
Abstract
Major depressive disorder (MDD) is a chronic, burdensome, highly prevalent disease that is characterized by depressed mood and anhedonia. MDD is especially burdensome as approved monoamine antidepressant treatments have weeks-long delays before clinical benefit and low remission rates. In the past 2 decades, a promising target emerged to improve patient outcomes in depression treatment: glutamatergic signaling. This narrative review provides a high-level overview of glutamate signaling in synaptogenesis and neural plasticity and the implications of glutamate dysregulation in depression. Based on this preclinical evidence implicating glutamate in depression and the rapid improvement of depression with ketamine treatment in a proof-of-concept trial, a range of N-methyl-d-aspartate (NMDA)-targeted therapies have been investigated. While an array of treatments has been investigated in registered phase 2 or 3 clinical trials, the development of most of these agents has been discontinued. Multiple glutamate-targeted antidepressants are actively in development, and two are approved. Nasal administration of esketamine (Spravato®) was approved by the US Food and Drug Administration (FDA) in 2019 to treat adults with treatment-resistant depression and in 2020 for adults with MDD with acute suicidal ideation or behavior. Oral combination dextromethorphan–bupropion (AXS-05, Auvelity® extended-release tablet) was FDA approved in 2022 for the treatment of MDD in adults. These approvals bolster the importance of glutamate in depression and represent an exciting breakthrough in contemporary psychiatry, providing new avenues of treatment for patients as first-line therapy or with either poor response or unacceptable side effects to monoaminergic antidepressants.
Graphical Abstract

Plain Language Summary
Major depressive disorder (MDD) is a common disease defined by sadness and a loss of interest or pleasure in activities. Depression treatments include therapy and antidepressant medication. Most available antidepressants affect the same types of chemicals that communicate in the brain (neurotransmitters) called monoamines. Unfortunately, these medicines can take weeks to work for some people and many still have depression symptoms with treatment. To provide more treatment options, new medicines have been studied that impact a different neurotransmitter in the brain, that is, glutamate. Glutamate, the most abundant neurotransmitter, is important for the growth of new brain cell connections, and there are changes in glutamate in people with depression versus people without depression. In 2000, the first small study in people showed that ketamine, a medicine targeting glutamate, quickly improved depression symptoms. Safety risks, including dissociation and sedation, require supervised administration and management guidance. Since 2000, about 20 glutamate-targeted medicines have been tested in human clinical trials. Two of these are approved as antidepressants. The medicine esketamine, which is inhaled up the nose at a clinic under medical supervision, is approved for the adjunctive treatment of people with MDD whose symptoms do not improve with other treatments or who have suicidal thoughts or actions (brand name Spravato®). The combination medicine dextromethorphan–bupropion is a swallowed tablet taken twice daily that is an approved monotherapy to treat adults with MDD (brand name Auvelity®). The approval of these drugs helps show the importance of glutamate in depression and provides more options for people with depression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Most antidepressants that are currently available target monoamine neurotransmitters in the brain but take weeks to see clinical benefits, and many patients do not respond to these treatments |
Since 2000, clinical trials have investigated antidepressants targeting a different neurotransmitter, glutamate |
Two glutamate-targeted antidepressants, esketamine nasal spray and dextromethorphan–bupropion tablet, have now been approved to treat depression and have potential efficacy and safety advantages over monoamine-targeted antidepressants |
1 Introduction
Major depressive disorder (MDD) is a highly prevalent disease, and depressive disorders have been a leading cause of chronic disease burden worldwide for the past 3 decades [1, 2]. MDD is characterized by core symptoms of frequent depressed mood and/or loss of interest or pleasure in activities (anhedonia), which causes meaningful distress or impairment [3]. Anhedonia is experienced by approximately 75% of people with depression and is reported to be one of the most bothersome symptoms [4, 5]. MDD is accompanied by a range of other symptoms, is associated with comorbidities, and is a risk factor for both development and worsening of cardiovascular disease, metabolic disorders, diabetes, and substance use disorders [3, 6]. People with MDD are at a 20 times higher risk for suicide than the general population, with a higher suicide risk than most mental disorders, including schizophrenia and bipolar disorder [7, 8].
A principal goal in MDD treatment is to reach full remission and return to normal functioning, in which functional impairment at work and/or in the home has also resolved [9]. To this aim, treatment guidelines recommend shared decision-making with patients to select psychotherapy and/or pharmacotherapy, with a tailored approach for severe, recurrent, or persistent MDD [10,11,12].
The majority of currently approved antidepressants directly modulate monoamine pathways [13, 14]. Depression is especially burdensome due to the low rates of remission associated with current monoamine antidepressant treatments [15, 16]. Unfortunately, while mechanistically distinct treatment options are available, including monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), serotonin–norepinephrine reuptake inhibitors (SNRIs), and atypical antidepressants (e.g., bupropion, mirtazapine), inadequate or partial response is still common in MDD treatment [13, 14, 17]. In a recent reanalysis of the pivotal STAR*D trial, 41% (1261/3110 patients) of patients with MDD treated with a first-line SSRI responded and 70% (2172/3110) did not achieve remission [18]. In patients who underwent multiple rounds of therapy, the probability of response or remission decreased with subsequent treatments, with only 21% response (22/106) and 10% remission (11/106) by the fourth therapy option [18]. This incomplete response reflects the neurobiological heterogeneity of depression, inviting the need for alternative therapeutics that target non-monoaminergic systems. While a variety of peripheral biomarkers, such as C-reactive protein and inflammatory markers, are under investigation to assist in treatment selection, they are not yet validated or used in widespread clinical practice [19, 20].
The burden of depression is compounded, as current monoamine-targeted agents have weeks-long to months-long delays between the increase in monoamine levels and the onset of therapeutic benefits [15, 21]. In patients who do achieve relief of depression symptoms, between 25 and 60% experience recurrence of depression [22, 23]. High relapse rates correlate with increased public health burden, hospitalization, and costs [24]. Additionally, patients treated with antidepressants often do not have significant improvements in quality of life, which does not return to normal for most people, even in those experiencing improvements in depression symptoms [25,26,27]. These efficacy limitations with monoamine antidepressants may also demoralize patients and contribute to the increased risk of suicide in people with depression [8]. Demoralization increases the risk of suicidal ideation or behavior, which may be associated with dysregulation in the hypothalamic–pituitary–adrenal axis that regulates the stress response [28, 29].
The unmet need with monoamine therapies is further highlighted as approximately 30% of patients undergoing pharmacotherapy for MDD have treatment-resistant depression (TRD) [17, 30]. A universal definition for TRD is not accepted, but TRD is commonly defined as a failure to respond or achieve remission after two or more mechanistically dissimilar antidepressants [9, 17]. In clinical practice, these mechanistically dissimilar antidepressants likely both target monoamine pathways, as most available antidepressants target monoamines [21]. The central nervous system can adapt to sequential monoamine antidepressant exposure via acquired or oppositional tolerance, which may contribute to resistance [31, 32]. This population has substantially lower quality of life and more activity impairment than people with MDD who respond to treatment [30, 33].
Real-world long-term use of current monoaminergic treatments for depression is complicated by low adherence rates: approximately half of people prescribed antidepressants discontinue treatment within 3 months [34]. This low adherence may be impacted not only by slow or incomplete response but also by adverse side effect profiles. Common side effects with SSRIs or SNRIs include increased risk of hyponatremia, gastrointestinal side effects, and sexual side effects, while TCAs are associated with weight gain, sedation, orthostatic hypotension, sexual dysfunction, and anticholinergic effects [35]. Additionally, adherence to antidepressants may be negatively impacted by high medication costs, as evidenced by improved adherence with generic antidepressants with lower out-of-pocket cost [36].
In an effort to overcome slow or inadequate response and tolerability issues and improve patient outcomes, new antidepressant targets have been under investigation, with a heavy emphasis on receptors and downstream antidepressant effects [21]. One promising target investigated for the past 2 decades for depression treatment is glutamatergic signaling, which plays a vital role in the release of neurotrophic factors and neuroplasticity [14, 21]. Neuroplasticity allows for learning and memory and is essential for neuronal systems to adapt to environmental stimuli and tailor appropriate responses [37].
This narrative review aims to summarize the current theory of glutamate in MDD and its validity as a therapeutic target. It then provides a historical perspective on glutamate-targeted agents investigated for MDD or TRD thus far, with an emphasis on AXS-05 [dextromethorphan–bupropion (Auvelity® extended-release tablet)], the only oral approved N-methyl-d-aspartate (NMDA) receptor antagonist and monoamine modulator for the treatment of MDD in adults.
2 Glutamate Signaling
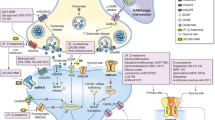
Glutamate signaling plays a complex role in synaptogenesis and neural plasticity and is reviewed briefly here (Niciu et al. [38] provides an in-depth review of glutamatergic neurotransmission). While glutamate was known to be present in the central nervous system, it was not acknowledged as a true neurotransmitter until 1984 [38, 39]. Since then, glutamate has been established as the major excitatory neurotransmitter of the nervous system, mediating fast excitatory transmission in the brain [40, 41]. Glutamate is both the most common amino acid in the brain [39] and the most abundant neurochemical agonist in the central nervous system [38]. Glutamate signaling involves not only traditional presynaptic to postsynaptic transmission but also recycling and regulation of glutamate by glial cells via excitatory amino acid transporters (Fig. 1) [38]. Vesicular glutamate transporters package presynaptic glutamate into vesicles within the neuron prior to release into the synaptic cleft [38].

Modified from Lener et al. 2017 [162] and Henter et al. 2021 [14]. Figure illustration elements modified from Servier Medical Art (2022) by Servier (https://smart.servier.com, used under a Creative Commons Attribution 4.0 License)
Glutamate signaling in neuroplasticity and synaptogenesis. Akt protein kinase B, AMPAR α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, BDNF brain-derived neurotrophic factor, EAAT excitatory amino acid transporter, EF2 eukaryotic elongation factor 2, ERK extracellular signal-regulated kinases, Gln glutamine, Glu glutamate, mGluR metabotropic glutamate receptor, mTOR mammalian target of rapamycin, NMDAR N-methyl-d-aspartate receptor, P phosphorylated, TrkB tropomyosin receptor kinase B, vGLUT vesicular glutamate transporter.
Receptors in the glutamate signaling cascade include ionotropic receptors (which act quickly, opening upon agonist binding and enable ion flow) and metabotropic receptors (which are G-protein-coupled receptors that help modulate synaptic activity and plasticity over a longer timescale) (Fig. 1) [14, 38]. These receptor types are complex with a broad range of effects and downstream targets [13].
The ionotropic multi-subunit NMDA receptor has the highest affinity for glutamate [38] (Hanson et al. [42] provide a review of the therapeutic implications of the NMDA receptor’s structure, functional diversity, and expression). The NMDA receptor is tightly regulated, requiring glutamate and glycine co-agonists for activation [13, 38]. Upon activation, the NMDA receptor ion channel is no longer blocked by the voltage-dependent pore blocker Mg2+, allowing for the influx of Ca2+ and Na+ cations into the postsynaptic neuron [38]. This influx of calcium can trigger multiple signaling cascades that promote cell survival and growth, including the phosphoinositide 3 kinase–protein kinase B (PI3K-AkT) pathway, mammalian target of rapamycin (mTOR) pathway, and brain-derived neurotrophic factor (BDNF) release (Fig. 1) [43, 44].
The mTOR pathway regulates protein translation and synaptic plasticity. mTOR is inhibited by tuberous sclerosis complexes and may be activated when PI3K–AkT or extracellular signal-regulated kinases (ERK) inhibit these complexes [45]. mTOR interacts downstream with ribosomal protein S6 kinases, which regulate protein biosynthesis by inhibiting phosphorylation of eukaryotic elongation factor 2 (eEF2), allowing for protein translation [45]. mTOR activation thus increases the expression of synaptic proteins vital for the formation, maturation, and function of synaptic spines and can lead to increases in the density of dendritic spines [44,45,46,47]. Additionally, mTOR processes impact behavioral plasticity, cognition, and neuronal excitability and survival [48]. BDNF is particularly important in the survival of neurons in adult brains and in synaptic plasticity [37]. Binding of BDNF to tropomyosin receptor kinase B (TrkB) receptors enhances neuronal survival, synaptogenesis, and plasticity and further activates kinase pathways to increase mTOR signaling [44, 49].
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (AMPAR) is also a multi-unit ionotropic receptor that is widely expressed in the central nervous system [38]. Glutamate has a lower affinity for ionotropic AMPARs than for NMDA receptors, which results in quicker inactivation of AMPARs than of NMDA receptors [38]. Activation of the AMPAR results in changes in AMPAR trafficking and localization within the cell, which can lead to long-term potentiation [40]. Additionally, activation of AMPAR by glutamate activates calcium channels and triggers the mTOR pathway (Fig. 1) [46].
Metabotropic glutamate receptors (mGluRs) trigger downstream effects by recruiting and activating G-proteins [38]. Glial cells can express mGluRs, in addition to metabolizing glutamate to glutamine and releasing the NMDA receptor co-agonist d-serine [50]. mGluRs are often located outside of the synapses on neurons and glia, but they can still modulate synaptic activity and plasticity [38]. Both ionotropic and metabotropic receptors interact with postsynaptic scaffolding proteins, including postsynaptic density protein of 95 kDA (PSD-95), which mechanically stabilizes synapses [38]. γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter and is essential to balance excitatory glutamate neurotransmission [51]. Excessive glycine, which can be released by both glial cells and glutamatergic neurons, can induce NMDA receptor endocytosis and depress NMDA receptor response [52].
3 Glutamate Dysregulation in Depression
A preclinical study in 1990 first indicated that NMDA receptor antagonists mimicked the clinical effects of antidepressants in murine models, leading to the glutamate hypothesis of depression [53]. Subsequent animal models have further supported that glutamatergic dysfunction plays a role in the mechanism of depression [54,55,56].
Chronic stress models in rodents are used to understand how the brain translates stress into depressive-like symptoms and physical changes in brain structure [51, 57]. Under chronic stress, neurons have impaired synaptic number and function as well as diminished plasticity, which increases susceptibility to depression [44]. The synaptic alterations from chronic stress may include loss of mature spines and synaptic connections [57]. Altered synaptic structure has been observed in people with depression [44]. In a rodent model, upon treatment with the NMDA receptor antagonist ketamine, synaptic protein expression was rapidly restored, reversing structural and functional deficits within 24 h [57]. Stress and depression can also dysregulate GABA function, which causes imbalances between excitatory and inhibitory activity [51].
BDNF signaling, downstream of glutamate activation of AMPAR, is decreased in the brains of people with depression [14, 37]. Traditional antidepressants slowly induce BDNF expression, which enhances synaptic plasticity and subsequent symptom response [37]. Rodent models support the importance of AMPAR activation and increased BDNF as essential for depression improvement with ketamine, as antidepressant effects are not observed if BDNF is neutralized or AMPAR inhibited [58, 59]. BDNF also plays a role in anxiety, schizophrenia, and other neurodegenerative disorders; a detailed summary of BDNF in antidepressant treatments is reviewed elsewhere [49].
Human imaging and tissue studies confirm there are changes in the glutamatergic system in people with depression. Positron emission tomography (PET) imaging studies and postmortem tissue analyses have revealed abnormalities in NMDA receptor and mGluR subunit/protein expression in the prefrontal cortex in patients with MDD [60, 61]. Additionally, a meta-analysis of proton magnetic resonance spectroscopy neuroimaging studies found decreased glutamatergic metabolite concentrations in the medial frontal cortex of people with depression compared with healthy people [62]. Glutamatergic dysregulation in MDD is also reflected by a significant (P = 8.5 × 10−5) elevation in blood glutamate levels in people with MDD [63].
The fundamental role of glutamatergic signaling in depression in humans was clinically supported in 2000 in the first small (N = 7), placebo-controlled, double-blind clinical trial of ketamine that demonstrated significant (P ≤ 0.001 at 72 h), rapid improvement in major depression symptoms compared with placebo [64]. The mechanism underlying this rapid action has been heavily investigated in preclinical models [65]. As a high-affinity NMDA receptor antagonist, it is theorized that ketamine treatment creates a surge of presynaptic glutamate release that then activates AMPARs; activation of AMPARs starts an intracellular signaling cascade that increases BDNF and mTOR activation, promoting neuronal recovery (Fig. 1) [66]. Clinically, a wide range of imaging studies, predominantly functional magnetic resonance imaging, have confirmed that ketamine treatment is associated with alterations within and between intrinsic connectivity networks implicated in depression, as reviewed in detail in Demchenko et al. [67]. The discovery of ketamine’s antidepressant activity led to further investigation of glutamatergic compounds that act via NMDA receptor antagonism or agonism.
4 Glutamatergic-Targeted Therapies
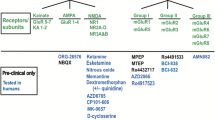
Here, we provide a brief historical perspective of NMDA-targeted therapies for depression, focusing on treatments that progressed into phase 3 clinical trials (Fig. 2). While a systematic literature search was not conducted, a methodical search was conducted of registered clinical trials on clinicaltrials.gov to document phase 2 and 3 trials for glutamate-targeted agents in MDD or TRD (except ketamine, which has been reviewed elsewhere). Subsequently, each NCT number was used as a search term for peer-reviewed study results using PubMed and Google Scholar. In the absence of peer-reviewed literature, study results were compiled from clinicaltrials.gov or any relevant industry press releases, where available. All trials included are documented in Online Resource 1, Supplementary Table 1.

Timeline of registered trials of glutamate-targeted drugs for treatment of depressiona. aFigure includes phase 2, 3, and 4 trials registered on ClinicalTrials.gov for the treatment of MDD or TRD as detailed in Online Resource 1, Supplementary Table 1. bThe first placebo-controlled, double-blind trial of ketamine in seven people with depression, conducted in 2000, was not registered on ClinicalTrials.gov [64]. cIntranasal esketamine is approved by the FDA, TPD, EMA, and TGA as an adjunctive treatment for TRD and/or for MDD with suicidal ideation/behavior or psychiatric emergency. dAXS-05 is approved to treat MDD in adults by the FDA. EMA European Medicines Agency, FDA US Food and Drug Administration, MDD major depressive disorder, NMDA N-methyl d-aspartate, TGA Australia’s Therapeutic Goods Administration, TPD Canada’s Therapeutic Products Directorate, TRD treatment-resistant depression
4.1 Historical Perspective
The first and perhaps most widely investigated NMDA antagonist for depression is ketamine; the historical perspective of ketamine in depression treatment is reviewed elsewhere [68,69,70]. Currently, off-label intravenous ketamine may provide rapid relief of symptoms in TRD and has been shown to be noninferior to electroconvulsive therapy; however, safety concerns require supervised administration [66, 71]. The most notable adverse events with ketamine are psychiatric events of dissociation, derealization, depersonalization, and perceptual disturbances [66]. Other adverse events with ketamine include dizziness, drowsiness, increased heart rate and blood pressure, and lower-urinary-tract symptoms [66]. In TRD, the effectiveness of ketamine does not decline with repeated treatment, although effectiveness varies in clinical practice [72, 73], and there is insufficient evidence on its long-term efficacy and tolerability [66]. The bioavailability of ketamine is highest with intravenous administration, and it is the most widely researched formulation for the acute treatment of depression [66]. While there have been fewer studies investigating intramuscular, oral, or subcutaneous ketamine formulations, initial evidence is suggestive of efficacy and safety when used under close medical supervision [74,75,76,77]. Ketamine, a dissociative anesthetic, is a Schedule III non-narcotic agent in the United States under the Controlled Substances Act, creating an additional burden to accessing it for treatment of depression [66]. Criticisms of the current clinical use of ketamine include limited and mixed data on repeated long-term ketamine administration, including how to maintain long-term response or remission, potential adverse events over time (e.g., changes in cognition or memory, urinary tract symptoms), and strategies to successfully transition off treatment [78, 79]. Additionally, unregulated off-label clinics administering ketamine may not abide by clinical recommendations [80].
The efficacy of ketamine was foundational to the glutamate hypothesis of depression and began a wave of investigations into NMDA-targeted therapies (Fig. 2) [69]. In the 2 years after the publication of proof of principle with ketamine, registered clinical trials were initiated investigating the oral therapies riluzole and memantine, which were initially developed for other indications (Fig. 2). Riluzole was US Food and Drug Administration (FDA) approved to treat amyotrophic lateral sclerosis in 1994 and is approved for this indication by multiple regulatory agencies [Canada’s Therapeutic Products Directorate (TPD), European Medicines Agency (EMA), Australia’s Therapeutic Goods Administration (TGA)] [81,82,83]. Riluzole can inhibit the release of presynaptic glutamate and interact with ionotropic receptors [84]. While initial studies indicated preliminary antidepressant effects, double-blind trials in MDD had inconsistent findings [84]. A 2020 systematic review and meta-analysis found no significant antidepressant activity with riluzole compared with placebo [85]. Memantine is a low- to moderate-affinity uncompetitive NMDA receptor antagonist that is approved as a symptomatic treatment for Alzheimer’s disease dementia by the FDA, TPD, EMA, TGA, and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) [86,87,88]. In a meta-analysis of 11 double-blind, randomized controlled trials (RCTs) investigating memantine for depression treatment, memantine improved depression symptoms; however, the effect size was small, and there was no significant improvement in response or remission [89]. It has been theorized that the inconsistent antidepressant effects with memantine may be due to its low trapping within NMDA receptors [90]. Neither riluzole nor memantine has ongoing clinical trials for depression treatment.
Despite these early failures, several unique glutamate-targeting antidepressant agents were subsequently investigated (Fig. 2); unfortunately, many did not progress past phase 2 clinical development. A single phase 2 RCT conducted from 2004 to 2005 (NCT00163059) investigated the efficacy of traxoprodil (an NMDA receptor NR2B subunit antagonist) to treat treatment-refractory MDD, but results were not reported, and no subsequent studies were conducted [91]. Another NMDA receptor N2B subunit antagonist, EVT 101 [92], was investigated in one phase 2 RCT (NCT01128452) that was terminated early due to a clinical hold issued by the FDA, with no results reported. An initial phase 2 trial (NCT00809562) indicated that basimglurant (an mGlu5-negative allosteric modulator) was well tolerated in people with TRD, but formal results were not published. In a phase 2 study, adjunctive basimglurant to treat MDD (NCT01437657) did not meet its primary efficacy endpoint, and development was discontinued [93].
Efficacy results were mixed in four phase 2 trials investigating the efficacy of lanicemine, a low-trapping NMDA channel blocker, to treat MDD [94]. Two phase 2 trials demonstrated rapid, significant improvement with lanicemine compared with placebo in people with treatment-resistant MDD [NCT00986479, P < 0.001 150 mg lanicemine versus placebo (N = 22 crossover design)] [95] or moderate to severe MDD with a history of poor response [NCT00781742, P < 0.01 for 100 mg lanicemine (n = 51) versus placebo (n = 50), P < 0.05 for 150 mg lanicemine (n = 51) versus placebo] [94]. However, in the other two studies, there was not significant improvement with lanicemine compared with placebo at 24 h in people with treatment-resistant MDD (NCT00491686) [94] nor at week 6 in people with MDD and inadequate response (NCT01482221).
Multiple clinical trial programs of NMDA receptor-targeted antidepressants have also been initiated and eventually discontinued in the past decade (Fig. 2). Adjunctive rislenemdaz (an NMDA receptor subunit 2B antagonist) did not demonstrate efficacy on the primary efficacy endpoint in two phase 2 RCTs in people with severe depression and recent active suicidal ideation (NCT01941043) [96] or MDD with a history of inadequate response (NCT02459236) [97]. Similarly, in two phase 2 trials, AV-101 (an NMDA receptor antagonist at the glycine site) did not meet the primary efficacy endpoint, with no differentiation versus placebo as adjunctive treatment in MDD (NCT03078322) [98] or monotherapy in TRD (NCT02484456) [99]. The oral NMDA receptor positive allosteric modulator zelquistinel (AGN-241751, GATE-251) [100] was investigated in a phase 1/2 trial (NCT03726658) and a phase 2 trial (NCT03586427) for the treatment of MDD, but the studies did not meet their primary efficacy endpoints.
Rapastinel (GLYX-13), an intravenously administered partial agonist of the NMDA receptor glycine binding site, reached phase 3 clinical studies for depression [101]. An early proof-of-concept study indicated there was a significant antidepressant response in people with TRD, within 2 h with rapastinel treatment [P < 0.05 at day 7 for 5 mg/kg (n = 20) and 10 mg/kg (n = 17) versus placebo (n = 33)] [101]. However, in three subsequent large phase 3 clinical trials, while adjunctive rapastinel was well tolerated, it did not reach the primary or key secondary endpoints [102]. A derivative of rapastinel, apimostinel (NRX-1074, AGN-241660), which has more potent binding at the NMDA receptor glycine site, was investigated for depression treatment in phase 1 and 2 clinical trials from 2014 to 2015 [14]. Results of these studies have not been published; however, a recent phase 1 trial (NCT05597241) indicates apimostinel development may be resuming.
Dextromethorphan, an approved over-the-counter antitussive, is an uncompetitive NMDA receptor antagonist and sigma-1 receptor agonist that has been evaluated in several formulations as an antidepressant [103, 104]. Additionally, dextromethorphan inhibits serotonin reuptake, which may also contribute to antidepressant effects [104]. Preclinical mouse models established a statistically significant improvement in depressive-like behavior with dextromethorphan treatment, prompting further investigation [104]. Dextromethorphan has activity at similar receptors as ketamine, and in vitro assays indicate that dextromethorphan has higher NMDA receptor antagonist activity and higher potency for sigma-1 than ketamine [21]. Unfortunately, dextromethorphan is metabolized quickly by the cytochrome P450 liver enzyme CYP2D6, which precludes therapeutic plasma levels with oral administration [21, 105].
To improve the bioavailability of dextromethorphan, it has been investigated in conjunction with quinidine, a potent inhibitor of cytochrome P450 2D6 that prevents the metabolic degradation of dextromethorphan into dextrorphan [105]. Nuedexta® is a commercially available combination of dextromethorphan (20 mg) and quinidine (10 mg) that is FDA approved to treat pseudobulbar affect [106]. In a small, open-label phase 2A study in 20 patients with TRD, oral dextromethorphan (45 mg)–quinidine (10 mg) improved depression symptoms from baseline, with 45% response and 35% remission rates, although the open-label design and lack of a control arm limit interpretation of the results [107]. Subsequently, no registered clinical trials have investigated Nuedexta® or any other dextromethorphan-quinidine combination in MDD or TRD. Deudextromethorphan (AVP-786), a similar weak NMDA receptor antagonist combination of deuterated dextromethorphan and quinidine, was investigated in a 2016 phase 2 study for treatment of MDD (NCT02153502); however, the sponsor reported that it halted development of this agent in depression because results of the phase 2 trial did not provide sufficient evidence of efficacy to justify continued development [142]. The only FDA-approved oral NMDA-targeted drug for depression is combination dextromethorphan–bupropion, discussed in more detail below [103].
In light of the negative trials with many glutamate-targeted agents, criticisms have arisen indicating that complementary or alternative pathways may be responsible for the antidepressant effects of NMDA-targeted agents [108]. Mechanistic studies to understand the therapeutic effects of glutamate-targeted agents currently in use will inform development of future antidepressant agents. For example, a 2018 small (N = 12) double-blind, crossover study in patients with TRD suggested that ketamine’s acute antidepressant effects require opioid system activation [109], highlighting another consideration in drug development.
4.2 Ongoing Clinical Candidates
Clinical trials are ongoing for two new potential NMDA receptor-antagonist antidepressants, esmethadone and MIJ821 (Table 1). Esmethadone (REL-1017, d-methadone), the opioid inactive (S)-enantiomer of methadone, is an uncompetitive NMDA receptor antagonist oral treatment that is currently in phase 3 clinical trials [90]. Esmethadone has low NMDA receptor affinity, demonstrates ketamine-like trapping in the NMDA receptor channel pore, and can undock from the NMDA receptor in the open conformation [90]. In phase 1 and 2 trials establishing preliminary safety and efficacy, esmethadone did not induce ketamine-like dissociation or opioid-like euphoria [90]. Additionally, there was no evidence of withdrawal upon abrupt discontinuation of 10 days of esmethadone treatment in a multiple ascending dose trial [90]. In two RCTs evaluating abuse potential in recreational drug users, esmethadone had no meaningful abuse potential, with significantly lower (P < 0.001) drug-liking scores than oxycodone or ketamine at up to six times the therapeutic dose [90].
A phase 2 randomized, double-blind, placebo-controlled 7-day trial (NCT03051256) showed a favorable safety and tolerability profile of esmethadone in adults with MDD and inadequate response. While the study was not powered for efficacy detection, there were improvements in depression symptoms with esmethadone 25 mg or 50 mg compared with placebo by day 4, which continued through day 14 follow-up (Table 1) [110]. Preliminary results from a 1-year open-label study of adjunctive esmethadone to treat MDD (NCT04855760, RELIANCE-OLS) were publicized via press release, reporting favorable tolerability, a 77.2% response rate (44/57 patients), and a 54.4% remission rate (31/57 patients) at month 12 in previously untreated participants (Table 1) [111]. However, two phase 3 double-blind placebo-controlled clinical trials investigating esmethadone as monotherapy (NCT05081167; RELIANCE-III) or adjunctive therapy (NCT04688164; RELIANCE-I) completed in 2022 did not meet their primary efficacy endpoints (Table 1) [112, 113]. In 2022, esmethadone as monotherapy for MDD was granted Fast Track designation by the FDA [114]; clinical development is ongoing, with two double-blind phase 3 RCTs of adjunctive esmethadone in MDD recruiting (NCT04855747; NCT06011577).
The other NMDA receptor antagonist in ongoing clinical development is MIJ821 (onfasprodil), which is being investigated for administration via intravenous infusion or subcutaneous injection [115]. MIJ821 is a potent, selective, reversible NR2B-NMDA negative allosteric modulator [116]. While earlier in development than other compounds discussed, the first trial in healthy volunteers indicated favorable safety and tolerability, with mild dissociative adverse events that were transient and dose dependent [116]. In an initial proof-of-concept RCT (NCT03756129), MIJ821 significantly improved (P < 0.05) depression symptoms within 24 h in adults with TRD compared with placebo (P < 0.05) within 24 h and was generally tolerable (Table 1) [115]. Three clinical trials are ongoing to investigate MIJ821 receptor occupancy in the brain (NCT05666687) and safety and efficacy in people with TRD (NCT05454410) or MDD with suicidal ideation with intent (NCT04722666).
An AMPA receptor positive allosteric modulator, TAK-653 (NBI-1065845) [117], is in phase 2 clinical trials for depression. In a phase 1 trial (NCT03792672) in healthy volunteers, TAK-653 was well tolerated, with no serious adverse events [118]. A phase 2 study in TRD (NCT03312894) was planned but withdrawn, and a phase 2 RCT is recruiting to investigate TAK-653 for treatment of adults with MDD (NCT05203341) (Table 1). In addition, an mGlu 2/3 receptor antagonist, TS-161, is in development. The first-in-human trial investigating TS-161 in 54 healthy subjects indicated it was orally bioavailable and has a good safety and tolerability profile [119]; a phase 2 RCT is recruiting to investigate TS-161 for TRD (NCT04821271) (Table 1).
4.3 Approved Glutamate-Targeted Treatments
Frequent failures in the glutamate space may have cast doubt on the glutamate theory of depression; however, the approvals of intranasal esketamine [120] and oral combination dextromethorphan–bupropion [103] have reinforced the importance of glutamate in depression. It has not been mechanistically established why esketamine and dextromethorphan–bupropion have significant, rapid antidepressant efficacy while other NMDA receptor-targeted drugs have struggled to show clinical improvement. Note that, while ketamine is used off label to treat depression, it is only approved as an anesthetic (by FDA, TPD, TGA) and, as such, is not discussed here [68, 121, 122].
4.3.1 Esketamine (Spravato® nasal spray)
Intranasal administration of the (S)-enantiomer of ketamine, esketamine (Spravato®), was initially approved by the FDA in 2019 for the treatment of adults with TRD and in 2020 for depression symptoms in adults with MDD with acute suicidal ideation or behavior (but not for treatment of the actual suicidality) [66, 120]. Intranasal esketamine has subsequently been approved as an adjunctive treatment for TRD by the TPD, EMA, and TGA and to treat MDD with psychiatric emergency by the TPD and EMA [123,124,125]. In a meta-analysis of eight double-blind RCTs investigating intranasal esketamine (placebo N = 661, esketamine N = 827), there were rapid, significant improvements in depression symptoms compared with placebo within 2–4 h of administration in people with TRD (P < 0.01) or MDD (P < 0.00001)[126]. These improvements were maintained until the end of the 28-day study period (TRD P < 0.001, MDD P < 0.00001) [126]. In the pivotal TRANSFORM-2 double-blind active-controlled trial of flexibly dosed intranasal esketamine versus placebo to treat TRD (Table 2), both adjunctive to a newly initiated oral SNRI or SSRI antidepressant, there was significantly greater improvement in the esketamine group at day 28 [P < 0.05 for adjunctive esketamine (n = 114) versus adjunctive placebo (n = 109)] [127]. There was also numerically higher response and remission with esketamine versus placebo at day 28 (Fig. 3) and numerically improved patient-reported function on the Sheehan Disability Scale (SDS). Additionally, in a recent phase 3 RCT, esketamine nasal spray adjunctive to a continuing SSRI or SNRI was superior to adjunctive extended-release quetiapine for the acute and continuation treatment of adults with TRD [remission at week 8; esketamine 27.1% (91/336) versus quetiapine 17.6% (60/340), adjusted P < 0.01] (Table 2) [128]. Adverse reactions with esketamine are similar to those seen with ketamine, with a risk for dissociation, abuse/misuse, increased blood pressure, sedation, and other adverse events [66, 120].

Response and remission rates with adjunctive intranasal esketamine in TRANSFORM-2 triala,b. aTRANSFORM-2 (NCT02418585) investigated adjunctive flexibly dosed intranasal esketamine versus adjunctive placebo with a newly initiated oral SNRI or SSRI antidepressant, in patients with TRD in a 28-day double-blind treatment phase. bPercentages shown are the proportion of patients remaining at visit [127]. ADT antidepressant, IN intranasal, MADRS Montgomery-Åsberg Depression Rating Scale, SNRI serotonin-norepinephrine reuptake inhibitor, SSRI selective serotonin reuptake inhibitor, TRD treatment-resistant depression, NR not reported
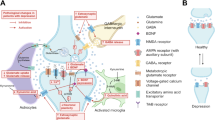
In addition to acting as an NMDA receptor antagonist, esketamine is a sigma-1 agonist, with a weaker affinity for sigma-1 than the (R)-ketamine enantiomer [129]. While originally classified as an opioid receptor subtype, sigma-1 receptors are chaperonins located on the endoplasmic reticulum and expressed mainly in the central nervous system in neurons and glial cells in the brain [130, 131]. These receptors are implicated in the development of not only depression but also other psychiatric and neurological diseases, including schizophrenia, anxiety, cognitive disorders, Alzheimer’s disease, ischemic stroke, and Parkinson’s disease [130]. Sigma-1 receptors may directly impact glutamatergic signaling by translocating to interact with NMDA receptors on the plasma membrane and modulating calcium influx (Fig. 4) [130]. High calcium then activates the calcium calmodulin-dependent protein kinase/ERK/mTOR pathways, which results in BDNF synthesis and PSD-95 scaffold synthesis [130]. Sigma-1 receptors can also exert antidepressant effects through modulating serotonin 5-HT1A function or modulation of the endoplasmic reticulum stress response, thus facilitating BDNF expression [130]. Additionally, sigma-1 may directly and indirectly balance dysregulated glutamate excitation and GABA inhibition [130]. Several treatments in clinical practice with antidepressant activity modulate sigma-1 as agonists (quetiapine, venlafaxine, bupropion, dextromethorphan–bupropion, desipramine, fluoxetine, ketamine, esketamine) or antagonists (sertraline) [131].

Figure illustration elements modified from Servier Medical Art (2022) by Servier (https://smart.servier.com, under a Creative Commons Attribution 4.0 License)
Antidepressant mechanisms of sigma-1 receptorsa. aActivated sigma-1 receptors may: translocate from the ER to ion channels, GPCR, or NMDAR at the cell membrane to modulate receptor activity; stabilize IP3R to maintain Ca2+ flow and ATP production; and modulate ER stress to enable downstream BDNF expression. ASIC1a acid-sensing ion channel, ATP adenosine triphosphate, BDNF brain-derived neurotrophic factor, CaMKII calmodulin-dependent protein kinase II, ER endoplasmic reticulum, ERK extracellular signal-regulated kinase, GPCR G-protein coupled receptor, IP3R inositol 1,4,5-trisphosphate receptor, IRE1 inositol-requiring enzyme 1, mTOR mammalian target of rapamycin, NMDAR N-methyl-d-aspartate receptor, P phosphorylated, XBP1 X-box-binding protein 1.
Intranasal esketamine must be administered at an approved facility, under the direct supervision of a healthcare provider, with at least a 2-h postdose observation period to monitor for adverse effects, such as dissociation, sedation, and blood pressure elevation [120]. A variety of RCTs are further investigating esketamine in people with TRD, MDD, bipolar disorder, and schizophrenia (Online Resource 1, Supplementary Table 1). Esketamine plays an important role as an FDA-approved, rapid-acting antidepressant. However, the administration and monitoring requirement may be burdensome on patients as multiple treatments are required each month during the maintenance phase [120]. Additionally, the activity of esketamine at opioid receptors may worsen abuse potential and some urge for caution in prescribing esketamine until its long-term effects are known, including relapse and suicide risk upon discontinuation [132]. As such, a more easily accessible treatment option with fewer safety concerns that still retains the rapid antidepressant effects is warranted. Adjunctive oral esketamine was investigated in a phase 2 (NCT04103892) trial in people with MDD but is not approved, and results of the study are not published.
Several recent studies indicate that real-world efficacy of esketamine treatment for TRD is consistent with clinical trials, reporting significant (P < 0.001) reductions in depression symptom scores for patients at a US clinic (N = 171) [133], 31.6% remission at week 4 in a French cohort (N = 66) [134], and 40.6% remission at month 3 in an Italian multicenter study (N = 116) [135]. No unexpected safety signals were identified in these studies [133,134,135].
4.3.2 Dextromethorphan–bupropion (AXS-05, Auvelity® extended-release tablet)
AXS-05 is an extended-release tablet of 45 mg dextromethorphan hydrobromide and 105 mg bupropion hydrochloride that was FDA approved in 2022 for the treatment of MDD in adults. As described earlier, dextromethorphan is an uncompetitive NMDA receptor antagonist and sigma-1 receptor agonist that is quickly metabolized [21]. As with quinidine, bupropion inhibits cytochrome P450 2D6, preventing the biotransformation of dextromethorphan and increasing dextromethorphan exposure [21]. In addition to serving as metabolic inhibitor of cytochrome P450 2D6 [21], bupropion enhances monoaminergic function as a norepinephrine–dopamine reuptake inhibitor and is approved to treat MDD (target dose 300 mg/day, maximum dose 450 mg/day) [136, 137]. While the main mechanism behind antidepressant effects with this combination is thought to be via NMDA receptor antagonism by dextromethorphan, bupropion inhibition of the neuronal reuptake of norepinephrine and dopamine may contribute [21]. Dextromethorphan inhibition of serotonin reuptake may also play a role in the antidepressant effects of AXS-05 [104]. Both dextromethorphan and bupropion are sigma-1 receptor agonists that can modulate glutamate and monoamine signaling (Fig. 4) [131].
When administered as AXS-05, the accumulation ratios of dextromethorphan based on maximum plasma concentration (Cmax) and area under the concentration–time curve (AUC0–12) are 20 and 32, respectively, compared with 1.3 based on Cmax and 1.4 based on AUC0–12 for dextromethorphan alone [103]. Of note, this pharmacokinetic profile has been established for the AXS-05 extended-release tablet, in which bupropion synergistically increases dextromethorphan exposure. This consistent pharmacokinetic profile may not be achieved with individual dextromethorphan and bupropion treatments of similar dosages, especially if bupropion concentration was too low to adequately inhibit cytochrome P450 2D6. As such, safety and efficacy results with individually administered dextromethorphan and bupropion would also vary. The single-pill formulation of AXS-05 (recommended dosage, twice daily) is likely to be advantageous for adherence, as single-pill regimens have demonstrated substantially better adherence and persistence in other chronic diseases than multi-treatment regimens [138,139,140].
Pivotal efficacy studies in MDD leading to approval of AXS-05 included two 6-week RCTs (Table 3). In the phase 3 placebo-controlled GEMINI study (NCT04019704), AXS-05 (n = 156) significantly improved symptoms of depression in adults with MDD compared with placebo (n = 162) at all timepoints, starting as early as week 1 (P < 0.01 weeks 1 and 6; P < 0.001 weeks 2–4) [141]. At week 6, significantly (P < 0.001) more participants treated with AXS-05 achieved remission (39.5%, 49/156 patients) and clinical response (54.0%, 67/156) compared with placebo [17.3% (26/162) and 34.0% (51/162), respectively] (Fig. 5) [141]. Additionally, at week 6, AXS-05 treatment improved patient reported outcomes of function and quality of life compared with placebo (SDS nominal P < 0.05; Quality of Life Enjoyment and Satisfaction Questionnaire – Short Form nominal P < 0.05) [141]. In the phase 2 parallel-group ASCEND study (NCT03595579), AXS-05 was compared with bupropion 105 mg alone in 97 adults with MDD (Table 3) [142]. There was significant improvement in depression symptoms with AXS-05 compared with bupropion by week 2, continuing throughout the study (P < 0.05 weeks 2 and 6; P < 0.01 weeks 3 and 4) [142]. Remission rates were also significantly (P < 0.01) better with AXS-05 compared with bupropion beginning at week 2, with 46.5% (20/43) and 16.2% (6/37) remission, respectively, at week 6 [142]. A pooled analysis of the GEMINI and ASCEND studies (N = 397) indicated that AXS-05 significantly improved depression compared with controls regardless of prior antidepressant treatment in the current MDE (P < 0.01 none, P < 0.05 ≥ 1 MDE), race (P < 0.01 white, P < 0.05 non-white), or sex (P < 0.01 female and male) [143]. In both studies, AXS-05 was well tolerated and was not associated with psychotomimetic effects, weight gain, or sexual dysfunction [141, 142]. The most common adverse events with AXS-05 across the two trials were dizziness, nausea, headache, somnolence, dry mouth, decreased appetite, and anxiety [141, 142].

Response and remission rates with oral AXS-05 in GEMINI trial. ***P < 0.001 versus control; analyzed via χ2 test. a GEMINI (NCT04019704) investigated AXS-05 versus placebo in patients with MDD for a 6-week double-blind treatment period. MADRS Montgomery-Åsberg Depression Rating Scale, MDD major depressive disorder, PO oral
The long-term safety and efficacy of AXS-05 was also investigated in two open-label trials, EVOLVE (NCT04634669) and COMET (NCT04039022) (Table 3). The EVOLVE study evaluated AXS-05 twice daily in 186 people with MDD who had at least one prior treatment for the current major depressive episode [144]. Improvements in depression symptoms from baseline were observed as soon as week 1 and continued at month 12 [144]. At week 6, remission was achieved by 46.0% of patients [144]. Long-term treatment was generally well tolerated, and the most common adverse events were similar to those seen in the short-term trials, with COVID-19 infection (8.9%) and insomnia (5.5%) also reported [144].
Long-term dosing was also well tolerated, with no new safety signals in the 1-year COMET study in 876 people with MDD, including TRD, as reported in a congress presentation [145]. Patients previously untreated with AXS-05 (n = 611) experienced rapid and durable improvements in depression symptoms, with 83% response and 69% remission at month 12 [145]. Improvements in function were also observed, with 76% SDS response at month 12 [145]. Additional analyses of the COMET study indicated that AXS-05 also improved depression symptoms and functioning in a subset of 70 participants with TRD [146] and rapidly resolved suicidal ideation in 37 participants with suicidal ideation at baseline [147]. AXS-05 development is ongoing for the treatment of Alzheimer’s disease-related agitation and for smoking cessation. To date, no RCTs have investigated long-term treatment outcomes or relapse prevention with AXS-05. Additionally, data on response in specific population subgroups have not been reported. As with all clinical trials, the conditions were tightly controlled, which may limit the generalizability of the results to real-world clinical conditions.
5 Conclusion
Current monoamine-targeted therapies do not rapidly improve depression symptoms, representing an unmet need for antidepressants with other mechanistic targets. The antidepressant effects of glutamatergic interventions were established by preclinical animal models and in clinical trials of ketamine. While a variety of other glutamate-targeting compounds have been investigated in clinical trials for depression since the proof of principle was established, few agents have demonstrated consistent efficacy. Off-label ketamine and FDA-approved intranasal esketamine provide rapid relief of depression symptoms with monitoring by a healthcare professional. AXS-05 (Auvelity), a recently approved oral NMDA receptor antagonist, sigma-1 receptor agonist, CYP2D6 inhibitor, and monoamine modulator, represents an important new treatment option for people with MDD, as it provides quick symptom relief without requiring intensive monitoring. Glutamate-targeted antidepressants provide potential advantages over classic SSRIs and SNRIs, including greater improvements in functional outcomes and rapid response and remission. The approval and clinical use of NMDA receptor antagonists support the importance of glutamate in depression and an advancement in contemporary psychiatry. However, as was the case with SSRIs and SNRIs, post-approval real-world data will be essential to confirm the safety profiles of the drugs and document potential adverse events or withdrawal syndrome that may arise with long-term use [148, 149]. After over 50 years of monoaminergic-targeted antidepressants, approval of glutamate-targeted therapies represents an important new mechanism for depression treatment. As such, these therapies provide another avenue of treatment for patients as first-line therapy or for those with poor response and/or issues of tolerability with monoaminergic antidepressant therapies.
References
Hasin DS, Sarvet AL, Meyers JL, et al. Epidemiology of adult DSM-5 major depressive disorder and its specifiers in the United States. JAMA Psychiatry. 2018;75(4):336–46. https://doi.org/10.1001/jamapsychiatry.2017.4602.
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2017;392(10159):1789–858. https://doi.org/10.1016/s0140-6736(18)32279-7.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington: American Psychiatric Publishing; 2013.
Franken IH, Rassin E, Muris P. The assessment of anhedonia in clinical and non-clinical populations: further validation of the Snaith–Hamilton Pleasure Scale (SHAPS). J Affect Disord. 2007;99(1–3):83–9. https://doi.org/10.1016/j.jad.2006.08.020.
Baune BT, Florea I, Ebert B, et al. Patient expectations and experiences of antidepressant therapy for major depressive disorder: a qualitative study. Neuropsychiatr Dis Treat. 2021;17:2995–3006. https://doi.org/10.2147/ndt.S325954.
Arnaud AM, Brister TS, Duckworth K, et al. Impact of major depressive disorder on comorbidities: a systematic literature review. J Clin Psychiatry. 2022;83(6):43390. https://doi.org/10.4088/JCP.21r14328.
Moitra M, Santomauro D, Degenhardt L, et al. Estimating the risk of suicide associated with mental disorders: a systematic review and meta-regression analysis. J Psychiatr Res. 2021;137:242–9. https://doi.org/10.1016/j.jpsychires.2021.02.053.
Chesney E, Goodwin GM, Fazel S. Risks of all-cause and suicide mortality in mental disorders: a meta-review. World Psychiatry. 2014;13(2):153–60. https://doi.org/10.1002/wps.20128.
McIntyre RS, Filteau M-J, Martin L, et al. Treatment-resistant depression: definitions, review of the evidence, and algorithmic approach. J Affect Disord. 2014;156:1–7. https://doi.org/10.1016/j.jad.2013.10.043.
Qaseem A, Barry MJ, Kansagara D, et al. Nonpharmacologic versus pharmacologic treatment of adult patients with major depressive disorder: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2016;164(5):350–9. https://doi.org/10.7326/M15-2570.
American Psychological Association. APA clinical practice guideline for the treatment of depression across three age cohorts. 2019. https://www.apa.org/depression-guideline/guideline.pdf. Accessed 18 Sept 2023.
US Department of Veterans Affairs. VA/DOD clinical practice guideline for the management of major depressive disorder. 2022. 4.0. https://www.healthquality.va.gov/guidelines/MH/mdd/VADoDMDDCPGFinal508.pdf. Accessed 18 Sept 2023.
Machado-Vieira R, Henter ID, Zarate CA Jr. New targets for rapid antidepressant action. Prog Neurobiol. 2017;152:21–37. https://doi.org/10.1016/j.pneurobio.2015.12.001.
Henter ID, Park LT, Zarate CA Jr. Novel glutamatergic modulators for the treatment of mood disorders: current status. CNS Drugs. 2021;35(5):527–43. https://doi.org/10.1007/s40263-021-00816-x.
Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. https://doi.org/10.1176/appi.ajp.163.1.28.
Huynh NN, McIntyre RS. What are the implications of the STAR*D trial for primary care? A review and synthesis. Prim Care Companion J Clin Psychiatry. 2008;10(2):91. https://doi.org/10.4088/pcc.v10n0201.
McIntyre RS, Alsuwaidan M, Baune BT, et al. Treatment-resistant depression: definition, prevalence, detection, management, and investigational interventions. World Psychiatry. 2023;22(3):394–412. https://doi.org/10.1002/wps.21120.
Pigott HE, Kim T, Xu C, et al. What are the treatment remission, response and extent of improvement rates after up to four trials of antidepressant therapies in real-world depressed patients? A reanalysis of the STAR*D study’s patient-level data with fidelity to the original research protocol. BMJ Open. 2023;13(7): e063095. https://doi.org/10.1136/bmjopen-2022-063095.
Gadad BS, Jha MK, Czysz A, et al. Peripheral biomarkers of major depression and antidepressant treatment response: current knowledge and future outlooks. J Affect Disord. 2018;233:3–14. https://doi.org/10.1016/j.jad.2017.07.001.
Orsolini L, Pompili S, Tempia Valenta S, et al. C-reactive protein as a biomarker for major depressive disorder? Int J Mol Sci. 2022;23(3):1616. https://doi.org/10.3390/ijms23031616.
Stahl SM. Dextromethorphan/bupropion: a novel oral nmda (N-methyl-d-aspartate) receptor antagonist with multimodal activity. CNS Spectr. 2019;24(5):461–6. https://doi.org/10.1017/S1092852919001470.
Sim K, Lau WK, Sim J, et al. Prevention of relapse and recurrence in adults with major depressive disorder: systematic review and meta-analyses of controlled trials. Int J Neuropsychopharmacol. 2016;19(2):pyv076. https://doi.org/10.1093/ijnp/pyv076.
Burcusa SL, Iacono WG. Risk for recurrence in depression. Clin Psychol Rev. 2007;27(8):959–85. https://doi.org/10.1016/j.cpr.2007.02.005.
Touya M, Lawrence DF, Kangethe A, et al. Incremental burden of relapse in patients with major depressive disorder: a real-world, retrospective cohort study using claims data. BMC Psychiatry. 2022;22(1):152. https://doi.org/10.1186/s12888-022-03793-7.
Almohammed OA, Alsalem AA, Almangour AA, et al. Antidepressants and health-related quality of life (HRQOL) for patients with depression: analysis of the Medical Expenditure Panel Survey from the United States. PLoS One. 2022;17(4): e0265928. https://doi.org/10.1371/journal.pone.0265928.
Saragoussi D, Christensen MC, Hammer-Helmich L, et al. Long-term follow-up on health-related quality of life in major depressive disorder: a 2-year European cohort study. Neuropsychiatr Dis Treat. 2018. https://doi.org/10.2147/NDT.S159276.
IsHak WW, Mirocha J, James D, et al. Quality of life in major depressive disorder before/after multiple steps of treatment and one-year follow-up. Acta Psychiatr Scand. 2015;131(1):51–60. https://doi.org/10.1111/acps.12301.
Costanza A, Vasileios C, Ambrosetti J, et al. Demoralization in suicide: a systematic review. J Psychosom Res. 2022;157: 110788. https://doi.org/10.1016/j.jpsychores.2022.110788.
Berardelli I, Serafini G, Cortese N, et al. The involvement of hypothalamus–pituitary–adrenal (hpa) axis in suicide risk. Brain Sci. 2020;10(9):653. https://doi.org/10.3390/brainsci10090653.
Zhdanava M, Pilon D, Ghelerter I, et al. The prevalence and national burden of treatment-resistant depression and major depressive disorder in the United States. J Clin Psychiatry. 2021;82(2):29169. https://doi.org/10.4088/JCP.20m13699.
Fava GA, Offidani E. The mechanisms of tolerance in antidepressant action. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(7):1593–602. https://doi.org/10.1016/j.pnpbp.2010.07.026.
Andrews PW, Amsterdam JD. A hormetic approach to understanding antidepressant effectiveness and the development of antidepressant tolerance—a conceptual view. Psychiatr Polska. 2020;11:78. https://doi.org/10.12740/PP/120084.
Jaffe DH, Rive B, Denee TR. The humanistic and economic burden of treatment-resistant depression in Europe: a cross-sectional study. BMC Psychiatry. 2019;19:1–11. https://doi.org/10.1186/s12888-019-2222-4.
Rossom RC, Shortreed S, Coleman KJ, et al. Antidepressant adherence across diverse populations and healthcare settings. Depress Anxiety. 2016;33(8):765–74. https://doi.org/10.1002/da.22532.
Pillinger T, Howes OD, Correll CU, et al. Antidepressant and antipsychotic side-effects and personalised prescribing: a systematic review and digital tool development. Lancet Psychiatry. 2023;10(11):860–76. https://doi.org/10.1016/S2215-0366(23)00262-6.
Bao Y, Ryan AM, Shao H, et al. Generic initiation and adherence to antidepressant therapy under Medicare Part D. Am J Manag Care. 2013;19(12):989.
Harmer CJ, Duman RS, Cowen PJ. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry. 2017;4(5):409–18. https://doi.org/10.1016/S2215-0366(17)30015-9.
Niciu MJ, Kelmendi B, Sanacora G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol Biochem Behavior. 2012;100(4):656–64. https://doi.org/10.1016/j.pbb.2011.08.008.
Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42(1):1–11. https://doi.org/10.1111/j.1471-4159.1984.tb09689.x.
Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62(1):63–77. https://doi.org/10.1016/j.neuropharm.2011.07.036.
Orrego F, Villanueva S. The chemical nature of the main central excitatory transmitter: a critical appraisal based upon release studies and synaptic vesicle localization. Neuroscience. 1993;56(3):539–55. https://doi.org/10.1016/0306-4522(93)90355-j.
Hanson JE, Yuan H, Perszyk RE, et al. Therapeutic potential of n-methyl-d-aspartate receptor modulators in psychiatry. Neuropsychopharmacol. 2023. https://doi.org/10.1038/s41386-023-01614-3.
Papadia S, Hardingham GE. The dichotomy of nmda receptor signaling. Neuroscientist. 2007;13(6):572–9. https://doi.org/10.1177/10738584070130060401.
Dwyer JM, Duman RS. Activation of mammalian target of rapamycin and synaptogenesis: role in the actions of rapid-acting antidepressants. Biol Psychiatry. 2013;73(12):1189–98. https://doi.org/10.1016/j.biopsych.2012.11.011.
Ignacio ZM, Reus GZ, Arent CO, et al. New perspectives on the involvement of mtor in depression as well as in the action of antidepressant drugs. Br J Clin Pharmacol. 2016;82(5):1280–90. https://doi.org/10.1111/bcp.12845.
Deyama S, Bang E, Wohleb ES, et al. Role of neuronal VEGF signaling in the prefrontal cortex in the rapid antidepressant effects of ketamine. Am J of Psychiatry. 2019;176(5):388–400. https://doi.org/10.1176/appi.ajp.2018.17121368.
Li N, Lee B, Liu RJ, et al. Mtor-dependent synapse formation underlies the rapid antidepressant effects of nmda antagonists. Science. 2010;329(5994):959–64. https://doi.org/10.1126/science.1190287.
Réus GZ, Quevedo J, Lúcia S, Rodrigues A. Mtor signaling in the neuropathophysiology of depression: current evidence. J Recept Ligand Channel Res. 2015;65:78.
Castrén E, Kojima M. Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol Dis. 2017;97:119–26. https://doi.org/10.1016/j.biopsych.2021.05.008.
Wilkinson ST, Sanacora G. A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/gaba neurotransmitter systems. Drug Discov Today. 2019;24(2):606–15. https://doi.org/10.1016/j.drudis.2018.11.007.
Duman RS, Sanacora G, Krystal JH. Altered connectivity in depression: Gaba and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102(1):75–90. https://doi.org/10.1016/j.neuron.2019.03.013.
Zhang X-Y, Ji F, Wang N, et al. Glycine induces bidirectional modifications in n-methyl-d-aspartate receptor-mediated synaptic responses in hippocampal ca1 neurons. J Biol Chem. 2014;289(45):31200–11. https://doi.org/10.1074/jbc.M114.570630.
Trullas R, Skolnick P. Functional antagonists at the nmda receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185(1):1–10. https://doi.org/10.1016/0014-2999(90)90204-j.
Lee Y, Son H, Kim G, et al. Glutamine deficiency in the prefrontal cortex increases depressive-like behaviours in male mice. J Psychiatry Neurosci. 2013;38(3):183–91. https://doi.org/10.1503/jpn.120024.
Tordera RM, Totterdell S, Wojcik SM, et al. Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (vglut1). Eur J Neurosci. 2007;25(1):281–90. https://doi.org/10.1111/j.1460-9568.2006.05259.x.
Hascup KN, Hascup ER, Stephens ML, et al. Resting glutamate levels and rapid glutamate transients in the prefrontal cortex of the flinders sensitive line rat: a genetic rodent model of depression. Neuropsychopharmacol. 2011;36(8):1769–77. https://doi.org/10.1038/npp.2011.60.
Li N, Liu RJ, Dwyer JM, et al. Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69(8):754–61. https://doi.org/10.1016/j.biopsych.2010.12.015.
Lepack AE, Fuchikami M, Dwyer JM, et al. Bdnf release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol. 2014. https://doi.org/10.1093/ijnp/pyu033.
Maeng S, Zarate CA Jr, Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–52. https://doi.org/10.1016/j.biopsych.2007.05.028.
Feyissa AM, Chandran A, Stockmeier CA, et al. Reduced levels of nr2a and nr2b subunits of nmda receptor and psd-95 in the prefrontal cortex in major depression. Progr Neuropsychopharmacol Biol Psychiatry. 2009;33(1):70–5. https://doi.org/10.1016/j.pnpbp.2008.10.005.
Deschwanden A, Karolewicz B, Feyissa AM, et al. Reduced metabotropic glutamate receptor 5 density in major depression determined by [11c] abp688 pet and postmortem study. Am J Psychiatry. 2011;168(7):727–34. https://doi.org/10.1176/appi.ajp.2011.09111607.
Moriguchi S, Takamiya A, Noda Y, et al. Glutamatergic neurometabolite levels in major depressive disorder: a systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol Psychiatry. 2019;24(7):952–64. https://doi.org/10.1038/s41380-018-0252-9.
Inoshita M, Umehara H, Watanabe S-Y, et al. Elevated peripheral blood glutamate levels in major depressive disorder. Neuropsychiatr Dis Treat. 2018;14:945–53. https://doi.org/10.2147/NDT.S159855.
Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–4. https://doi.org/10.1016/s0006-3223(99)00230-9.
Kadriu B, Musazzi L, Henter ID, et al. Glutamatergic neurotransmission: pathway to developing novel rapid-acting antidepressant treatments. Int J Neuropsychopharmacol. 2019;22(2):119–35. https://doi.org/10.1093/ijnp/pyy094.
McIntyre RS, Rosenblat JD, Nemeroff CB, et al. Synthesizing the evidence for ketamine and esketamine in treatment-resistant depression: an international expert opinion on the available evidence and implementation. Am J Psychiatry. 2021;178(5):383–99. https://doi.org/10.1176/appi.ajp.2020.20081251.
Demchenko I, Tassone VK, Kennedy SH, et al. Intrinsic connectivity networks of glutamate-mediated antidepressant response: a neuroimaging review. Front Psychiatry. 2022;13: 864902. https://doi.org/10.3389/fpsyt.2022.864902.
Hashimoto K. Rapid-acting antidepressant ketamine, its metabolites and other candidates: a historical overview and future perspective. Psychiatry Clin Neurosci. 2019;73(10):613–27. https://doi.org/10.1111/pcn.12902.
Kraus C, Wasserman D, Henter ID, et al. The influence of ketamine on drug discovery in depression. Drug Discov Today. 2019;24(10):2033–43. https://doi.org/10.1016/j.drudis.2019.07.007.
Corriger A, Pickering G. Ketamine and depression: a narrative review. Drug Des Dev Ther. 2019;13:3051–67. https://doi.org/10.2147/DDDT.S221437.
Anand A, Mathew SJ, Sanacora G, et al. Ketamine versus ect for nonpsychotic treatment-resistant major depression. N Engl J Med. 2023;388(25):2315–25. https://doi.org/10.1056/NEJMoa2302399.
Smith-Apeldoorn SY, Veraart JK, Spijker J, et al. Maintenance ketamine treatment for depression: a systematic review of efficacy, safety, and tolerability. Lancet Psychiatry. 2022;9(11):907–21. https://doi.org/10.1016/S2215-0366(22)00317-0.
Alnefeesi Y, Chen-Li D, Krane E, et al. Real-world effectiveness of ketamine in treatment-resistant depression: a systematic review and meta-analysis. J Psychiatr Res. 2022;151:693–709. https://doi.org/10.1016/j.jpsychires.2022.04.037.
Meshkat S, Haikazian S, Di Vincenzo JD, et al. Oral ketamine for depression: an updated systematic review. World J Biol Psychiatry. 2023;24:1–13. https://doi.org/10.1080/15622975.2023.2169349.
Lee W, Sheehan C, Chye R, et al. Subcutaneous ketamine infusion in palliative patients for major depressive disorder (SKIPMDD)—phase II single-arm open-label feasibility study. PLoS One. 2023;18(11): e0290876. https://doi.org/10.1371/journal.pone.0290876.
Cavenaghi VB, Da Costa LP, Lacerda ALT, et al. Subcutaneous ketamine in depression: a systematic review. Front Psychiatry. 2021;12: 513068. https://doi.org/10.3389/fpsyt.2021.513068.
Ahuja S, Brendle M, Smart L, et al. Real-world depression, anxiety and safety outcomes of intramuscular ketamine treatment: a retrospective descriptive cohort study. BMC Psychiatry. 2022;22(1):634. https://doi.org/10.1186/s12888-022-04268-5.
Jelen LA, Stone JM. Ketamine for depression. Int Rev Psychiatry. 2021;33(3):207–28. https://doi.org/10.1080/09540261.2020.1854194.
Andrade C. Ketamine for depression, 4: in what dose, at what rate, by what route, for how long, and at what frequency? J ClinC Psychiatry. 2017;78(7):10106. https://doi.org/10.4088/JCP.17f11738.
Nemeroff CB. Ketamine: Quo vadis? Am Psychiatric Assoc. 2018;175:297–9.
Sanofi winthrop industrie. Rilutek smpc. 2006. https://www.Ema.Europa.Eu/en/documents/product-information/rilutek-epar-product-information_en.Pdf. Sanofi Winthrop Industrie; 2006.
Mylan-riluzole [product monograph (Canada)]. Etobicoke, Ontario, Canada: Mylan Pharmaceuticals ulc; 2012. Mylan Pharmaceuticals ULC; 2012.
Pharmacor riluzole [Australian product information]. Chatswood, New South Wales, Australia: Pharmacor Pty ltd; 2018. Pharmacor Pty LTD; 2018.
Sakurai H, Dording C, Yeung A, et al. Longer-term open-label study of adjunctive riluzole in treatment-resistant depression. J Affect Disord. 2019;258:102–8. https://doi.org/10.1016/j.jad.2019.06.065.
Yao R, Wang H, Yuan M, et al. Efficacy and safety of riluzole for depressive disorder: a systematic review and meta-analysis of randomized placebo-controlled trials. Psychiatry Res. 2020;284: 112750. https://doi.org/10.1016/j.psychres.2020.112750.
Namenda [package insert]. St Louis, MO, USA: Forest Pharmaceuticals, Inc; 2012.
Pract memantine [product monograph (Canada)]. Toronto, Ontario, Canada: Teva Canada Limited; 2020. Teva Canada Limited; 2020.
Mylan Pharmaceuticals Limited. Memantine smpc. 2018. https://www.Ema.Europa.Eu/en/documents/product-information/memantine-mylan-epar-product-information_en.Pdf. Mylan Pharmaceuticals Limited; 2018.
Hsu T-W, Chu C-S, Ching P-Y, et al. The efficacy and tolerability of memantine for depressive symptoms in major mental diseases: a systematic review and updated meta-analysis of double-blind randomized controlled trials. J Affect Disord. 2022;306:182–9. https://doi.org/10.1016/j.jad.2022.03.047.
Fava M, Stahl SM, De Martin S, et al. Esmethadone-hcl (rel-1017): a promising rapid antidepressant. Eur Arch Psychiatry Clin Neurosci. 2023. https://doi.org/10.1007/s00406-023-01571-4.
Preskorn SH, Baker B, Kolluri S, et al. An innovative design to establish proof of concept of the antidepressant effects of the nr2b subunit selective n-methyl-d-aspartate antagonist, cp-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–7. https://doi.org/10.1097/JCP.0b013e31818a6cea.
Stroebel D, Buhl DL, Knafels JD, et al. A novel binding mode reveals two distinct classes of nmda receptor glun2b-selective antagonists. Mol Pharmacol. 2016;89(5):541–51. https://doi.org/10.1124/mol.115.103036.
Quiroz JA, Tamburri P, Deptula D, et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiat. 2016;73(7):675–84. https://doi.org/10.1001/jamapsychiatry.2016.0838.
Sanacora G, Smith M, Pathak S, et al. Lanicemine: A low-trapping nmda channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014;19(9):978–85. https://doi.org/10.1038/mp.2013.130.
Zarate CA Jr, Mathews D, Ibrahim L, et al. A randomized trial of a low-trapping nonselective n-methyl-d-aspartate channel blocker in major depression. Biol Psychiatry. 2013;74(4):257–64. https://doi.org/10.1016/j.biopsych.2012.10.019.
Paterson B, Fraser H, Wang C, et al. A randomized, double-blind, placebo-controlled, sequential parallel study of cerc-301 in the adjunctive treatment of subjects with severe depression and recent active suicidal ideation despite antidepressant treatment. National Network of Depression Centers Annual Conference; Ann Arbor, MI. 2015.
Avalo Therapeutics. Cerecor reports top-line data from cerc-301 phase 2 study for major depressive disorder. 2016.
VistaGen Therapeutics. Vistagen reports topline phase 2 results for av-101 as an adjunctive treatment of major depressive disorder. 2019.
Park LT, Kadriu B, Gould TD, et al. A randomized trial of the n-methyl-d-aspartate receptor glycine site antagonist prodrug 4-chlorokynurenine in treatment-resistant depression. Int J Neuropsychopharmacol. 2020;23(7):417–25. https://doi.org/10.1093/ijnp/pyaa025.
Pothula S, Liu R-J, Wu M, et al. Positive modulation of nmda receptors by agn-241751 exerts rapid antidepressant-like effects via excitatory neurons. Neuropsychopharmacol. 2021;46(4):799–808. https://doi.org/10.1038/s41386-020-00882-7.
Preskorn S, Macaluso M, Do Vishaal M, et al. Randomized proof of concept trial of glyx-13, an n-methyl-d-aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. J Psychiatr Pract. 2015;21(2):140–9. https://doi.org/10.1097/01.pra.0000462606.17725.93.
Allergan. Allergan announces phase 3 results for rapastinel as an adjunctive treatment of major depressive disorder (mdd). 2019.
Auvelity [package insert]. New York, NY, USA: Axsome Therapeutics, Inc.; 2022. Axsome Therapeutics Inc.; 2022.
Majeed A, Xiong J, Teopiz KM, et al. Efficacy of dextromethorphan for the treatment of depression: a systematic review of preclinical and clinical trials. Expert Opin Emerg Drugs. 2021;26(1):63–74. https://doi.org/10.1080/14728214.2021.1898588.
Taylor CP, Traynelis SF, Siffert J, et al. Pharmacology of dextromethorphan: relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacol Ther. 2016;164:170–82. https://doi.org/10.1016/j.pharmthera.2016.04.010.
Nudexta [package insert]. Aliso Viejo, CA, USA: Avanir Pharmaceutical, Inc. 2010.
Murrough JW, Stade E, Sayed S, et al. Dextromethorphan/quinidine pharmacotherapy in patients with treatment resistant depression: a proof of concept clinical trial. J Affect Disord. 2017;218:277–83. https://doi.org/10.1016/j.jad.2017.04.072.
Sanacora G, Schatzberg AF. Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacol. 2015;40(2):259–67. https://doi.org/10.1038/npp.2014.261.
Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175(12):1205–15. https://doi.org/10.1038/s41380-019-0503-4.
Fava M, Stahl S, Pani L, et al. Rel-1017 (esmethadone) as adjunctive treatment in patients with major depressive disorder: a phase 2a randomized double-blind trial. Am J Psychiatry. 2022;179(2):122–31. https://doi.org/10.1176/appi.ajp.2021.21020197.
Relmada Therapeautics Inc. Relmada therapeutics announces efficacy and safety results from the phase 3 long-term study of rel-1017 in major depressive disorder.
Relmada Therapeutics Inc. Relmada Therapeutics announces topline results from phase 3 Reliance I trial for REL-1017 as an adjunctive treatment for major depressive disorder. 2022.
Relmada Therapeutics Inc. Relmada Therapeutics announces top-line results from phase 3 Reliance III trial for REL-1017 as a monotherapy for the treatment of major depressive disorder. 2022.
Relmada Therapeutics Inc. Relmada Therapeutics receives FDA fast track designation for REL-1017 as a monotherapy for the treatment of major depressive disorder. 2022.
Ghaemi N, Sverdlov A, Shelton R, et al. Efficacy and safety of mij821 in patients with treatment-resistant depression: results from a randomized, placebo-controlled, proof-of-concept study. Eur Psychiatry. 2021;64(S1):S334–5. https://doi.org/10.1192/j.eurpsy.2021.897.
Gomez-Mancilla B, Levy JA, Ganesan S, et al. Mij821 (onfasprodil) in healthy volunteers: first-in-human, randomized, placebo-controlled study (single ascending dose and repeated intravenous dose). Clin Transl Sci. 2023;16(11):2236–52. https://doi.org/10.1111/cts.13623.
Dijkstra F, O’Donnell P, Klaassen E, et al. Central nervous system effects of TAK-653, an investigational alpha-amino-3-hydroxy-5-methyl-4-isoxazole receptor (AMPAR) positive allosteric modulator in healthy volunteers. Transl Psychiatry. 2022;12(1):408. https://doi.org/10.1038/s41398-022-02148-w.
O’Donnell P, Dijkstra FM, Damar U, et al. Transcranial magnetic stimulation as a translational biomarker for ampa receptor modulation. Transl Psychiatry. 2021;11(1):325. https://doi.org/10.1038/s41398-021-01451-2.
Watanabe M, Marcy B, Hiroki A, et al. Evaluation of the safety, tolerability, and pharmacokinetic profiles of tp0473292 (ts-161), a prodrug of a novel orthosteric mglu2/3 receptor antagonist tp0178894, in healthy subjects and its antidepressant-like effects in rodents. Int J Neuropsychopharmacol. 2022;25(2):106–17. https://doi.org/10.1093/ijnp/pyab062.
Spravato [package insert]. Titusville, NJ, USA: Janssen Pharmaceuticals, Inc.; 2020. Janssen Pharmaceuticals Inc; 2020.
N ketamine injection [product monograph (Canada)]. Mississauga, Ontario, Canada: Baxter Corp; 2022. Baxter Corp; 2022.
Ketamine baxter [Australian product information]. Old Toongabbie, New South Wales, Australia: Baxter Healthcare Pty Ltd; 2020. Baxter Healthcare Pty Ltd; 2020.
Spravato [product monograph (Canada)]. Toronto, Ontario, Canada: Janssen Inc.; 2023. Janssen Inc.; 2023.
Janssen-Cilag International NV. Spravato smpc. 2023. Available from: https://www.Ema.Europa.Eu/en/documents/product-information/spravato-epar-product-information_en.Pdf Janssen-Cilag International NV; 2023.
Spravato [Australian product information]. Macquarie Park, New South Wales, Australia: Janssen-Cilag Pty Ltd; 2024. Janssen-Cilag Pty Ltd; 2024.
Wang SM, Kim NY, Na HR, et al. Rapid onset of intranasal esketamine in patients with treatment resistant depression and major depression with suicide ideation: a meta-analysis. Clin Psychopharmacol Neurosci. 2021;19(2):341–54. https://doi.org/10.9758/cpn.2021.19.2.341.
Popova V, Daly EJ, Trivedi M, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. 2019;176(6):428–38. https://doi.org/10.1176/appi.ajp.2019.19020172.
Reif A, Bitter I, Buyze J, et al. Esketamine nasal spray versus quetiapine for treatment-resistant depression. N Engl J Med. 2023;389(14):1298–309. https://doi.org/10.1056/NEJMoa2304145.
Bonaventura J, Lam S, Carlton M, et al. Pharmacological and behavioral divergence of ketamine enantiomers: implications for abuse liability. Mol Psychiatry. 2021;26(11):6704–22. https://doi.org/10.1038/s41380-021-01093-2.
Ren P, Wang J, Li N, et al. Sigma-1 receptors in depression: Mechanism and therapeutic development. Front Pharmacol. 2022;13: 925879. https://doi.org/10.3389/fphar.2022.925879.
Sałaciak K, Pytka K. Revisiting the sigma-1 receptor as a biological target to treat affective and cognitive disorders. Neurosci Biobehav Rev. 2022;132:1114–36. https://doi.org/10.1016/j.neubiorev.2021.10.037.
Schatzberg AF. A word to the wise about intranasal esketamine. Am J Psychiatry. 2019;176(6):422–4. https://doi.org/10.1176/appi.ajp.2019.19040423.
Brendle M, Ahuja S, Valle MD, et al. Safety and effectiveness of intranasal esketamine for treatment-resistant depression: a real-world retrospective study. J Comp Eff Res. 2022;11(18):1323–36. https://doi.org/10.2217/cer-2022-0149.
Samalin L, Rothärmel M, Mekaoui L, et al. Esketamine nasal spray in patients with treatment-resistant depression: the real-world experience in the French cohort early-access programme. Int J Psychiatry Clin Pract. 2022;26(4):352–62. https://doi.org/10.1080/13651501.2022.2030757.
Martinotti G, Vita A, Fagiolini A, et al. Real-world experience of esketamine use to manage treatment-resistant depression: a multicentric study on safety and effectiveness (real-esk study). J Affect Disord. 2022;319:646–54. https://doi.org/10.1016/j.jad.2022.09.043.
Stahl SM, Pradko JF, Haight BR, et al. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim Care Companion J Clin Psychiatry. 2004;6(4):159. https://doi.org/10.4088/pcc.v06n0403.
Wellbutrin sr [package insert]. Durham, NC, USA: Glaxosmithkline; 2022. GlaxoSmithKline; 2022.
Altice F, Evuarherhe O, Shina S, et al. Adherence to hiv treatment regimens: systematic literature review and meta-analysis. Patient Prefer Adherence. 2019;13:475–90. https://doi.org/10.2147/PPA.S192735.
Parati G, Kjeldsen S, Coca A, et al. Adherence to single-pill versus free-equivalent combination therapy in hypertension: A systematic review and meta-analysis. Hypertension. 2021;77(2):692–705. https://doi.org/10.1161/HYPERTENSIONAHA.120.15781.
Weisser B, Predel H-G, Gillessen A, et al. Single pill regimen leads to better adherence and clinical outcome in daily practice in patients suffering from hypertension and/or dyslipidemia: results of a meta-analysis. High Blood Press Cardiovasc Prev. 2020;27:157–64. https://doi.org/10.1007/s40292-020-00370-5.
Iosifescu DV, Jones A, O’Gorman C, et al. Efficacy and safety of AXS-05 (dextromethorphan-bupropion) in patients with major depressive disorder: a phase 3 randomized clinical trial (GEMINI). J Clin Psychiatry. 2022;83(4):41226. https://doi.org/10.4088/JCP.21m14345.
Tabuteau H, Jones A, Anderson A, et al. Effect of axs-05 (dextromethorphan-bupropion) in major depressive disorder: a randomized double-blind controlled trial. Am J of Psychiatry. 2022;179(7):490–9. https://doi.org/10.1176/appi.ajp.21080800.
Jones A, O'Gorman C, Clayton AH, et al. Analysis of efficacy of AXS-05 in the treatment of major depressive disorder based on gender, race, and prior antidepressant use. [poster presentation]. LSCTM Annual Meeting, New York, NY, USA; April 6-9 2021. https://isctm.org/public_access/17th_Annual/Poster/Jones_poster.pdf.
Jones A, Streicher C, Alter S, et al. Axs-05 (dextromethorphan-bupropion) improves depressive symptoms and functioning in patients with one prior treatment failure: results from the evolve long-term, open label study. CNS Spectr. 2023;28(2):247. https://doi.org/10.1017/S1092852923001840.
O'Gorman C, Jones A, Tabuteau H. Sustained efficacy with long-term treatment with AXS-05: Results from the comet phase 3 trial, a long-term, open open-label study evaluating the efficacy and safety of AXS-05 for the treatment of MDD. American Society of Clinical Psychopharmacology Annual Meeting; 2021; Miami Beach, FL; 2021.
Jones A, O'Gorman C, Tabuteau H. Sustained effects of AXS-05, an oral nmda receptor antagonist, in treatment resistant depression patients: Results from the COMET-TRD trial. American Society of Clinical Psychopharmacology Annual Meeting; 2021; Miami Beach, FL; 2021.
O'Gorman C, Jones A, Tabuteau H. Rapid reduction in suicidal ideation in patients treated with AXS-05, an oral nmda receptor antagonist with multimodal activity: results from the COMET-SI trial. American Society of Clinical Psychopharmacology Annual Meeting; 2021; Miami Beach, FL; 2021.
Carvalho AF, Sharma MS, Brunoni AR, et al. The safety, tolerability and risks associated with the use of newer generation antidepressant drugs: a critical review of the literature. Psychother Psychosom. 2016;85(5):270–88. https://doi.org/10.1159/000447034.
Cosci F, Chouinard G. Acute and persistent withdrawal syndromes following discontinuation of psychotropic medications. Psychother Psychosom. 2020;89(5):283–306. https://doi.org/10.1159/000506868.
Relmada Therapeutics Inc. Relmada Therapeutics announces top-line results from phase 3 Reliance I trial for REL-1017 as an adjunctive treatment for major depressive disorder.
Relmada Therapeutics Inc. Relmada Therapeutics announces top-line results from phase 3 Reliance III trial for rel-1017 as a monotherapy for the treatment of major depressive disorder.
Canuso CM, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind, randomized, placebo-controlled study. Am J Psychiatry. 2018;175(7):620–30. https://doi.org/10.1176/appi.ajp.2018.17060720.
Takahashi N, Yamada A, Shiraishi A, et al. Efficacy and safety of fixed doses of intranasal esketamine as an add-on therapy to oral antidepressants in Japanese patients with treatment-resistant depression: a phase 2b randomized clinical study. BMC Psychiatry. 2021;21(1):526. https://doi.org/10.1186/s12888-021-03538-y.
Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiat. 2018;75(2):139–48. https://doi.org/10.1001/jamapsychiatry.2017.3739.
Wajs E, Aluisio L, Holder R, et al. Esketamine nasal spray plus oral antidepressant in patients with treatment-resistant depression: Assessment of long-term safety in a phase 3, open-label study (sustain-2). J Clin Psychiatry. 2020. https://doi.org/10.4088/JCP.19m12891.
Fu DJ, Ionescu DF, Li X, et al. Esketamine nasal spray for rapid reduction of major depressive disorder symptoms in patients who have active suicidal ideation with intent: double-blind, randomized study (ASPIRE I). J Clin Psychiatry. 2020. https://doi.org/10.4088/JCP.19m13191.
Ionescu DF, Fu DJ, Qiu X, et al. Esketamine nasal spray for rapid reduction of depressive symptoms in patients with major depressive disorder who have active suicide ideation with intent: Results of a phase 3, double-blind, randomized study (ASPIRE II). Int J Neuropsychopharmacol. 2021;24(1):22–31. https://doi.org/10.1093/ijnp/pyaa068.
Chen X, Hou X, Bai D, et al. Efficacy and safety of flexibly dosed esketamine nasal spray plus a newly initiated oral antidepressant in adult patients with treatment-resistant depression: a randomized, double-blind, multicenter, active-controlled study conducted in China and USA. Neuropsychiatr Dis Treat. 2023;19:693–707. https://doi.org/10.2147/ndt.S391096.
Fedgchin M, Trivedi M, Daly EJ, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressant in treatment-resistant depression: results of a randomized, double-blind, active-controlled study (TRANSFORM-1). Int J Neuropsychopharmacol. 2019;22(10):616–30. https://doi.org/10.1093/ijnp/pyz039.
Daly EJ, Trivedi MH, Janik A, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2019;76(9):893–903. https://doi.org/10.1001/jamapsychiatry.2019.1189.
Axsome Therapeutics Inc. Axsome Therapeutics announces topline results of the STRIDE-1 phase 3 trial in treatment resistant depression and expert call to discuss clinical implications.
Lener MS, Niciu MJ, Ballard ED, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. 2017;81(10):886–97. https://doi.org/10.1016/j.biopsych.2016.05.005.
Acknowledgements
Sarah Engelberth, Ph.D., of Nucleus Global, an Inizio company, provided editorial support for the manuscript, including drafting of the initial outline and revision at all stages, under the direction of the authors, funded by Axsome Therapeutics, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Development of this review was funded by Axsome Therapeutics., Inc., the manufacturer of Auvelity® (dextromethorphan–bupropion). The sponsor was involved in the original conception of the study; however, the authors were responsible for the critical appraisal of the work and decision to submit the manuscript for publication.
Conflict of interest
R. Jain received speakers’ fees from Alkermes, Corium, Intra-Cellular Therapies, Ironshore Pharmaceuticals, Lilly, Lundbeck, Merck, Neos Therapeutics, Otsuka, Pamlab, Pfizer, Shire, Sunovion, Takeda, and Tris Pharmaceuticals; consultant fees from AbbVie, Acadia, Adamas, Alfasigma, Cingulate Therapeutics, Eisai, Evidera, Impel, Janssen, Lilly, Lundbeck, Merck, Neos Therapeutics, Neurocrine Biosciences, Osmotica, Otsuka, Pamlab, Pfizer, Shire, Sunovion, Supernus, Takeda, and Teva; is an advisory board member for Adamas, Alkermes, Janssen, Lilly, Lundbeck, Merck, Neos Therapeutics, Neurocrine Biosciences, Otsuka, Pamlab, Pfizer, Shire, Sunovion, Supernus, Takeda, and Teva; and received research support from AbbVie, Lilly, Lundbeck, Otsuka, Pfizer, Shire, and Takeda. R. S. McIntyre has received research grant support from CIHR/GACD/National Natural Science Foundation of China (NSFC); and speaker/consultation fees from Lundbeck, Janssen, Alkermes, Neumora Therapeutics, Boehringer Ingelheim, Sage, Biogen, Mitsubishi Tanabe, Purdue, Pfizer, Otsuka, Takeda, Neurocrine, Sunovion, Bausch Health, Axsome, Novo Nordisk, Kris, Sanofi, Eisai, Intra-Cellular Therapies, Inc., NewBridge Pharmaceuticals, AbbVie, and Atai Life Sciences. He is CEO of Braxia Scientific Corp (a company focused on ketamine treatment and psychedelic medicine for advanced mental health treatments).
Availability of data and material
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
Both authors contributed to the manuscript conception and design, read and critically reviewed and revised the work, approved the final submitted manuscript, and agree to be accountable for the work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
McIntyre, R.S., Jain, R. Glutamatergic Modulators for Major Depression from Theory to Clinical Use. CNS Drugs (2024). https://doi.org/10.1007/s40263-024-01114-y
Accepted:
Published:
DOI: https://doi.org/10.1007/s40263-024-01114-y