
Abstract
Purpose of Review
Over many decades, researchers have been designing studies to investigate the relationship between genotypes and phenotypes to gain an understanding about the effect of genetics on disease. Recently, a high-throughput approach called phenome-wide associations studies (PheWAS) have been extensively used to identify associations between genetic variants and many diseases and traits simultaneously. In this review, we describe the value of PheWAS along with methodological issues and challenges in interpretation for current applications of PheWAS.
Recent Findings
PheWAS have uncovered a paradigm to identify new associations for genetic loci across many diseases. The application of PheWAS has been effective with phenotype data from electronic health records, epidemiological studies, and clinical trials data.
Summary
The key strength of PheWAS is to identify the association of one or more genetic variants with multiple phenotypes, which can showcase interconnections among the phenotypes due to shared genetic associations. While the PheWAS approach appears promising, there are a number of challenges that need to be addressed to provide additional robustness to PheWAS findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In an emerging clinical discipline called precision medicine, the primary focus is to use an individual’s clinical data along with genetic, environmental, and lifestyle information to tailor clinical care. The steps toward discovery for precision medicine involve enrollment of individuals into studies to then link their genotype and phenotype data to identify clinically relevant genetic associations. The most common methodology to determine such genotype-phenotype connections is called genome-wide association studies (GWAS) [1•, 2], where tests for association are performed between single-nucleotide polymorphisms (SNPs) across the genome (usually over 500,000 SNPs) and a single disease outcome or trait. There is now growing evidence demonstrating the validity of some of these genetic associations [3••]. However, there is still a limited impact of GWAS due to its focus on a single phenotype, and hence, the exploration of the effect of a given SNP across multiple phenotypes is not feasible. For instance, there are a number of GWAS that have identified associations between loci in the fat mass and obesity (FTO) gene and loci in the body mass index (BMI) [4,5,6]. There are known predispositions to various diseases due to variation in BMI [7, 8]. However, by design, the focus of the GWAS case-control studies of BMI limits their ability to identify links between variations in the FTO gene with other diseases in a high-throughput manner.
An alternative approach called phenome-wide association study (PheWAS) has shown some success by simultaneously scanning genome-wide significant variants over hundreds or thousands of phenotypes [9••, 10••]. For example, using PheWAS, Cronin et al. examined the aforementioned genome-wide significant FTO locus across a number of diseases. They identified not only BMI-mediated disorders such as obesity and type 2 diabetes but also associations with sleep apnea, fibrocystic breast disease, nonalcoholic liver disease, and gram-positive bacterial infections [11]. PheWAS is a high-throughput way to identify such cross-phenotype associations, i.e., an association of genetic variant with multiple phenotypes, diseases, or traits. Such findings have the potential to uncover pleiotropy or an underlying genetic architecture of disease comorbidities. In early 2010, Denny et al. demonstrated the first successful application of the PheWAS methodology using phenotypes derived from an electronic health record (EHR) [9]. Before this study, researchers in the Electronic Medical Records and Genomics (eMERGE) network had developed approaches to identify the relationship between genetic variants and a few phenotypes derived from EHRs [10]. The eMERGE network is a collection of biorepositories with genetic data linked to EHRs within different healthcare systems across the USA [12]. The first PheWAS analysis illustrated the value of using billing codes within EHR for retrospective genomic studies. Since then, the utility of EHR data has exponentially grown for genomic studies from understanding the underlying biology of complex diseases to novel drug targets and their side effects [13,14,15,16]. The genetic component of PheWAS is not limited to SNPs; we can use structural variations (copy number variations), mitochondrial variation [17], and gene regions for low-frequency and rare variants (population allele frequency < 1%) [18•] as well as non-genetic measures such as clinical laboratory measures [19] and quantitative measures derived from biomarkers.
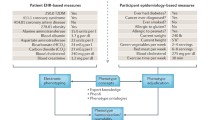
Here, we review the current scope, application, and key association findings of the PheWAS methodology (Fig. 1). We provide an introduction to the different data types used for PheWAS and the developments in algorithmic approaches to define phenotypes for clinical research. We then review different methods and tools available to perform PheWAS (Table 1). We subsequently provide a brief overview of upcoming methods and tools in the development for PheWAS analysis. We end our review by providing current limitations and a commentary on the future direction of PheWAS applications.

Current scope of phenome-wide associations study (PheWAS). A methodology to link genetic variations with a broad spectrum of phenotypes using statistical tests to identify genetic associations with a single trait or phenotype as well as multiple phenotypes. SNPs (single-nucleotide polymorphisms) are the most commonly used genetic variations in PheWAS. The phenotypes in PheWAS can be derived from electronic health records (ICD-9 billing codes, clinical laboratory measurements), epidemiology cohorts (self-reported disease status, clinical laboratory measures, health surveys), or clinical trials (clinical laboratory measurements). PheWAS is a high-throughput approach to scan thousands of phenotypes which can be further used to generate new hypothesis for a more focused analysis on specific phenotypes
Phenotype Data in PheWAS
The majority of PheWAS studies has used data from de-identified EHRs [9, 11, 17, 20,21,22,23,24,25,26,27,28] linked to genotype data, while a few have been performed in large-scale epidemiologic studies [29,30,31,32] and clinical trials [33, 34]. The representation of the phenome varies in each of these types of studies. For PheWAS in the EHR, the phenome can be represented as billing codes, PheCodes [9••], clinical lab measurements, or comprehensive electronic phenotyping algorithms. In the epidemiology and clinical trial-based PheWAS, the phenome can be represented by the data types collected in the study which may include lab measurements, biomarker assays, self-report health and disease history, and environmental surveys. The representation of the phenome drives the selection of the statistical technique for the PheWAS analyses, thus is a critical component of the PheWAS design.
To date, the billing codes within EHR are most extensively used in EHR-based PheWAS. The World Health Organization (WHO) maintains these billing codes, also commonly known as international classification of disease (ICD) codes, to classify human diseases in standard units [35••]. In a health system, ICD codes are primarily referred to as billing codes since historically their primary purpose was to use them for insurance claims. ICD codes consist of codes related to signs, symptoms, disease diagnoses, and procedures, as well as injuries and related conditions. These codes are a reflection of an individual’s health over the time, and their frequent presence within the EHR has made them a valuable tool for research. There are multiple versions of ICD codes, and new revisions replace the previous versions. ICD version 10 (ICD-10) is the most recent. Up until October 2015, all health systems within the USA used version 9 of the ICD codes (ICD-9). The ICD-9 codes are alphanumeric codes ranging from three- to five-digit codes, where the first three digits are the category of the condition and each digit after the decimal could represent anatomical location or severity of the condition. For example, a three-digit code “440” is used for “atherosclerosis,” the fourth digit of the code provides more specificity about location of the disease (440.0—atherosclerosis of aorta; 440.1—atherosclerosis of renal artery), and lastly, the fifth digit provides additional specificity (440.21—atherosclerosis of native arteries of the extremities with rest pain; 440.23—atherosclerosis of native arteries of the extremities with ulceration). Whereas, ICD-10 codes have considerably different disease concepts and structure than ICD version 9. The length of ICD-10 code ranges from 3 to 7 characters, with first three characters represent disease category, and then each character after the decimal provides more detail on the disease etiology, anatomic site, severity, and laterality. For example, ICD-9 code 440.23 translates to “I70.209” (unspecified atherosclerosis of native arteries of extremities, unspecified extremity) in ICD-10. There are over 68,000 ICD-10 codes in comparison to approximately 14,000 ICD-9 codes, and this pose some challenges to translate existing ICD-9 codes in EHRs to the ICD-10 equivalents. There are several pitfalls in mapping codes between the two versions, where there are both one-to-many and many-to-one mappings. For example, ICD-9 code 250.81 (diabetes with other specified manifestations, type I (juvenile type), not stated as uncontrolled) maps to ten different ICD-10 codes. There are a handful of conversion tools available now to map ICD-9 to ICD-10 [36]. However, it is a significant undertaking to convert existing ICD-9 codes in the EHR systems to ICD-10 codes and make them research ready, especially when there are no research-acceptable ICD-10 diagnosis codes as of yet.
To date in the area of PheWAS research, ICD-9 diagnosis codes are most commonly used to design a case control-based study. There can be instances of misdiagnosis for an ICD-9 code or short visit history of a patient within a health system, which can create ambiguity. The individuals in a study using EHR data are defined as cases when there are at least multiple instances, such as three, for a given code at different time points (this is referred to as rule-of-three). The threshold varies across different studies; in most cases, it is agreed upon to use at least two or more instances to define a case. All the individuals with the absence of that code are assigned as controls. The ICD-9 codes can be used in the raw form as well as grouped into custom groupings. Since ICD-9 codes are categorized in a hierarchy, we can group four- and five-digit codes to three digits, which can be one way to combine correlated phenotypes. Although, such grouping of individual ICD-9 codes requires careful consideration, for example, subcategories of ICD-9 code “250” (diabetes mellitus) consist of two clinically different diseases type 1 diabetes (250.1*) and type II diabetes (250.2*) and they should be investigated independently. There is another custom grouping of ICD-9 codes developed by Denny et al. called PheCodes (or PheWAS Codes) [9••]. The PheCodes are a curated list of diagnosis codes where similar disease codes are combined into custom higher category codes. The replication of SNP-ICD-9 code association across different EHRs can be effective using PheCodes [13]. These PheCodes are currently established for ICD-9 only and has not yet been extended or adapted to include ICD-10.
In EHRs, the combination of different measures can be used to develop well-informed algorithms to understand the genetic etiology of diseases better and elucidate our understanding of the architectural differences due to raw phenotypes and processed phenotype data. Here, processed phenotypes refer to the phenotypes derived through electronic phenotype algorithms which utilize ICD-9 codes, clinical notes, clinical laboratory measures, demographic, and lifestyle information in combination to define the phenotype. Researchers in the eMERGE network have developed a catalog called PheKB, which consists of over 30 phenotype algorithms to derive phenotypes from EHRs [37].
There are also studies which have demonstrated the use of clinical laboratory measures from EHR as well as epidemiological studies and clinical trials in PheWAS [29, 32,33,34, 38] . The laboratory measures are quantitative values collected from patients during the routine check-ups as well as for the diagnosis and monitoring of the diseases. The phenome derived from epidemiological cohorts also includes self-reported disease status, nutrition diet supplements, psychiatric traits, use of alcohol, smoking habits, and more. Table 1 shows the different phenotype categories used for different PheWAS studies, which is an important component to then select the analysis method used.
Candidate Variants PheWAS
To date, the bulk of PheWAS analyses has centered on candidate variant analyses, where researchers have assessed the variants identified through GWAS against a comprehensive list of phenotype measurements. Researchers at Vanderbilt University performed an initial PheWAS application in their EHR through a combination approach to first perform a GWAS on an EHR-derived phenotype and then further extending it to a PheWAS on the variants identified as genome-wide significant [9••, 10••]. This study reported variants associated with electrocardiographic QRS duration in GWAS, and subsequently, the variations in SCN5A/10A were also associated with atrial fibrillation and cardiac arrhythmias [10••]. In a similar approach, a new genetic locus rs965513 in forkhead box E1 (FOXE1) was associated with primary hypothyroidism (through GWAS) as well as new diseases derived from ICD-9 billing codes for thyroiditis, nutritional deficiency of anemia, and non-toxic nodular goiter, among others [55]. The FOXE1 locus also has known connections with thyroid cancer through GWAS [56, 57]. Additionally, a systematic application of PheWAS using EHR-derived phenotypes replicated 66% of associations previously reported in the NHGRI GWAS catalog [20]. This study demonstrates the accuracy and reliability of associations identified by PheWAS methodology. All of these studies highlight the key feature of PheWAS which is to expand the search space of diseases that are not considered before for known disease-related genetic variants.
Researchers have used epidemiological cohorts linked with genetic information in PheWAS to capture novel associations with self-reported disease status; clinical laboratory measures (LDL, HDL, glucose, and more); nutrition diet supplements; and environmental measures. These studies include data from cohorts such as Population Architecture using Genomics and Epidemiology network and National Health and Nutrition Examination Survey. The PheWAS on ethnically diverse individuals in these cohorts identified many novel genetic associations that are ancestry-specific as well as significant in one or more race/ethnicity groups [29, 32, 58]. Overall, these studies illustrate that PheWAS using epidemiological study data can also be used to improve the characterization of disease and health outcomes.
The choice of statistical method in PheWAS to investigate genetic associations with complex diseases is conceptually similar to GWAS. In its simplest form, the allele distribution of genetic variants across a given population is compared against case and control status of each phenotype. For binary outcomes, logistic regression is a popular method, and the statistical model can be adjusted for confounding effects such as age, sex (male or female), population structure, and others. For quantitative variables such as clinical laboratory measures, linear regression or analysis of variance are commonly used for analysis. Researchers use different software packages for the implementation of these regression tests for PheWAS analysis such as PLINK [59, 60]; PLATO [61]; and R-PheWAS [62], SAS software©, and STATA [63].
Genome-Wide PheWAS
Evaluating a range of phenotypes or traits in PheWAS shows its advantage over traditional GWAS on a single trait and the transition to investigating all the variants across the genome is shown in a handful of PheWAS publications. In the first genome-wide PheWAS, researchers used 27 laboratory measurements from antiretroviral therapy-naive individuals enrolled in AIDS (acquired immunodeficiency syndrome) clinical trials [33]. In this proof-of-concept study, authors demonstrated that the identified cross-phenotype associations highlight the important interrelationships between the phenotypes from treatment-naive individuals. The study also highlights that PheWAS can be used to create new hypotheses to analyze intermediate phenotypes, subphenotypes, and endophenotypes, and to identify pharmacogenomic associations to better under understand the pharmacokinetics of the drugs [34].
Recently, researchers at Geisinger Health System and the Michigan Genomics Initiative conducted separate genome-wide PheWAS using clinical data from EHR [50, 51]. Verma et al. used PheWAS to investigate all common variants on the Illumina HumanCoreExome chip and clinical laboratory measures from ~ 12,000 European American individuals [50]. Subsequently, they tested the significant SNPs from the clinical lab PheWAS with 541 diagnosis codes [50]. Dey et al. demonstrated the application of a new statistical method (Table 1) for PheWAS and tested ~ 30 million imputed SNPs with 1500 EHR-based PheWAS codes [51]. Dey et al. also proposed a new method for binary outcomes, called SPAtest, which is a variation of logistic regression that estimates p values using saddlepoint approximation. The authors demonstrate that this approximation method is computationally efficient than traditional regression methods [51]. This approach can be computationally efficient for large-scale genome-wide PheWAS, especially for studies with an unbalanced case-control ratio [51].
Rare-Variant PheWAS
As the cost of DNA sequencing over the years continues to decrease, whole genome and exome sequencing are discovering large numbers of low-frequency genetic variations commonly known as rare variants. These new technologies present an excellent opportunity to identify rare-variant associations with clinical phenotypes and diseases. The traditional statistical methods such as logistic regression, linear regression for GWAS, and PheWAS approaches are usually underpowered for rare-variant analysis due to low sample size for each rare variant, although considerable progress has been made in method development to identify disease associations with rare variants such as gene burden tests, dispersion tests, and variance-based tests, among others [64]. Application of rare-variant tests has been limited in PheWAS where multiple phenotypes are simultaneously studied. A study from Basile et al. proposed an approach to create bins of rare variants based on the prior biological knowledge and further test for association with phenotypes using dispersion test such as SKAT (sequence kernel association test). In the study, they performed rare-variant PheWAS on variants with MAF < 0.01 in 82 known pharmacogenes and nine phenotypes derived from EHR-based phenotype algorithms [18•].
The use of different statistical methods to perform rare-variant analysis has been available for quite some time. Lee et al. present an extensive review of different methodology available for rare-variant association studies [64]. Rare-variant analysis in PheWAS is still in an early stage, and there are only a handful of out-of-the-box tools that allow the investigation of multiple phenotypes in a high-throughput manner such as BioBin [65], RV-test [66], and PLINK/SEQ [67].
PheWAS Using Non-genetic Information
The majority of PheWAS to date still focuses on identifying the associations of SNPs with phenotypes across the phenome. However, recently, a few studies have demonstrated the application of PheWAS using non-genetic measures. For rheumatoid arthritis (RA), Doss et al. utilized serologic tests to group RA patients into two groups: seropositive RA and seronegative RA individuals, to derive a binary independent variable [52]. Then they tested the independent variable against disease PheCodes [52] (custom ICD-9 PheCode grouping). In this study, an association between fibromyalgia and seronegative RA was most significant, and seropositive RA had associations with chronic airway obstruction and tobacco use. In an independent work, Liao et al. presented a more targeted approach to investigate comorbidities in patients with RA [19]. They used quantitative measures from 36 autoantibodies grouped by ten antigens with known connections to RA and comprehensively tested these measures against PheCodes. The key finding was between autoantibody fibronectin and obesity as well as between fibrinogen and pneumonopathy. These studies utilized logistic and linear regression implemented in PLINK and R PheWAS packages for association analysis. Both of these studies highlight the importance of analyzing subphenotypes within RA patients and the patterns of different diseases that occur in these groups. We can expect to see more use of lab measures to define patient subgroups for investigating phenotype heterogeneity in a study population.
Conclusions
Challenges and Future Directions
Current development and infrastructure support for biobanks linked to genetic information suggest that there will be an increase in the collection of genomic and patient health data in the coming years. The use of biobanks for retrospective case-control studies has already shown some success, and it will continue to play a critical role in identifying novel genetic associations. The PheWAS methodology will become a run-of-the-mill approach to generate new hypotheses to study the interconnection between a wide range of disorders and associations across the genome. Although, there can be some challenges with the genome-wide PheWAS analysis such as multiple hypothesis testing and computational burden, which lead to a challenge in identifying true pleiotropic associations, biologically relevant associations, and interpreting the results in a high-throughput manner [13,14,15,16].
There are several challenges in the current scope of PheWAS which need additional development to enhance the robustness of PheWAS association findings. One such challenge in an association study with a large number of statistical tests between SNPs and phenotypes is the multiple hypothesis testing burden. Most commonly, a Bonferroni correction is applied to account for false positives due to multiple hypothesis testing [68]. The Bonferroni correction is an overly conservative approach, because it assumes that all the tests are independent. However, on many occasions, the SNPs included in a study may be correlated due to underlying linkage disequilibrium. In GWAS, a p value of 5 × 10−8 is considered as the genome-wide significant for common variants when tested with one phenotype or trait. As the field is moving toward genome-wide PheWAS, it will increase the number of SNP-phenotype models tested and thereby increase the Bonferroni threshold. For example, in an EHR, there are ~ 14,000 ICD-9 diagnoses codes, and if we test them with all the common variants (e.g., 1 million SNPs), then the threshold will increase to 3.57 × 10−12. Identifying independent SNPs through LD pruning can lower the p value threshold to some extent [69••]. There may also be correlation among the phenotypes; however, more methods development is required to identify independent phenotypes from EHR data. For example, calculating the pairwise correlation between the phenotypes to estimate the number of independent phenotypes to generate a more appropriate denominator for a Bonferroni correction could be explored. Developing more robust strategies for dealing with the multiple testing burdens that control the type I error rate (false positives), while also controlling the type 2 error rate (false negatives—missing true signals), are essential to the future of these endeavors.
The GWAS PheWAS will affect not only the multiple testing burden but also the computational burden. Even multithreading and parallel computing options in some of the current packages (PLINK, PLATO) might not be adequate since those will be limited to available computational resources. Cloud platforms such as Amazon web services (AWS), Google Cloud Platform, and Microsoft Azure can be used to develop new tools or extend existing tools to perform large-scale PheWAS in a more efficient and less time-consuming manner. Currently, there are a handful of cloud-based tools to perform GWAS such as Google BigQuery [70], easyGWAS [71], and CloudAssoc [72] but there are none currently available for PheWAS. There are also platforms built upon different clouds that can be used to perform association testing such as DNAnexus [73]. It is important to note that cloud computing can incur higher computation costs than the traditional high-performance cluster computing in a local computing environment.
PheWAS have identified genetic associations across different complex diseases and traits. However, the interpretation of results in a high-throughput manner is currently limited. There are two aspects of results analysis in PheWAS: (1) understanding the clinical relationship when there is a SNP associated with two or more phenotypes and (2) identifying biologically relevant associations based on the functional implications of the SNP. Although, identification of cross-phenotype associations is a strength of PheWAS, it can also be challenging to distinguish cross-phenotype associations which can be due to pleiotropy, comorbidity, or confounding phenotype patterns. Few statistical tests can help distinguish between these different types of cross-phenotype associations; Solovieff et al. provides a review on such methods [74•]. It is also important to investigate the functional implications of the SNPs for the statistically significant PheWAS associations, a similar challenge that we experience in GWAS. As explained earlier, the FTO gene association with obesity-related traits was discovered and replicated by many GWAS. The PheWAS approach led to identifying cross-phenotype associations with BMI, including obesity and type 2 diabetes (T2D) that could potentially also be due to the effect of FTO variants. Although, most significant correlations were observed in FTO gene, however, the direct functional implication of this gene with metabolic traits was not confirmed by association studies. Smemo et al. showed in their expression analysis of human brain that variants mapping to the FTO gene interacted with the promoter region of the IRX3 gene and observed an increase in the expression of IRX3 due to variants in FTO gene with known BMI association [75]. This finding highlights the importance of including functional information such as gene expression, eQTL, chromatin marks, and variant annotations to fine-map findings from association studies. Several existing statistical and functional methods for fine mapping can be applied to better understand the causality of the genetic variants identified through PheWAS such as PAINTOR [76], RIVERA [77], CAVIAR [78], IDEAS [79], and CHROMHMM [80], among others.
Conclusions
In this review, we presented the strengths and advantages of PheWAS as well as the challenges based on the current scope of these high-throughput association studies. Among many challenges, PheWAS also delivers tremendous opportunities for validating the robustness of associations and discovering cross-phenotype associations. Testing of multiple phenotypes together not only helps in identifying the same variants linked to multiple diseases but also helps in discovery and new hypothesis generation. Analyzing, different components of an individual’s information ranging from disease diagnosis, laboratory measures and demographic data help in elucidating the genetic architecture of complex traits that exist in both common and rare genetic variations in study populations. For future studies, rare-variant analyses utilizing multiple phenotypes, a better understanding through functional implication analyses, and optimized methods for high-throughput analyses are likely to strengthen PheWAS methodology and enhance our understanding of complex traits.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Bush WS, Moore JH. Chapter 11: genome-wide association studies. Lewitter F, Kann M, editors. PLoS Comput Biol. 2012;8(12):e1002822. https://doi.org/10.1371/journal.pcbi.1002822. This provides a basic understanding on designing genome-wide association studies
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24.
•• Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22. This is an excellent review of the successes and challenges with GWAS over past decade
Loos RJF, Yeo GSH. The bigger picture of FTO—the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2013;10:51–61.
Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–6.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007 ;316(5826):889–94.
Jacobsen S, Sonne-Holm S. Increased body mass index is a predisposition for treatment by total hip replacement. Int Orthop. 2005;29:229–34.
Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–51.
•• Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–10. This is the first PheWAS performed using EHRs data
•• Ritchie MD, Denny JC, Crawford DC, Ramirez AH, Weiner JB, Pulley JM, et al. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am J Hum Genet. 2010;86:560–72. This study demonstrates the application of EHRs data in genomic studies
Cronin RM, Field JR, Bradford Y, Shaffer CM, Carroll RJ, Mosley JD, et al. Phenomewide association studies demonstrating pleiotropy of genetic variants within FTO with and without adjustment for body mass index. Front Genet 2014;5.
McCarty CA, Chisholm RL, Chute CG, Kullo IJ, Jarvik GP, Larson EB, et al. The eMERGE network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genet. 2011 Jan 26;4:13. https://doi.org/10.1186/1755-8794-4-13.
Denny JC, Bastarache L, Roden DM. Phenome-wide association studies as a tool to advance precision medicine. Annu Rev Genomics Hum Genet. 2016;17:353–73.
Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome–phenome relationship using phenome-wide association studies. Nat Rev Genet. 2016;17:129–45.
Roden DM. Phenome-wide association studies: a new method for functional genomics in humans: Phenome-wide association studies. J Physiol. 2017;595:4109–15. Updated References
Pendergrass SA, Ritchie MD. Phenome-wide association studies: leveraging comprehensive phenotypic and genotypic data for discovery. Curr Genet Med Rep. 2015;3:92–100.
Mitchell SL, Hall JB, Goodloe RJ, Boston J, Farber-Eger E, Pendergrass SA, et al. Investigating the relationship between mitochondrial genetic variation and cardiovascular-related traits to develop a framework for mitochondrial phenome-wide association studies. BioData Min 2014;7.
• Basile AO, Wallace JR, Peissig P, McCarty CA, Brilliant M, Ritchie MD. Knowledge driven binning and PheWAS analysis in Marshfield personalized medicine research project using BIOBIN. Pac Symp Biocomput. 2016;21:249–60. This is the first published rare-variant PheWAS analysis
Liao KP, Sparks JA, Hejblum BP, Kuo I-H, Cui J, Lahey LJ, et al. Phenome-wide association study of autoantibodies to Citrullinated and Noncitrullinated Epitopes in rheumatoid arthritis: PheWAS FOR AUTOANTIBODIES IN RA. Arthritis Rheumatol. 2017;69:742–9.
Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–10.
Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet. 2011;89:529–42.
Hebbring SJ, Schrodi SJ, Ye Z, Zhou Z, Page D, Brilliant MH. A PheWAS approach in studying HLA-DRB1*1501. Genes Immun. 2013;14:187–91.
Verma A, Verma SS, Pendergrass SA, Crawford DC, Crosslin DR, Kuivaniemi H, et al. eMERGE Phenome-Wide Association Study (PheWAS) identifies clinical associations and pleiotropy for stop-gain variants. BMC Med. Genomics. 2016;9. Available from: http://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-016-0191-8
Verma A, Basile AO, Bradford Y, Kuivaniemi H, Tromp G, Carey D, et al. Phenome- wide association study to explore relationships between immune system related genetic loci and complex traits and diseases. Yao Y-G, editor. PLoS One. 2016;11(8):e0160573. https://doi.org/10.1371/journal.pone.0160573. eCollection 2016
Verma SS, Lucas AM, Lavage DR, Leader JB, Metpally R, Krishnamurthy S, et al. Identifying genetic associations with variability in metabolic health and blood count laboratory values: diving into the quantitative traits by leveraging longitudinal data from an ehr. Pac Symp Biocomput Pac Symp Biocomput. 2016;22:533–44.
Shameer K, Denny JC, Ding K, Jouni H, Crosslin DR, de Andrade M, et al. A genomeand phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet. 2014;133(1):95–109. https://doi.org/10.1007/s00439-013-1355-7.
Namjou B, Marsolo K, Caroll RJ, Denny JC, Ritchie MD, Verma SS, et al. Phenomewide association study (PheWAS) in EMR-linked pediatric cohorts, genetically links PLCL1 to speech language development and IL5-IL13 to Eosinophilic Esophagitis. Front Genet 2014; 5. Available from: http://journal.frontiersin.org/article/10.3389/fgene.2014.00401/abstract
Ye Z, Mayer J, Ivacic L, Zhou Z, He M, Schrodi SJ, et al. Phenome-wide association studies (PheWASs) for functional variants. Eur J Hum Genet. 2015;23:523–9.
Pendergrass SA, Brown-Gentry K, Dudek S, Frase A, Torstenson ES, Goodloe R, et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the population architecture using genomics and epidemiology (PAGE) network. PLoS Genet. 2013;9(1):e1003087. https://doi.org/10.1371/journal.pgen.1003087.
Pendergrass SA, Brown-Gentry K, Dudek SM, Torstenson ES, Ambite JL, Avery CL, et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet Epidemiol. 2011;35:410–22.
Polimanti R, Kranzler HR, Gelernter J. Phenome-wide association study for alcohol and nicotine risk alleles in 26394 women. Neuropsychopharmacology. 2016;41:2688–96.
Hall MA, Verma A, Brown-Gentry KD, Goodloe R, Boston J, Wilson S, et al. Detection of Pleiotropy through a Phenome-wide association study (PheWAS) of epidemiologic data as part of the environmental architecture for genes linked to environment (EAGLE) study. PLoS Genet. 2014;10(12):e1004678. https://doi.org/10.1371/journal.pgen.1004678. eCollection 2014 Dec.
Moore CB, Verma A, Pendergrass S, Verma SS, Johnson DH, Daar ES, et al. Phenomewide association study relating Pretreatment laboratory parameters with human genetic variants in AIDS Clinical Trials Group protocols. O Open Forum Infect Dis. 2015;2(1):ofu113. https://doi.org/10.1093/ofid/ofu113. eCollection 2015 Jan
Verma A, Bradford Y, Verma SS, Pendergrass SA, Daar ES, Venuto C, et al. Multiphenotype association study of patients randomized to initiate antiretroviral regimens in AIDS Clinical Trials Group protocol A5202: Pharmacogenet. Genomics. 2017;27:101–11.
• World Health Organization. History of the development of the ICD [Internet]. Available from: http://www.who.int/classifications/icd/en/HistoryOfICD.pdf. A great review of ICD codes.
Krive J, Patel M, Gehm L, Mackey M, Kulstad E, Li J “John,” et al. The complexity and challenges of the international classification of diseases, ninth revision, clinical modification to international classification of diseases, 10th revision, clinical modification transition in EDs. Am J Emerg Med 2015;33:713–718.
Kirby JC, Speltz P, Rasmussen LV, Basford M, Gottesman O, Peissig PL, et al. PheKB: a catalog and workflow for creating electronic phenotype algorithms for transportability. J Am Med Inform Assoc. 2016;23:1046–52.
Crawford DC, Goodloe R, Brown-Gentry K, Wilson S, Roberson J, Gillani NB, et al. Characterization of the Metabochip in diverse populations from the international HapMap project in the epidemiologic architecture for genes linked to environment (EAGLE) project. Pac Symp Biocomput. 2013:188–99.
Emdin CA, Khera AV, Natarajan P, Klarin D, Won H-H, Peloso GM, et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;68:2761–72.
Hebbring SJ, Rastegar-Mojarad M, Ye Z, Mayer J, Jacobson C, Lin S. Application of clinical text data for phenome-wide association studies (PheWASs). Bioinformatics. 2015;31:1981–7.
Karaca S, Civelek E, Karaca M, Sahiner UM, Ozgul RK, Kocabas CN, et al. Allergyspecific Phenome-wide association study for Immunogenes in Turkish children. Sci Rep. 2016 Sep 14;6:33152. https://doi.org/10.1038/srep33152.
Klarin D, Emdin CA, Natarajan P, Conrad MF, INVENT consortium, Kathiresan S. Genetic analysis of venous Thromboembolism in UK biobank identifies the ZFPM2 locus and implicates obesity as a causal risk factor. Circ Cardiovasc Genet. 2017;10(2). https://doi.org/10.1161/CIRCGENETICS.116.001643.
Liu J, Ye Z, Mayer JG, Hoch BA, Green C, Rolak L, et al. Phenome-wide association study maps new diseases to the human major histocompatibility complex region. J Med Genet. 2016;53:681–9.
Millwood IY, Bennett DA, Walters RG, Clarke R, Waterworth D, Johnson T, et al. A phenome-wide association study of a lipoprotein-associated phospholipase A2 loss-offunction variant in 90 000 Chinese adults. Int J Epidemiol. 2016;45:1588–99.
Namjou B, Marsolo K, Lingren T, Ritchie MD, Verma SS, Cobb BL, et al. A GWAS study on liver function test using eMERGE network participants. Prokunina-Olsson L, editor. PLoS One. 2015;10:e0138677.
Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–85.
Simonti CN, Vernot B, Bastarache L, Bottinger E, Carrell DS, Chisholm RL, et al. The phenotypic legacy of admixture between modern humans and Neandertals. Science. 2016;12:737–41.
Wang X, Pandey AK, Mulligan MK, Williams EG, Mozhui K, Li Z, et al. Joint mousehuman phenome-wide association to test gene function and disease risk. Nat Commun. 2016;7:10464.
Millard LAC, Davies NM, Timpson NJ, Tilling K, Flach PA, Smith GD. MR-PheWAS: hypothesis prioritization among potential causal effects of body mass index on many outcomes, using Mendelian randomization. Sci Rep. 2015;5:16645. https://doi.org/10.1038/srep16645.
Verma A, Leader JB, Verma SS, Frase A, Wallace J, Dudek S, et al. Integrating clinical laboratory measures and ICD-9 code diagnoses in phenome-wide association studies. Pac Symp Biocomput. 2016;21:168–79.
Dey R, Schmidt EM, Abecasis GR, Lee S. A fast and accurate algorithm to test for binary phenotypes and its application to PheWAS. Am J Hum Genet. 2017;101:37–49.
Doss J, Mo H, Carroll RJ, Crofford LJ, Denny JC. Phenome-wide association study of rheumatoid arthritis subgroups identifies association between Seronegative disease and fibromyalgia: PheWAS of rheumatoid arthritis subgroups. Arthritis Rheumatol. 2017;69:291–300.
Liao KP, Kurreeman F, Li G, Duclos G, Murphy S, Guzman R, et al. Associations of autoantibodies, autoimmune risk alleles, and clinical diagnoses from the electronic medical records in rheumatoid arthritis cases and non-rheumatoid arthritis controls. Arthritis Rheum. 2013;65:571–81.
Neuraz A, Chouchana L, Malamut G, Le Beller C, Roche D, Beaune P, et al. Phenome- wide association studies on a quantitative trait: application to TPMT enzyme activity and Thiopurine therapy in Pharmacogenomics. Slonim DK, editor. PLoS Comput Biol. 2013;9:e1003405.
Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical Records for Genome- and Phenome-Wide Studies. Am J Hum Genet. 2011;89:529–42.
Takahashi M, Saenko VA, Rogounovitch TI, Kawaguchi T, Drozd VM, Takigawa-Imamura H, et al. The FOXE1 locus is a major genetic determinant for radiation-related thyroid carcinoma in Chernobyl. Hum Mol Genet. 2010;19:2516–23.
Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat. Genet. 2009;41:460–4.
Bush WS, Boston J, Pendergrass SA, Dumitrescu L, Goodloe R, Brown-Gentry K, et al. Enabling high-throughput genotype-phenotype associations in the epidemiologic architecture for genes linked to environment (EAGLE) project as part of the population architecture using genomics and epidemiology (PAGE) study. Pac Symp Biocomput. 2013:373–84.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4. Available from: https://academic.oup.com/gigascience/articlelookup/doi/10.1186/s13742-0150047-8
Hall MA, Wallace J, Lucas AM, Kim D, Verma SS, McCarty CA, et al. PLATO software provides analytic framework for investigating complexity beyond genome-wide association studies. Nat. Commun. 2017. https://doi.org/10.1038/s41467-017-00802-2
Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinforma Oxf Engl. 2014;30:2375–6.
StataCorp. Stata statistical software: release 12. College Station, TX: StataCorp LP; 2011.
Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95:5–23.
Moore CB, Wallace JR, Frase AT, Pendergrass SA, Ritchie MD. BioBin: a bioinformatics tool for automating the binning of rare variants using publicly available biological knowledge. BMC Med Genet. 2013;6(Suppl 2):S6. https://doi.org/10.1186/1755-8794-6-S2-S6.
Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data: table 1. Bioinformatics. 2016;32:1423–6.
PLINK/SEQ: A library for the analysis of genetic variation data. at [Internet]. Available from: <http://atgu.mgh.harvard.edu/plinkseq/>.
Armstrong RA. When to use the Bonferroni correction. Ophthalmic Physiol Opt. 2014;34:502–8.
•• Sobota RS, Shriner D, Kodaman N, Goodloe R, Zheng W, Gao Y-T, et al. Addressing population-specific multiple testing burdens in genetic association studies: population-specific genome-wide thresholds. Ann Hum Genet. 2015;79:136–47. In this study, the authors describe an in-depth approach to identify multiple hypothesis correction thresholds based on different population linkage disequilibrium structures
Google BigQuery [Internet]. Available from: https://cloud.google.com/bigquery/what-isbigquery
Grimm DG, Roqueiro D, Salomé PA, Kleeberger S, Greshake B, Zhu W, et al. easyGWAS: a cloud-based platform for comparing the results of genome-wide association studies. Plant Cell. 2017;29:5–19.
Dai W, Wang Q, Gao M, Zhang L. CloudAssoc: A pipeline for imputation based genome wide association study on cloud. IEEE; 2012 [cited 2017 Sep 16]. p. 1435–8. Available from: http://ieeexplore.ieee.org/document/6526190/
DNAnexus Cloud Platform: A Global Network for Genomics [Internet]. Available from: https://www.dnanexus.com/company
• Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013 Jul;14(7):483–95. https://doi.org/10.1038/nrg3461. This is a review on challenges with differentiating true vs false pleiotropy and a review on statistical methods to address such challenges
Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, et al. Obesityassociated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–5.
Kichaev G, Yang W-Y, Lindstrom S, Hormozdiari F, Eskin E, Price AL, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. Di Rienzo a, editor. PLoS Genet. 2014;10:e1004722.
Li Y, Kellis M. Joint Bayesian inference of risk variants and tissue-specific epigenomic enrichments across multiple complex human diseases. Nucleic Acids Res. 2016;44:e144–4.
Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E. Identifying causal variants at loci with multiple signals of association. Genetics. 2014;198:497–508.
Zhang Y, An L, Yue F, Hardison RC. Jointly characterizing epigenetic dynamics across multiple human cell types. Nucleic Acids Res. 2016;44:6721–31.
Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9:215–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Anurag Verma and Marylyn D. Ritchie each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article contains no studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Genetic Epidemiology
Rights and permissions
About this article

Cite this article
Verma, A., Ritchie, M.D. Current Scope and Challenges in Phenome-Wide Association Studies. Curr Epidemiol Rep 4, 321–329 (2017). https://doi.org/10.1007/s40471-017-0127-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40471-017-0127-7