
Abstract
The worldwide prevalence of obesity has nearly doubled, with an increase in obesity-related cardiovascular disease and mortality. Several factors are involved in the genesis of hypertension and hypertensive heart disease (HHD) in overweight/obesity. This review is focused on bridging factors between excessive adiposity and HHD, presenting a unifying hypothesis of vascular–metabolic syndrome, where an “handicap” of the natriuretic peptide system has a central role both in adipocyte dysmetabolism as well as in increased blood pressure and HHD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 The Increasing of Obesity-Related Metabolic and Cardiovascular Health Problems
The worldwide prevalence of obesity has nearly doubled in the last decades, reaching a 35 % of adults (aged 20+) with overweight (BMI ≥25 kg/m2), a total of more than half a billion adults worldwide [1], together with medical care costs that are staggering [2].The prevalence of overweight/obesity is also increasing in our Country from 52.0 to 55.3 % in men and from 33.6 to 34.5 % in women between 2001 and 2008, particularly in Southern Italy and in less educated people [3]. In Italy the prevalence of metabolic syndrome is 23 % and it increases with age, comparable to the US data (both evaluated with the ATP III criteria) [4, 5].
Many years ago, Stamler et al. [6] in the Chicago Heart Association Detection Project in Industry, which enrolled more than 38,000 individuals from 1967 to 1973, highlighted the joint risk between obesity and hypertension. Thirty two-year CVD death rates were higher for patients with higher BMI at baseline and no hypertension (when hypertension was defined by a higher blood pressure (BP) levels than today’s). For those with hypertension at baseline, CVD death rates were substantially higher overall, and increased in a stepwise fashion for patients with higher baseline BMI levels. A similar pattern of results was observed for individual outcomes of CHD death and stroke death rates, as well as for hospitalizations for CHD, stroke, and HF during follow-up using Medicare data.
The increased obesity-related cardiovascular disease and mortality is not “a bolt from the blue” but it is associated with cardiovascular damage that is most often subclinical and that can be present from a young age. In a study of 260,000 overweight and obese children in Germany and Switzerland, 35 % had hypertension with increased ventricular mass or arterial stiffness [7]. In four cohort studies followed for a mean of 23 years, overweight or obese children who remained obese as adults had substantially increased risk of hypertension, diabetes, dyslipidemia, and carotid atherosclerosis [8].
In Europe, the number of children who are overweight or obese is growing at a rate of about 400,000 new cases per year. An European child out of four is overweight or obese. Italy holds the negative distinction in Europe.
2 Overweight/Obesity and Hypertension
It is estimated that about 75 % of essential hypertensive have an overweight/obese phenotype [9]. It is less known that also night-time BP, that correlates better with target organ damage and cardiovascular events [10], is closely related with adiposity [11].
Data from 1,827 hypertensive, referred to our Hypertension Excellence Centre, showed indeed that BMI is linearly correlated with night-time systolic BP and night-time pulse pressure (PP). Moreover night-time systolic BP and night-time PP increase with increasing degree of adiposity, particularly in untreated hypertensive or uncontrolled hypertensive (Sarzani et al. 2013, manuscript in preparation). In addition, sleep apnea−hypopnea syndrome is very common in obesity, contributing to a non-dipper hypertensive pattern [12], leading to more severe organ damage and increased mortality [13].
Conversely, weight reduction leads to BP reduction. Systematic reviews and meta-analysis consistently report a decrease in systolic BP (SBP) of about 1 mmHg per kg of weight loss with a follow-up of 2–3 years [14, 15]. There is an attenuation in the longer-term, with a decrease of “only” about 6 mmHg in SBP per 10 kg of weight loss, keeping in mind that a reduction of 2 mmHg corresponds, in the medium-long term, to a reduction of 10 % in stroke mortality and 7 % in cardiac ischemic mortality in some populations [16].
3 Pathogenesis of Increased Blood Pressure in Overweight and Obesity: The Role of Sodium Intake
For hundreds of millions of years of evolution, dietary intake of salt in terrestrial animals was <1 g/day. Selected genetic and physiological systems developed during evolution in order to withhold “the sea” inside terrestrial organisms (being the sea an external environment rich in mineral salts in which life has originally developed) despite the very low dietary intake of salt [17, 18]. Today this genotype has become redundant in the presence of our sedentary lifestyle and a diet low in fibre and high in animal fats and glucose. Indeed, in the last 5,000 years the intake of salt has increased dramatically, until the current average intake of about 10–15 g of salt/day especially in overweight/obese. The Minisal study data [19] indicate an average salt intake of 10 g in man and of 8 g in woman with a positive association between increasing BMI and sodium intake that can average 12 g in obesity. In obese patients, a higher mean BP is required to obtain appropriate natriuresis in presence of medium-high sodium intake [20].
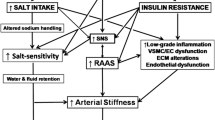
The excess of sodium intake is certainly important in obesity-related hypertension, but other factors increase the effect of salt on BP (Table 1). Despite many factors involved in the genesis of hypertension in overweight/obesity, there is at least a common final mechanism: a “natriuretic” handicap.
4 Renin–Angiotensin–Aldosterone System
Aldosterone excretion rates in the Yanomamo Indians are high [21]. They consumed an extremely low sodium diet, probably typical of the early diets of human evolution, when regulation of aldosterone secretion evolved to optimize the conservation of sodium for purposes of “standing” and survival in a salt-scarce world. Today, with diets containing an excess of salt, aldosterone excretion may exceed what is required, because humans did not have time to adapt their genetic/biological systems, leading to an inappropriately elevated level of aldosterone and increased of its “armed arm” epithelial sodium channel (ENaC) activity despite high sodium intake [22]. Sodium intake and sodium in the body are therefore essential cofactors to make “pathological” the renin–angiotensin–aldosterone system that, when increased in sodium-wasting conditions such as Gitelman and Bartter syndromes, do not lead to any cardiovascular damage.
Moreover, an “escape” of aldosterone secretion from “real life” inhibition with ACEI or ARBs appears to be present with increasing in fat mass, suggesting the attractive hypothesis of a BMI threshold for aldosterone breakthrough in treated essential hypertensive [23].
5 Hypertensive Heart Disease
Cardiomyocyte hypertrophy leading to LV wall thickening and LV mass (LVM) increase is the primary mechanism by which the heart reduces stress on the LV wall imposed by the unrelenting pressure overload. However, a proportion of LVM variation may be determined by other biological influences (Table 2) although, even well-defined “non-hemodynamic factors” such as obesity, metabolic syndrome and obstructive sleep apnea syndrome may indeed act through hemodynamic overload. Hypertensive myocardial remodeling involves also increased rates of cardiomyocyte apoptosis, perivascular and interstitial fibrosis, and microcirculatory changes, important components that can also be affected by non-hemodynamic factors, leading to pathological myocardial remodeling not only in the LV, but also in the left atrium and right ventricle where is clearly documented perivascular fibrosis [24].
Myocardial fibrosis is one of the key features of hypertensive myocardial remodeling that contributes to the increased risk of adverse cardiac events in hypertensive patients with LVH through different pathways: LV diastolic dysfunction/failure [25] (in particular, the decreased “elastic recoil” of the contracted ventricle impairs the early phase of ventricular filling characterizing diastolic dysfunction and leading to increased meso-telediastolic intraventricular pressure); impaired coronary flow reserve [26]; ventricular arrhythmias [27].
Perivascular fibrosis is the pathological hallmark in myocardial tissue of hypertensive patients, fibrosis that is also present in the non-hypertrophied right ventricle and that contributes to abnormal right ventricular function too.
Non hemodynamic factors contribute to ventricular changes in human hypertension, particularly in obese subjects, by the predominant expression and/or activity of local and/or circulating molecules that stimulate remodeling mechanisms over molecules that inhibit remodeling.
6 Obesity, Metabolic Syndrome, and Cardiac Damage
The metabolic syndrome (MetS) is a cluster of cardiovascular risk factors that exposes to an increased cardiovascular risk. A substantial part of the metabolic syndrome-related CV risk is mediated by LV hypertrophy that is not uncommon in these patients [28].
Experimental evidence indicates that inflammation and fibrosis could play a critical role in the development of cardiac damage in hypertension. An increase in reactive oxygen species (ROS) production and several cytokines (such as TNF-α and TGF-β) are implicated in the development of endothelial dysfunction and cardiomyocyte hypertrophy in vitro [29–31] and they are elevated in pressure overload states leading to the development of cardiac fibrosis and diastolic dysfunction in vivo [32]. The relationship between inflammation and CD in MetS has also been characterized in a clinical setting. Sciarretta et al. [33] in 2007 showed that hypertensives with MetS had increased levels of CRP, TNF- α, and TGF-β, and also of PICP, a marker of fibrosis, as compared with patients without MetS. All these markers were significantly related to increased LVM and to impaired diastolic and systolic functions, independently from the known MS parameters and from the MetS itself, and these correlations were enhanced in patients with MetS.
However, in our recent paper we showed that MetS loses its independent relationship with LVH when BMI is taken into account, suggesting that the effects of MetS on LV parameters are mainly driven by the degree of adiposity [34].
In obese patients, plasma volume is increased and more tissue needs to be perfused, resulting in an increased cardiac output. A high-calories high-salt diet increases BP, which in turn is the main promoter towards LVH. The systemic, circulating RAAS is also dysregulated in patients with visceral obesity [35] and the effects of angiotensin II and aldosterone may be pathological on the heart and kidney depending on salt intake with food [36], even though the BP factor is always present and difficult to keep separated in animal models. Nevertheless, the growth of the non-cardiomyocytes myocardial fraction, although sensible to BP levels being mostly perivascular, is not only regulated by the hemodynamic load and angiotensin II as well as aldosterone appear to be involved in promoting the adverse structural remodeling of the myocardial collagen matrix [37]. The activation of cardiac fibroblasts with accumulation of fibrillar type I and type III collagens and the remodeling of the cardiac interstitium represents a major determinant of pathological hypertrophy, being responsible of the abnormal myocardial stiffness, leading to ventricular diastolic dysfunction. Hypertension and aging can also affect the expression of several vascular growth factors and components of the extracellular matrix [38–41], that can mediate vascular and myocardial tissue changes.
7 A Unifying Hypothesis: A Cardiac Natriuretic Peptide System Deficit at the Basis of the Metabolic and Cardiovascular Derangements of Obesity
Excessive adiposity, especially when visceral, is strongly associated with deranged metabolism and cardiovascular disease. Visceral adipose tissue surrounds and infiltrates even “noble organs” such as the heart itself. Moreover, transgenic murine models with forced expression of diacylglycerol acyltransferase (DGAT1) in cardiomyocytes, exhibited increased cardiomyocyte lipid accumulation, cardiac fibrosis, ventricular remodeling, and cardiac contractile dysfunction and decreased mitochondrial biogenesis in the absence of obesity, insulin resistance or systemic dyslipidemia [42]. Therefore, a unifying hypothesis of a “vascular–metabolic” syndrome characterized by high-normal BP or hypertension associated with glucose and lipid dysmetabolism, secondary to the accumulation of visceral fat, becomes plausible [43]. We like to propose this terminology because the classical definition of “metabolic syndrome” is too much toward metabolic derangement only, whereas the cardiovascular system is deeply involved in this common condition.
Which molecular mechanism is likely to be the key mediator of the vascular–metabolic syndrome? Our hypothesis is that at the basis of this syndrome there is a “natriuretic peptide handicap”.
The cardiac natriuretic peptides (NPs), atrial natriuretic peptide (ANP) and the ventricular peptide BNP, are key hormones in fluid and hemodynamic homeostasis. Their actions are mediated by NP Receptor A (NPRA), whose intracellular domain possesses guanylyl cyclase activity and generates the second messenger cGMP. Another receptor, NPRC, which is referred to as the ‘clearance’ receptor, binds ANP and BNP to remove them from the circulation [44]. Unexpectedly, NPRA and NPRC were found to be present in adipose tissue of both rats and humans [45, 46] and to be nutritionally regulated. Together these observations suggested that cardiac NPs have an active metabolic role on adipocytes and that, conversely, there was a potential role for adipose tissue in the clearance of these peptides from the circulation, which, in obesity, may contribute to hypertension.
The discovery that NPRC levels were reduced in adipose tissue by fasting [47] and increased by insulin [48] suggests a more complex picture leading to the idea that NPs may exhibit their lipolytic action through the local balance of NPRA and NPRC expressions in adipocytes. Obese and hypertensive patients are characterized by low NPRA/NPRC ratio with low ANP circulating levels [49], an influential factor determining the metabolic switch between lipolysis versus lipogenesis. In another study of our group [50] the BP fall induced by a single infusion of ANP was greater and more sustained after low calorie diet when compared with that obtained in basal condition. Even the increase in cyclic GMP was greater after diet, reflecting a greater biological activity of NPs, also confirmed by the increased diuresis and natriuresis. Moreover, gene expression of the A receptor was not affected by the fasting instead. This modulation of gene expression also appeared to be tissue specific: fasting did not modify C receptor expression in the kidney. Therefore it is evident as effects of fasting were specific for C receptor and adipose tissue.
Previous reports have confirmed and extended this finding showing that low NPs levels are associated with MetS and its individual components (waist circumference, fasting glucose, HDL-cholesterol and triacylglycerols), as well as with insulin resistance, even after adjustment for body mass index (BMI) [51], this reduction attributable to the increased clearance in adipose tissue.
Low NPs levels in MetS predispose to insulin resistance and given the direct anti-inflammatory effect of NPs (through the reduction in inflammatory mediators such as TNF-α, COX-2 (cyclo-oxygenase-2), MCP-1 (monocyte chemoattractant protein-1) and through the direct stimulus to the secretion of the adipocyte-specific anti-inflammatory hormone with insulin-sensitizing properties, adiponectin) promote a “pro-inflammatory” state [52].
Moreover accumulating evidence suggest that NPRC is not only a clearance receptor, but may be involved in some biological actions of NPs [53]. It has been demonstrated that NPRC is a negative regulator of adenylate cyclase/cAMP system. It has been suggested that all functions of ANP that are not fully explained by an increase in intracellular cGMP levels, i.e. inhibition of aldosterone, renin and vasopressin secretion, as well as the antiproliferative effects, could be mediated by NPRC stimulation. As a matter of fact, cAMP has been found to be involved in the above mentioned effects of ANP. BNP has been shown to play antiproliferative actions in cardiac fibroblasts through a non-cGMP-mediated mechanism involving the NPRC. When physiologically activated, NPRC seems to exert protective vascular effects, thus suggesting a direct antihypertensive role. For example, NaCl loading exerts a significant impact on the NPRC population in renal and vascular endothelial cells. A marked reduction of NPRC number has been observed in salt-loaded Dahl salt-sensitive hypertensive rats, suggesting a role of this mechanism in the pathophysiology of hypertension in this animal model.
However in the well-known NPRC-KO mice, all the cardiovascular and metabolic changes have been well explained and considered to be secondary to the increased biological activity of the NP system through the classic biological active receptor NPRA that is no more opposed by the clearance receptor NPRC [54].
The collection of our data together with many other studies suggest that, similar to the “biologically active” peptides (ANP and BNP), also their corresponding NT peptides may be increasingly removed from the circulation with an increase of adiposity. Effective treatments aimed at the reduction of adipose mass, produce also an increase in the circulation of all of these peptides with beneficial hemodynamic and metabolic effects. Moreover, it must be recalled that mice defective for BNP or NPRA have cardiac hypertrophy, fibrosis and arrhythmias [55, 56], showing that a handicap of natriuretic peptides activities is directly involved also in HDD.
Finally, in our recent paper [57] we have shown that cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. In our work we demonstrated also that mice lacking NPRC have smaller adipocytes and greater expression of UCP1, even in white fat depots suggesting an activation of thermogenic program also in white adipose tissue. NPs and catecholamines showed to share a common final thermogenic pathway.
8 Conclusions
In conclusion, cardiac natriuretic peptides appear to be “master regulators” of adipocytes as well as of the cardiovascular system: they are natriuretic, anti-hypertensive, anti-hypertrophic, anti-fibrotic and anti-arrhythmic, but they are also the first true “cardiometabolic hormones” with a potential anti-obesity and anti-dysmetabolic role (Fig. 1).

Cardiac natriuretic peptides: a key link between heart and adipose tissue in hypertension and metabolic syndrome
References
World health statistics 2013. World Health Organization; 2013.
Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff. 2009;28:w822–31.
Gallus S, Odone A, Lugo A, Bosetti C, Colombo P, Zuccaro P, La Vecchia C. Overweight and obesity prevalence and determinants in Italy: an update to 2010. Eur J Nutr. 2013;52(2):677–85.
Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356–9.
The Italian Cardiovascular Epidemiological Observatory. It Heart J. 2004; (suppl 3):49S–52S.
Stamler J, Dyer AR, Shekelle RB, Neaton J, Stamler R. Relationship of baseline major risk factors to coronary and all-cause mortality, and to longevity: findings from long-term follow-up of Chicago cohorts. Cardiology. 1993;82:191–222.
l’Allemand-Jander D. Clinical diagnosis of metabolic and cardiovascular risks in overweight children: early development of chronic diseases in the obese child. Int J Obes (Lond). 2010;34(suppl 2):S32–6.
Juonala M, Magnussen CG, Berenson GS, Venn A, Burns TL, Sabin MA, Srinivasan SR, Daniels SR, Davis PH, Chen W, Sun C, Cheung M, Viikari JS, Dwyer T, Raitakari OT. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med. 2011;365:1876–85.
Bramlage P, Pittrow D, Wittchen HU, Kirch W, Boehler S, Lehnert H, Hoefler M, Unger T, Sharma AM. Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am J Hypertens. 2004;17:904–10.
Hansen TW, Li Y, Boggia J, Thijs L, Richart T, Staessen JA. Predictive role of the night-time blood pressure. Hypertension. 2011;57:3–10.
Kotsis V, Stabouli S, Bouldin M, Low A, Toumanidis S, Zakopoulos N. Impact of obesity on 24-hour ambulatory blood pressure and hypertension. Hypertension. 2005;45:602–7.
Mokhlesi B. Obesity hypoventilation syndrome: a state-of-the-art review. Resp Care. 2010;55:1347–62.
The Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). ESH/ESC Guidelines for the management of arterial hypertension. J Hypertens. 2013;2013(31):1281–357.
Aucott L, Rothnie H, McIntyre L, Thapa M, Waweru C, Gray D. Long-term weight loss from lifestyle intervention benefits blood pressure? A systematic review. Hypertension. 2009;54:756–62.
Siebenhofer A, Jeitler K, Berghold A, Waltering A, Hemkens LG, Semlitsch T, Pachler C, Strametz R, Horvath K. Long-term effects of weight-reducing diets in hypertensive patients. Cochrane Database Syst Rev. 2011;7(9):CD008274.
Lewington S, Clarke R, Qiziibash N, Peto R, Collins R, Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–13.
Gleiberman L. Blood pressure and dietary salt in human populations. Ecol Food Nutr. 1973;2:143–56.
Dyer AR, Elliott P, Marmot M, Kesteloot H, Stamler R, Stamler J. Commentary: strength and importance of the relation of dietary salt to blood pressure. Intersalt Steering and Editorial Committee. BMJ. 1996;29(312):1661–4.
Donfrancesco C, Ippolito R, Lo Noce C, Palmieri L, Iacone R, Russo O, Vanuzzo D, Galletti F, Galeone D, Giampaoli S, Strazzullo P. Excess dietary sodium and inadequate potassium intake in Italy: results of the MINISAL study. Nutr Metab Cardiovasc Dis. 2013;23:850–6.
Rocchini AP, Key J, Bondie D, Chico R, Moorehead C, Katch V, Martin M. The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med. 1989;321:580–5.
Oliver WJ, Cohen EL, Neel JV. Blood pressure, sodium intake, and sodium related hormones in the Yanomamo Indians, a “no-salt” culture. Circulation. 1975;52:146–51.
Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. 2005;16:3154–9.
Sarzani R, Guerra F, Mancinelli L, Buglioni A, Franchi E, Dessì-Fulgheri P. Plasma aldosterone is increased in class 2 and 3 obese essential hypertensive patients despite drug treatment. Am J Hypertens. 2012;25:818–26.
Díez J, Frohlich ED. A translational approach to hypertensive heart disease. Hypertension. 2010;55:1–8.
Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27:341–8.
Schwartzkopff B, Motz W, Frenzel H, Vogt M, Knauer S, Strauer BE. Structural and functional alterations of the intramyocardial coronary arterioles in patients with arterial hypertension. Circulation. 1993;88:993–1003.
Boldt A, Wetzel U, Lauschke J, Weigl J, Gummert J, Hindricks G, Kottkamp H, Dhein S. Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart. 2004;90:400–5.
De Simone G, Devereux RB, Chinali M, Roman MJ, Lee ET, Resnick HE, Howard BV. Metabolic syndrome and left ventricular hypertrophy in the prediction of cardiovascular events: the Strong Heart Study. Nutr Metab Cardiovasc Dis. 2009;19:98–104.
Bauersachs J, Bouloumie A, Fraccarollo D, Hu K, Busse R, Ertl G. Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: role of enhanced vascular superoxide production. Circulation. 1999;100:292–8.
Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation. 1997;95:1247–52.
Lim JY, Prk SJ, Hwang HY, Park EJ, Nam JH, Kim J, Park SI. TGF-beta1 induces cardiac hypertrophic responses via PKC-dependent ATF-2 activation. J Mol Cell Cardiol. 2005;39:627–36.
Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. 2004;43:739–45.
Sciarretta S, Ferrucci A, Ciavarella GM, De Paolis P, Venturelli V, Tocci G, De Biase L, Rubattu S, Volpe M. Markers of inflammation and fibrosis are related to cardiovascular damage in hypertensive patients with metabolic syndrome. Am J Hypertens. 2007;20:784–91.
Guerra F, Mancinelli L, Angelini L, Fortunati M, Rappelli A, Dessì-Fulgheri P, Sarzani R. The association of left ventricular hypertrophy with metabolic syndrome is dependent on body mass index in hypertensive overweight or obese patients. PLoS ONE. 2011;6(1):e16630.
Sarzani R, Salvi F, Dessì-Fulgheri P, Rappelli A. Renin–angiotensin system, natriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in humans. J Hypertens. 2008;26:831–43.
Dluhy RG, Williams GH. Aldosterone—villain or bystander? N Engl J Med. 2004;351:8–10.
Brilla CG, Rupp H, Funck R, Maisch B. The renin–angiotensin–aldosterone system and myocardial collagen matrix remodelling in congestive heart failure. Eur Heart J. 1995;16 Suppl O:107–9.
Sarzani R, Brecher P, Chobanian AV. Growth factor expression in aorta of normotensive and hypertensive rats. J Clin Invest. 1989;83:1404–8.
Takasaki I, Chobanian AV, Sarzani R, Brecher P. Effect of hypertension on fibronectin expression in the rat aorta. J Biol Chem. 1990;265:21935–9.
Sarzani R, Arnaldi G, Chobanian AV. Hypertension-induced changes of platelet-derived growth factor receptor expression in rat aorta and heart. Hypertension. 1991;17:888–95.
Sarzani R, Arnaldi G, Takasaki I, Brecher P, Chobanian AV. Effects of hypertension and aging on platelet-derived growth factor and platelet-derived growth factor receptor expression in rat aorta and heart. Hypertension. 1991;18:III93–9.
Glenn DJ, Wang F, Nishimoto M, Cruz MC, Uchida Y, Holleran WM, Zhang Y, Yeghiazarians Y, Gardner DG. A murine model of isolated cardiac steatosis leads to cardiomyopathy. Hypertension. 2011;57:216–22.
Sarzani R. The clinical significance of metabolic syndrome in hypertension: metabolic syndrome increases cardiovascular risk: the contrary position. High Blood Press Cardiovasc Prev. 2008;15:59–62.
Maack T, Suzuki M, Almeida FA, Nussenzveig D, Scarborough RM, McEnroe GA, Lewicki JA. Physiological role of silent receptors of atrial natriuretic factor. Science. 1987;238:675–8.
Sarzani R, Paci VM, Dessi-Fulgheri P, Espinosa E, Rappelli A. Comparative analysis of atrial natriuretic peptide receptor expression in rat tissues. J Hypertens Suppl. 1993;11(5):S214–5.
Sarzani R, Dessì-Fulgheri P, Paci VM, Espinosa E, Rappelli A. Expression of natriuretic peptide receptors in human adipose and other tissues. J Endocrinol Invest. 1996;19:581–5.
Sarzani R, Paci VM, Zingaretti CM, Pierleoni C, Cinti S, Cola G, Rappelli A, Dessì-Fulgheri P. Fasting inhibits natriuretic peptides clearance receptor expression in rat adipose tissue. J Hypertens. 1995;13:1241–6.
Nakatsuji H, Maeda N, Hibuse T, Hiuge A, Hirata A, Kuroda Y, Kishida K, Kihara S, Funahashi T, Shimomura I. Reciprocal regulation of natriuretic peptide receptors by insulin in adipose cells. Biochem Biophys Res Commun. 2010;392:100–5.
Dessi-Fulgheri P, Sarzani R, Tamburrini P, Moraca A, Espinosa E, Cola G, Giantomassi L, Rappelli A. Plasma atrial natriuretic peptide and natriuretic peptide receptor gene expression in adipose tissue of normotensive and hypertensive obese patients. J Hypertens. 1997;15:1695–9.
Dessì-Fulgheri P, Sarzani R, Serenelli M, Tamburrini P, Spagnolo D, Giantomassi L, Espinosa E, Rappelli A. Low caloric diet enhances renal, hemodynamic, and humoral effects of exogenous atrial natriuretic peptide in obese hypertensives. Hypertension. 1999;33:658–62.
Wang TJ, Larson MG, Keyes MJ, Levy D, Benjamin EL, Vasan RS. Association of plasma natriuretic peptide levels with metabolic risk factors in ambulatory individuals. Circulation. 2007;115:1345–53.
Savoia C, Volpe M, Alonzo A, Rossi C, Rubattu S. Natriuretic peptides and cardiovascular damage in the metabolic syndrome: molecular mechanisms and clinical implications. Clin Sci. 2010;118:231–40.
Rubattu S, Sciarretta S, Morriello A, Calvieri C, Battistoni A, Volpe M. NPR-C: a component of the natriuretic peptide family with implications in human diseases. J Mol Med. 2010;88:889–97.
Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, Smithies O. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci. 1999;96:7403–8.
Tamura N, Ogawa Y, Chusho H, Nakamura K, Nakao K, Suda M, Kasahara M, Hashimoto R, Katsuura G, Mukoyama M, Itoh H, Saito Y, Tanaka I, Otani H, Katsuki H, Nakao K. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci. 2000;97:4239–44.
Knowles JW, Esposito G, Mao L, Hagaman JR, Fox JE, Smithies O, Rockman HA, Maeda N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest. 2001;107(8):975–84.
Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessì-Fulgheri P, Zhang C, Takahashi N, Sarzani R, Collins S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122:1022–36.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sarzani, R., Bordicchia, M., Spannella, F. et al. Hypertensive Heart Disease and Obesity: A Complex Interaction Between Hemodynamic and Not Hemodynamic Factors. High Blood Press Cardiovasc Prev 21, 81–87 (2014). https://doi.org/10.1007/s40292-014-0054-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40292-014-0054-3