessed by the primary depression measure, the 21-item HAM-D. Adverse effects included nausea/vomiting, headache, and anxiety. Although no standardized scale was administered to evaluate psychotomimetic effects, none of the participants subjectively reported these symptoms. It should be noted, however, that as with the ketamine studies, 50 % nitrogen gas may be an inadequate placebo, given that N2O causes acute euphoria and has a sweet smell and taste not found in nitrogen gas. N2O’s antidepressant efficacy was not as robust as that of intravenous ketamine; specifically, N2O was associated with a 20 % response rate and a 15 % remission rate 24 h post-inhalation, compared with response and remission rates of 71 and 29 %, respectively, for ketamine [7]. Nevertheless, this trial adds credence to the idea that NMDA receptor antagonism is a major mechanism of action in rapid antidepressant response. In addition, N2O has a broad mechanism of action similar to that of ketamine. As an example, N2O induces corticotropin-releasing hormone, which activates opioidergic neurons, and indirectly inhibits GABAergic interneurons [33].
Dextromethorphan
Like ketamine and N2O, dextromethorphan is a non-competitive NMDA receptor antagonist. One 12-week randomized, double-blind, placebo-controlled trial of dextromethorphan as adjunctive therapy studied 309 subjects with BD taking valproic acid and 123 healthy subjects. No significant group differences were seen between groups as assessed by mean Young Mania Rating Scale (YMRS) score and HAM-D score [34]. Interestingly, blood concentrations for the group receiving 60 mg/day of dextromethorphan were 65.2 ± 15.1 ng/mL, which is equivalent to 0.24 μM; however, studies have reported that 4 μM peripheral dextromethorphan is required to occupy 50 % of central NMDA receptors [35], suggesting that the negative result may have been attributable to insufficient blood concentration. Another retrospective chart review of 22 patients with either BD-II or BD not otherwise specified (BD-NOS) found that the addition of 20 mg dextromethorphan and 10 mg quinidine (a cytochrome 2D6 inhibitor) once or twice daily to the current medication regimen significantly improved Clinical Global Impression (CGI) score by 1.66 (1 = slightly improved, 2 = much improved) [36]. This dextromethorphan-quinidine combination, Nuedexta, which is approved for the treatment of pseudobulbar affect, is now being studied for TRD (ClinicalTrials.gov identifier: NCT01882829). No published studies have explored dextromethorphan as depression monotherapy.
Memantine
Memantine is an FDA-approved treatment for moderate-to-severe Alzheimer’s-like dementia. Clinical and preclinical studies of memantine for the treatment of depression, however, have shown mixed results. One 8-week, double-blind, placebo-controlled study of MDD patients found that memantine (5–20 mg/day) did not separate from placebo on the primary antidepressant measure [37]. In another 8-week, randomized, placebo-controlled trial of 31 MDD outpatients, adjunctive memantine (5–20 mg/day) did not separate from placebo on the primary outcome measure, the MADRS [38]. In contrast, a 3-year, naturalistic, mirror-image assessment of 30 BD patients found that adjunctive memantine (20–30 mg/day) significantly decreased total time spent manic or depressed, decreased global severity of symptoms, shortened duration of illness, and reduced number of episodes per year [39•]. Finally, an in vitro electrophysiological study found that memantine, unlike ketamine, did not phosphorylate eEF2 kinase or augment BDNF production. As noted previously, both are hypothesized to be major mechanisms of antidepressant action in NMDA receptor antagonists [40].
Lanicemine (AZD6765)
The proprietary low-trapping NMDA receptor antagonist lanicemine has been and continues to be studied in TRD. A randomized, placebo-controlled, crossover study found that a single infusion of lanicemine 150 mg resulted in a 32 % response rate (7/22) as compared to 15 % (3/22) for placebo [41]. Lanicemine was not associated with increased psychosis or dissociation, but its antidepressant effect was transient; it separated from placebo only at 80 and 110 min post-infusion. In a 3-week, placebo-controlled trial, six repeated infusions of lanicemine (100 or 150 mg) adjunctive to current treatment had significant antidepressant effects without ketamine-like side effects [42]. However, in a 6-week phase IIb study, repeated-dose (50 and 150 mg) lanicemine failed to separate from placebo (39 % placebo response rate at trial end) [43].
Subunit-Selective (NR2B) NMDA Receptor Antagonists
The NMDA receptor complex is a tetramer composed of two GluN1 (NR1) subunits and two GluN2 (NR2) subunits. The pharmacological and biophysical properties of the NMDA receptor are largely controlled by the NR2 subunit [44]. Glutamate binds to NR2 while glycine or D-serine binds to the NR1 and NR3 subunits [45, 46]. In an in vivo study of GluN2B knockout in cortical principal neurons, mice showed depression-like behaviors in the tail suspension and open field tests. These knockout mice also did not respond to ketamine; mammalian target of rapamycin (mTOR) signaling and synaptic protein synthesis were also blocked in the knockout mice [47]. Interestingly, a statistically significant difference in the rs1805502 polymorphism was noted within GRIN2B, the gene encoding GluN2B (NR2B), in a study looking at 178 subjects with TRD, 612 subjects with non-TRD, and 779 healthy controls [48]. Several NR2B subunit selective antagonists (reviewed below) have been explored in the treatment of MDD.
Ro 25-6981
In rodents exposed to unpredictable foot shock for 21 days, Ro 25-6981 injection reversed despair-like behaviors, as assessed via the sucrose preference test and novelty suppressed feeding test, which are thought to mirror symptoms of anhedonia and anxiety [13]. To date, no published clinical trials of Ro 25-6981 in MDD have been conducted.
Traxoprodil (CP101-606)
In a randomized, double-blind, placebo-controlled study, the intravenous NR2B selective antagonist CP-101606 had significant antidepressant effects (60 % response rate compared to 20 % in the placebo group), and 78 % of treatment responders maintained this antidepressant effect for at least 1 week [49]. Unfortunately, further development of the drug was stopped due to potential cardiovascular toxicity (specifically, QTc prolongation). CP-101606 has also demonstrated PCP-like reinforcing effects in preclinical models of drug discrimination and self-administration and, hence, abuse potential [50].
MK-0657
The oral NR2B antagonist MK-0657 (4–8 mg/day) was tested for antidepressant efficacy in a randomized, double-blind, placebo-controlled, cross-over pilot study. Only 5 of 21 planned subjects completed the study because the drug manufacturer discontinued its production. MK-0657 monotherapy had antidepressant effects only on the secondary outcome measures, and no psychotomimetic side effects were reported [51]. Cerecor continues to develop this compound under the name CERC-301. Recently, a randomized, double-blind, placebo-controlled sequential parallel study of adjunctive CERC-301 was completed in patients with severe depression and recent active suicidal ideation (ClinicalTrials.gov identifier: NCT01941043); these results are not yet available.
NMDA Receptor Glycine Site Modulators
As mentioned above, the NMDA receptor requires co-agonist binding of glutamate and glycine for its activation. Glycine and D-serine bind to NR1 and NR3 while glutamate binds to NR2.
D-cycloserine
D-cycloserine (DCS), a broad-spectrum antibiotic, is a partial agonist at the NMDA receptor’s glycine site and a functional NMDA receptor antagonist at doses ≥100 mg/day [52]. In an initial 6-week, placebo-controlled, cross-over trial of 250 mg/day as adjunctive treatment in TRD, DCS failed to separate from placebo (p = 0.51) [53]. The same group recruited more patients (n = 26) into their study to assess the efficacy of escalating-dose DCS (up to 1000 mg/day) [54]. Indeed, high-dose DCS had significant antidepressant effects as measured by the HAM-D (p = 0.005) and self-reported Beck Depression Inventory (BDI) (p = 0.046). It was well tolerated and caused no psychotomimetic effects. An 8-week, open-label trial of DCS in patients with schizophrenia or BD, followed by a 6-week, double-blind, placebo-controlled study is ongoing (ClinicalTrials.gov identifier: NCT02304432). This study is specifically looking at subjects who have four copies of the glycine decarboxylase gene mutation that results in unusually low baseline glycine levels [55]. The study will explore both the clinical effectiveness of DCS as well as the effects of this mutation on glycine metabolism in the brain.
GLYX-13
GLYX-13 is a glycine site partial agonist with a short half-life (7 min) that readily crosses the blood–brain barrier. In preclinical studies, both peripheral administration and direct infusion of GLYX-13 into the medial prefrontal cortex had antidepressant-like effects without psychotomimetic effects. During a placebo-controlled safety and efficacy trial of GLYX-13, 33 unmedicated TRD inpatients were randomized. Subjects who received 5 or 10 mg/kg of GLYX-13 displayed a significant antidepressant response while those who received either 1 or 30 mg/kg did not separate from the placebo group [56•]. The manufacturers of GLYX-1, Naurex Inc., will be conducting an open-label extension study for 36 weeks to examine the potential long-term side effects of repeated GLYX-13 infusion in individuals with MDD (ClinicalTrials.gov identifier: NCT02192099).
NRX-1074
This compound is an analogue of GLYX-13 that is reportedly several thousand-fold more potent at the NR1 glycine partial agonist site with potentially minimal CNS side effects [57]. A randomized, double-blind, placebo-controlled efficacy and safety study of a single dose of intravenous NRX-1074 is currently ongoing (Clinicaltrials.gov identifier: NCT02067793), and there are plans to develop this compound for oral administration.
AMPA Receptor Agonists
As discussed above, ketamine’s antidepressant efficacy is hypothesized to occur via glutamate-induced AMPA receptor stimulation with downstream effects on signal transduction/second messenger cascades and transcription/translation of synaptic and neurogenic proteins [58]. Pretreating rodents with the AMPA receptor antagonist NBQX mitigated the antidepressant-like effects of ketamine, MK-801 (another noncompetitive NMDA receptor antagonist), and Ro 25-6981 (an NR2B subunit-selective antagonist) [59–63]. One of the potential benefits of AMPA receptor modulators is that they likely lack the psychotomimetic and dissociative side effects observed with direct NMDA receptor antagonists. Despite the substantial amount of evidence from preclinical studies that AMPA receptor activation is implicated in ketamine’s rapid antidepressant response, few positive clinical findings exist. This may be because (1) AMPA receptors are heterogeneous, due to the combination of various subunits (GluR1-4); (2) they are further diversified by splice variants; and (3) posttranslational modifications and the presence of accessory proteins make it difficult to create in vitro models in the laboratory [64].
ORG-26576
ORG-26576 is an AMPA receptor positive allosteric modulator (PAM) that demonstrated a dose-dependent antidepressant response in a small (n = 30), randomized, placebo-controlled trial [65]. Although the medication groups (100 or 400 mg bid) showed greater change from MADRS baseline relative to placebo, the results were highly variable and there was no consistent pattern of response in drug vs. placebo groups throughout the 28-day period. Nevertheless, the higher ORG-26576 dose was associated with improved information processing speed and executive functioning.
Drugs Targeting Metabotropic Glutamate Receptors
The available drugs that modulate mGluRs are often allosteric modulators that change the affinity of natural ligands. As these modulators typically “fine-tune” receptor activity, one would theoretically expect fewer side effects than those associated with full agonist or antagonist binding [66].
Group I mGluR NAMs
mGluR5 is extrasynaptically/perisynaptically localized at postsynaptic densities, and its activation potentially limits excitotoxic glutamate synaptic spillover. In mood disorders research, mGluR5 has been preferentially targeted over mGluR1 receptors because mGluR5 concentrations are positively correlated with NMDA receptor levels and because these two receptor groups are linked by a variety of intracellular mechanisms, thus allowing for indirect modulation of the NMDA receptor by mGluR5 manipulation [9•, 67, 68]. One study found that MDD patients had less mGluR5 binding in vivo as well as fewer mGluR5 proteins in postmortem brain than healthy subjects [69]. mGluR5 also regulates AMPA receptor internalization [70]. A 14-day administration of 10 mg/kg 2-methyl-6-(phenylethynyl)-pyridine (MPEP) [71, 72] increased BDNF mRNA levels in the hippocampus but not in the cortex of Wistar adult male rats, thus suggesting that mGluR5 antagonism may be linked to BDNF production [73]. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (MTEP) also exhibited anxiolytic and antidepressant-like activity in preclinical rodent models of despair [74–76].
AZD2066
A three-arm (placebo, duloxetine 30–60 mg/day, and the oral mGluR5 antagonist AZD2066 12–18 mg/day) randomized trial recruited 131 depressed patients, but the study was terminated prematurely. The preliminary findings revealed no beneficial results on the primary outcome measure (MADRS), depression response, or depression remission (ClinicalTrials.gov identifier: NCT01145755).
Ro4917523 (Basimglurant, RG7090)
In the first completed phase II trial (NCT00809562), Ro4917523 monotherapy (at five different doses to assess safety and tolerability) was compared to oral placebo in a 10-day inpatient TRD protocol. To our knowledge, the results of this initial study have not been reported. A completed phase IIb study compared modified-release basimglurant (0.5 and 1.5 mg/day) to placebo as an adjunctive medication over 9 weeks in individuals with TRD (6 weeks double-blind treatment, 3 weeks post-treatment follow-up) [77•]. Subjects (N = 333) were randomized to three treatment arms (n = 108 adjunctive placebo, n = 112 adjunctive basimglurant 0.5 mg/day, n = 111 adjunctive basimglurant 1.5 mg/day). Two were excluded from the study due to the emergence of mania. No statistically significant differences were observed on the primary endpoint (mean MADRS change from baseline to 6 weeks). However, the placebo response rate was 47 %, which is well above the 40 % reported in other adjunctive MDD trials as a threshold that impedes separation [78]. Keeping this high placebo response rate in mind, higher dose basimglurant positively affected several secondary endpoints, including patient self-reported depression scores and CGI scale scores. The most common adverse events were dizziness (23 %) and two self-resolving cases of mania at the 1.5 mg/day dose.
Group II mGluR NAM/Antagonists
Glutamate has the strongest affinity for group II mGluRs [9•]. These receptors are mainly presynaptic with perisynaptic localization [79]. Thus, they are only activated in the presence of excessive synaptic glutamate/spillover from neighboring synapses. Both mGluR2/3 antagonists and NAMs have been studied for the treatment of MDD in preclinical models [80, 81].
The mGluR2/3 NAM Ro4491533 [82] was found to have antidepressant-like efficacy in preclinical models of depression. In a mouse model of despair, the mGluR2/3 NAM Ro4432717 reversed some of the prototypical cognitive impairments that may also be observed in MDD (e.g., short-term memory deficits and cognitive inflexibility) [83]. An initial clinical trial assessing the safety and tolerability of mGluR2/3 modulators (BCI-838, BCI-632) was completed in healthy volunteers, but results are not yet available (ClinicalTrials.gov identifiers: NCT01546051, NCT01548703).
Group III mGluR PAMs
Group III mGluRs include mGluR4, mGluR6, mGluR7, and mGluR8 and are predominantly located presynaptically. mGluR7 is the most abundant mGluR in the central nervous system [84]. mGluR7 null mice exhibited a resilient phenotype in standard preclinical depression and anxiety-provocation paradigms [85]. mGluR7 mRNA expression in the suprapyramidal portion of the dentate gyrus was significantly higher in stress-sensitive Wistar Kyoto rats compared to control (Sprague–Dawley) rats [86].
AMN082
AMN082, an mGluR7 PAM, showed dose-dependent antidepressant activity in the tail-suspension test in C57BL6 male mice. Furthermore, this particular study showed that AMN082 exerted its antidepressant effects by modulating AMPA and NMDA receptor phosphorylation in the hippocampus [87]. AMN082 also increased synaptic protein levels (synapsin I and GluR1, which is an AMPA receptor subunit) and mTOR phosphorylation in the prefrontal cortex of male Sprague–Dawley rats, suggesting synaptogenesis as a major antidepressant mechanism of action [88]. AMN082 had no antidepressant effect in mGluR7 knockout mice, further supporting the importance of glutamatergic neurotransmission in its antidepressant properties. Two studies, however, suggest that the mechanism of action of AMN082 involves serotonin rather than glutamate; specifically, N-benzhydrylethane-1,2-diamine (Met-1) is a major metabolite of AMN082 that has monoaminergic affinity [89, 90]. To date, no compounds with a primary action at mGluR7 have been investigated in clinical trials for neuropsychiatric disorders.
Conclusion
Preclinical and clinical data support the use of NMDA receptor antagonists, NR2B subunit-selective antagonists, NMDA receptor glycine site partial agonists, AMPA receptor agonists, and mGluR NAMs/PAMs as possible novel therapeutics for mood disorders. It is interesting to note that, despite almost 25 years of preclinical and 15 years of clinical efficacy data, ketamine is not often considered in treatment algorithms for TRD. This is partly due to the lack of large, multi-site, randomized, and psychoactive placebo-controlled trials assessing ketamine’s safety, efficacy, and tolerability. In light of the proven clinical success of ketamine, alternative routes of ketamine administration and safety and tolerability studies with repeated dosing are absolutely critical to its continued development.
It is also important to emphasize that high-quality clinical efficacy data are currently available only for ketamine. Studies of other NMDA receptor antagonists and non-NMDA receptor modulators are either just entering clinical studies, are supported by very small datasets, or have been marked by negative results. One hypothesis for this preferential success is that ketamine’s antidepressant efficacy requires increased synaptic glutamate release and the activation of AMPA receptors. As a result, glutamate modulators that do not increase synaptic glutamate release and/or activate non-NMDA receptors would likely not have antidepressant activity in clinical trials. In addition, ketamine has been found to activate second messenger/signal transduction cascades that other glutamatergic modulators do not, and this may also be responsible for its differential antidepressant efficacy. As noted above, one example of this is the preclinical report of ketamine’s ability to phosphorylate eEF2 kinase and increase local BDNF production; in contrast, the glutamatergic modulator memantine did not have these cellular and molecular effects [40].
It should also be noted that some leading investigators have questioned the importance of glutamate in ketamine’s antidepressant mechanism of action and have hypothesized that non-glutamatergic—for instance, monoaminergic, cholinergic, or opioidergic—mechanisms may contribute significantly to ketamine’s antidepressant efficacy in clinical studies [91]. Other more selective glutamate modulating drugs may not have these off-target effects. Nevertheless, without results from head-to-head clinical studies comparing ketamine and other glutamatergic modulators, no compelling conclusions can be drawn for any of the above hypotheses.
Moving forward, one must keep in mind that depression is a heterogeneous illness and, therefore, that well-characterized patient populations or subtypes of patient populations should be studied when testing the effectiveness of any novel drug. Nevertheless, as this article has underscored, recent developments in the search for new, rapid-acting antidepressant agents hold considerable promise for developing new treatments for mood disorders. In addition to being urgently needed, these novel findings have revolutionized the field, challenged old paradigms and current limitations, and brought hope to those who must live with these devastating disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. 2011;21(9):655–79.
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–105.
Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS, et al. Grand challenges in global mental health. Nature. 2011;475(7354):27–30.
Wijeratne C, Sachdev P. Treatment-resistant depression: critique of current approaches. Aust N Z J Psychiatry. 2008;42(9):751–62.
Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185(1):1–10.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–4.
Zarate Jr CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–64.
Niciu MJ, Kelmendi B, Sanacora G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol Biochem Behav. 2012;100(4):656–64.
Pilc A, Wieronska JM, Skolnick P. Glutamate-based antidepressants: preclinical psychopharmacology. Biol Psychiatry. 2013;73(12):1125–32. The article concisely summarizes glutamate-based antidepressants in pre-clinical studies and focuses on major concepts and mechanisms.
Monteggia LM, Zarate Jr C. Antidepressant actions of ketamine: from molecular mechanisms to clinical practice. Curr Opin Neurobiol. 2015;30:139–43.
Iadarola ND, Niciu MJ, Richards EM, Vande Voort JL, Ballard ED, Lundin NB, et al. Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: a perspective review. Ther Adv Chronic Dis. 2015;6(3):97–114.
Ohgi Y, Futamura T, Hashimoto K. Glutamate signaling in synaptogenesis and NMDA receptors as potential therapeutic targets for psychiatric disorders. Curr Mol Med. 2015;15(3):206–21.
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69(8):754–61.
Duman RS, Aghajanian GK. Neurobiology of rapid acting antidepressants: role of BDNF and GSK-3beta. Neuropsychopharmacology. 2014;39(1):233.
Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res. 2005;136(1–2):29–37.
Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res. 2009;199(3–4):203–34.
Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134–42. This is the largest clinical trial of ketamine to date. It validated the antidepressant effects of ketamine and used an active placebo, midazolam.
Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74(4):250–6. This is the largest trial of repeated ketamine infusion.
Shiroma PR, Albott CS, Johns B, Thuras P, Wels J, Lim KO. Neurocognitive performance and serial intravenous subanesthetic ketamine in treatment-resistant depression. Int J Neuropsychopharmacol. 2014;17(11):1805–13.
Rasmussen KG, Lineberry TW, Galardy CW, Kung S, Lapid MI, Palmer BA, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444–50.
Diamond PR, Farmery AD, Atkinson S, Haldar J, Williams N, Cowen PJ, et al. Ketamine infusions for treatment resistant depression: a series of 28 patients treated weekly or twice weekly in an ECT clinic. J Psychopharmacol. 2014;28(6):536–44.
Lai R, Katalinic N, Glue P, Somogyi AA, Mitchell PB, Leyden J, et al. Pilot dose–response trial of i.v. ketamine in treatment-resistant depression. World J Biol Psychiatry. 2014;15(7):579–84.
Lapidus KA, Levitch CF, Perez AM, Brallier JW, Parides MK, Soleimani L, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014;76(12):970–6. This is the first controlled study of intranasal ketamine.
Cusin C, Hilton GQ, Nierenberg AA, Fava M. Long-term maintenance with intramuscular ketamine for treatment-resistant bipolar II depression. Am J Psychiatry. 2012;169(8):868–9.
Lara DR, Bisol LW, Munari LR. Antidepressant, mood stabilizing and procognitive effects of very low dose sublingual ketamine in refractory unipolar and bipolar depression. Int J Neuropsychopharmacol. 2013;16(9):2111–7. This is the first clinical trial to report the robust and rapid antidepressant effect of sublingual ketamine.
Irwin SA, Iglewicz A. Oral ketamine for the rapid treatment of depression and anxiety in patients receiving hospice care. J Palliat Med. 2010;13(7):903–8.
Paslakis G, Gilles M, Meyer-Lindenberg A, Deuschle M. Oral administration of the NMDA receptor antagonist S-ketamine as add-on therapy of depression: a case series. Pharmacopsychiatry. 2010;43(1):33–5.
Irwin SA, Iglewicz A, Nelesen RA, Lo JY, Carr CH, Romero SD, et al. Daily oral ketamine for the treatment of depression and anxiety in patients receiving hospice care: a 28-day open-label proof-of-concept trial. J Palliat Med. 2013;16(8):958–65.
De Gioannis A, De Leo D. Oral ketamine augmentation for chronic suicidality in treatment-resistant depression. Aust N Z J Psychiatry. 2014;48(7):686.
Glue P, Gulati A, Le Nedelec M, Duffull S. Dose- and exposure-response to ketamine in depression. Biol Psychiatry. 2011;70(4):e9–10. author reply e11-2.
Domino EF. Taming the ketamine tiger. 1965. Anesthesiology. 2010;113(3):678–84.
Nagele P, Duma A, et al. Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Soc Biol Psychiatry. 2015. This study showed that nitrous oxide, a non-ketamine NMDA receptor antagonist, had rapid antidepressant effects. This finding implicates NMDA receptor antagonism in the mechanism of antidepressant activity.
Sanders RD, Weimann J, Maze M. Biologic effects of nitrous oxide: a mechanistic and toxicologic review. Anesthesiology. 2008;109(4):707–22.
Lee SY, Chen SL, Chang YH, Chen SH, Chu CH, Huang SY, et al. The DRD2/ANKK1 gene is associated with response to add-on dextromethorphan treatment in bipolar disorder. J Affect Disord. 2012;138(3):295–300.
Church J, Sawyer D, McLarnon JG. Interactions of dextromethorphan with the N-methyl-D-aspartate receptor-channel complex: single channel recordings. Brain Res. 1994;666(2):189–94.
Kelly TF, Lieberman DZ. The utility of the combination of dextromethorphan and quinidine in the treatment of bipolar II and bipolar NOS. J Affect Disord. 2014;167:333–5.
Zarate Jr CA, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163(1):153–5.
Smith EG, Deligiannidis KM, Ulbricht CM, Landolin CS, Patel JK, Rothschild AJ. Antidepressant augmentation using the N-methyl-D-aspartate antagonist memantine: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2013;74(10):966–73.
Serra G, Koukopoulos A, De Chiara L, Koukopoulos AE, Tondo L, Girardi P, et al. Three-year, naturalistic, mirror-image assessment of adding memantine to the treatment of 30 treatment-resistant patients with bipolar disorder. J Clin Psychiatry. 2015;76(1):e91–7. This article suggests that although memantine may not be an effective augmenting agent for major depressive disorder, it may be effective in bipolar depression.
Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A. 2014;111(23):8649–54.
Zarate Jr CA, Mathews D, Ibrahim L, Chaves JF, Marquardt C, Ukoh I, et al. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol Psychiatry. 2013;74(4):257–64.
Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014;19(9):978–85.
Sanacora G, Johnson M, Khan A, Atkinson S, Riesenberg R, Schronen J. Adjunctive lanicemine (AZD6765) in patients with major depressive disorder and a history of inadequate response to antidepressants: primary results from a randomized, placebo-controlled study (PURSUIT). 53rd Meeting of the ACNP. 2014. Hollywood, FL, USA.
Gielen M, Siegler RB, Mony L, Johnson JW, Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459(7247):703–7.
Furukawa H, Singh SK, Mancusso R, Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438(7065):185–92.
Yao Y, Mayer ML. Characterization of a soluble ligand binding domain of the NMDA receptor regulatory subunit NR3A. J Neurosci. 2006;26(17):4559–66.
Miller OH, Yang L, Wang CC, Hargroder EA, Zhang Y, Delpire E, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. 2014;3:e03581.
Zhang C, Li Z, Wu Z, Chen J, Wang Z, Peng D, et al. A study of N-methyl-D-aspartate receptor gene (GRIN2B) variants as predictors of treatment-resistant major depression. Psychopharmacology (Berl). 2014;231(4):685–93.
Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–7.
Nicholson KL, Mansbach RS, Menniti FS, Balster RL. The phencyclidine-like discriminative stimulus effects and reinforcing properties of the NR2B-selective N-methyl-D-aspartate antagonist CP-101 606 in rats and rhesus monkeys. Behav Pharmacol. 2007;18(8):731–43.
Ibrahim L, Diaz GN, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, et al. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32(4):551–7.
Millan MJ. N-methyl-D-aspartate receptor-coupled glycineB receptors in the pathogenesis and treatment of schizophrenia: a critical review. Curr Drug Targets CNS Neurol Disord. 2002;1(2):191–213.
Heresco-Levy U, Javitt DC, Gelfin Y, Gorelik E, Bar M, Blanaru M, et al. Controlled trial of D-cycloserine adjuvant therapy for treatment-resistant major depressive disorder. J Affect Disord. 2006;93(1–3):239–43.
Heresco-Levy U, Gelfin G, Bloch B, Levin R, Edelman S, Javitt DC, et al. A randomized add-on trial of high-dose D-cycloserine for treatment-resistant depression. Int J Neuropsychopharmacol. 2013;16(3):501–6.
Sumiyoshi T, Anil AE, Jin D, Jayathilake K, Lee M, Meltzer HY. Plasma glycine and serine levels in schizophrenia compared to normal controls and major depression: relation to negative symptoms. Int J Neuropsychopharmacol. 2004;7(1):1–8.
Moskal JR, Burch R, Burgdorf JS, Kroes RA, Stanton PK, Disterhoft JF, et al. GLYX-13, an NMDA receptor glycine site functional partial agonist enhances cognition and produces antidepressant effects without the psychotomimetic side effects of NMDA receptor antagonists. Expert Opin Investig Drugs. 2014;23(2):243–54. This article provides a quick overview of GLYX-13, including its mechanism of action, preclinical results, and clinical data.
Naurex Inc. NRX-1074. 2015. http://www.naurex.com/pipeline/nrx-1074.
Browne CA, Lucki I. Antidepressant effects of ketamine: mechanisms underlying fast-acting novel antidepressants. Front Pharmacol. 2013;4:161.
Maeng S, Zarate Jr CA, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–52.
Andreasen JT, Gynther M, Rygaard A, Bogelund T, Nielsen SD, Clausen RP, et al. Does increasing the ratio of AMPA-to-NMDA receptor mediated neurotransmission engender antidepressant action? Studies in the mouse forced swim and tail suspension tests. Neurosci Lett. 2013;546:6–10.
Koike H, Chaki S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav Brain Res. 2014;271:111–5.
O’Neill MJ, Witkin JM. AMPA receptor potentiators: application for depression and Parkinson’s disease. Curr Drug Targets. 2007;8(5):603–20.
Bleakman D, Alt A, Witkin JM. AMPA receptors in the therapeutic management of depression. CNS Neurol Disord Drug Targets. 2007;6(2):117–26.
Ward SE, Bax BD, Harries M. Challenges for and current status of research into positive modulators of AMPA receptors. Br J Pharmacol. 2010;160(2):181–90.
Nations KR, Bursi R, Dogterom P, Ereshefsky L, Gertsik L, Mant T, et al. Maximum tolerated dose evaluation of the AMPA modulator Org 26576 in healthy volunteers and depressed patients: a summary and method analysis of bridging research in support of phase II dose selection. Drugs R D. 2012;12(3):127–39.
Rondard P, Pin JP. Dynamics and modulation of metabotropic glutamate receptors. Curr Opin Pharmacol. 2015;20:95–101.
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23(3):583–92.
Alagarsamy S, Marino MJ, Rouse ST, Gereau RW, Heinemann SF, Conn PJ. Activation of NMDA receptors reverses desensitization of mGluR5 in native and recombinant systems. Nat Neurosci. 1999;2(3):234–40.
Deschwanden A, Karolewicz B, Feyissa AM, Treyer V, Ametamey SM, Johayem A, et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [(11)C]ABP688 PET and postmortem study. Am J Psychiatry. 2011;168(7):727–34.
Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104(39):15537–42.
Tatarczynska E, Klodzinska A, Chojnacka-Wojcik E, Palucha A, Gasparini F, Kuhn R, et al. Potential anxiolytic- and antidepressant-like effects of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist. Br J Pharmacol. 2001;132(7):1423–30.
Liu CY, Jiang XX, Zhu YH, Wei DN. Metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine produces antidepressant effects in rats: role of brain-derived neurotrophic factor. Neuroscience. 2012;223:219–24.
Legutko B, Szewczyk B, Pomierny-Chamiolo L, Nowak G, Pilc A. Effect of MPEP treatment on brain-derived neurotrophic factor gene expression. Pharmacol Rep. 2006;58(3):427–30.
Palucha A, Branski P, Szewczyk B, Wieronska JM, Klak K, Pilc A. Potential antidepressant-like effect of MTEP, a potent and highly selective mGluR5 antagonist. Pharmacol Biochem Behav. 2005;81(4):901–6.
Pomierny-Chamiolo L, Poleszak E, Pilc A, Nowak G. NMDA but not AMPA glutamatergic receptors are involved in the antidepressant-like activity of MTEP during the forced swim test in mice. Pharmacol Rep. 2010;62(6):1186–90.
Domin H, Szewczyk B, Wozniak M, Wawrzak-Wlecial A, Smialowska M. Antidepressant-like effect of the mGluR5 antagonist MTEP in an astroglial degeneration model of depression. Behav Brain Res. 2014;273:23–33.
Quiroz J, Tamburri P, Deptula D, Banken L, Beyer U, Fontoura P, et al. The efficacy and safety of basimglurant as adjunctive therapy in major depression: a randomized, double-blind, placebo-controlled study. Neuropsychopharmacology. 2014;39:S376–7. This article discusses the difficulties associated with clinical trials when placebo rates are high and explores possible solutions.
Stolk P, Ten Berg MJ, Hemels ME, Einarson TR. Meta-analysis of placebo rates in major depressive disorder trials. Ann Pharmacother. 2003;37(12):1891–9.
Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106(3):481–503.
Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, et al. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46(4):457–67.
Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. Int J Neuropsychopharmacol. 2012;15(4):429–34.
Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer-Urios I, Schneider M, et al. Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression. J Neurogenet. 2011;25(4):152–66.
Goeldner C, Ballard TM, Knoflach F, Wichmann J, Gatti S, Umbricht D. Cognitive impairment in major depression and the mGlu2 receptor as a therapeutic target. Neuropharmacology. 2013;64:337–46.
Bradley SR, Levey AI, Hersch SM, Conn PJ. Immunocytochemical localization of group III metabotropic glutamate receptors in the hippocampus with subtype-specific antibodies. J Neurosci. 1996;16(6):2044–56.
Cryan JF, Kelly PH, Neijt HC, Sansig G, Flor PJ, van Der Putten H. Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci. 2003;17(11):2409–17.
O’Mahony CM, Bravo JA, Dinan TG, Cryan JF. Comparison of hippocampal metabotropic glutamate receptor 7 (mGlu7) mRNA levels in two animal models of depression. Neurosci Lett. 2010;482(2):137–41.
Bradley SR, Uslaner JM, Flick RB, Lee A, Groover KM, Hutson PH. The mGluR7 allosteric agonist AMN082 produces antidepressant-like effects by modulating glutamatergic signaling. Pharmacol Biochem Behav. 2012;101(1):35–40.
Palucha-Poniewiera A, Szewczyk B, Pilc A. Activation of the mTOR signaling pathway in the antidepressant-like activity of the mGlu5 antagonist MTEP and the mGlu7 agonist AMN082 in the FST in rats. Neuropharmacology. 2014;82:59–68.
Sukoff Rizzo SJ, Leonard SK, Gilbert A, Dollings P, Smith DL, Zhang MY, et al. The metabotropic glutamate receptor 7 allosteric modulator AMN082: a monoaminergic agent in disguise? J Pharmacol Exp Ther. 2011;338(1):345–52.
Palucha-Poniewiera A, Branski P, Lenda T, Pilc A. The antidepressant-like action of metabotropic glutamate 7 receptor agonist N,N′-bis(diphenylmethyl)-1,2-ethanediamine (AMN082) is serotonin-dependent. J Pharmacol Exp Ther. 2010;334(3):1066–74.
Sanacora G, Schatzberg AF. Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacology. 2015;40(2):259–67.
Acknowledgments
Funding for this work was supported by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIA-MH002857), by a NARSAD Independent Investigator Award to Carlos A. Zarate, and by a Brain and Behavior Mood Disorders Research Award to Carlos A. Zarate. The authors thank the 7SE research unit and staff for their support, Ioline Henter for outstanding editorial assistance, and Matthew H. Sieber, PhD for help with the figures.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Funding for this work was supported by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIA-MH002857), by a NARSAD Independent Investigator Award to Carlos A. Zarate, and by a Brain and Behavior Mood Disorders Research Award to Carlos A. Zarate. A patent for the use of ketamine in depression has been awarded that lists Carlos A. Zarate among the inventors; he has assigned his rights on the patent to the U.S. government, but will share a percentage of any royalties that may be received by the government. Minkyung Park and Mark J. Niciu have no conflict of interest to disclose, financial or otherwise.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Mood and Anxiety Disorders
Rights and permissions
About this article

Cite this article
Park, M., Niciu, M.J. & Zarate, C.A. Novel Glutamatergic Treatments for Severe Mood Disorders. Curr Behav Neurosci Rep 2, 198–208 (2015). https://doi.org/10.1007/s40473-015-0050-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40473-015-0050-5