
Abstract
The advent of targeted therapeutics has greatly improved outcomes of chronic myeloid leukemia (CML) patients. Despite increased efficacy and better clinical responses over cytotoxic chemotherapies, many patients receiving targeted drugs exhibit a poor initial response, develop drug resistance, or undergo relapse after initial success. This inter-individual variation in response has heightened the interest in studying pharmacogenetics and pharmacogenomics (PGx) of cancer drugs. In this review, we discuss the influence of various germline and somatic factors on targeted drug response in CML. Specifically, we examine the role of genetic variants in drug metabolism genes, i.e. CYP3A family genes, and drug transporters, i.e. ABC and SLC family genes. Additionally, we focus on acquired somatic variations in BCR-ABL1, and the potential role played by additional downstream signaling pathways, in conferring resistance to targeted drugs in CML. This review highlights the importance of PGx of targeted therapeutics and its potential application to improving treatment decisions and patient outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Genetic makeup in both host body and cancer tissue affect tyrosine kinase inhibitors (TKIs)’ treatment effect in CML. |
Understanding and implementation of pharmacogenomics of targeted therapy can improve treatment outcomes of CML. |
1 Introduction
Anti-cancer treatment strategies have evolved in recent decades to reflect the new technologies and treatment options that have been developed in response to the wide variation and heterogeneity of patient outcomes with similar clinical and pathological characteristics. In many cases, patients treated with the same therapeutic regimens exhibit significant differences in drug response and survival outcomes. Thus, traditional treatment decision-making strategies that rely only on the histopathological and clinical factors of disease or environmental factors like age and sex are not fully effective for all patients [1, 2]. With the emergence of human genome sequencing technologies and high-throughput genetic analysis, it has become abundantly clear that an individual’s genetic makeup can affect response to drugs, thus giving rise to increased interest in closely examining genetic profiles in cancer patient care.
This heightened interest has resulted in an increase in studies focusing on the role of pharmacogenetics and pharmacogenomics (often jointly abbreviated as PGx) in treating cancers [3, 4]. PGx can be defined as the study of genetic factors that can influence a patient’s response to drugs. Here, genetic factors can refer to single nucleotide polymorphisms (SNPs), haplotypes or other heritable mutations, copy number variations (CNVs) and chromosomal alterations. Before the advent of genome-wide approaches, treatment approaches relied on studying candidate genetic variants important in drug response, which assumes the importance certain genes hold in drug sensitivity [3]. Examining and identifying both somatic and germline genetic factors that influence drug sensitivity is important to provide prognostic information, predict treatment outcomes, and improve drug efficacy and safety for individual patients [1]. PGx approaches incorporating a more comprehensive understanding of the genetic and molecular processes may also lead to the identification of new molecularly targeted agents that are safer and more effective for cancer patient care. Thus, by personalizing therapy tailored to an individual patient’s genotype, anti-cancer treatment strategies can maximize efficacy and minimize toxicity in patients [4].
Due to the nonspecific effects with a narrow therapeutic index of cytotoxic chemotherapeutic agents and resulting in serious toxicities, the relevance of PGx approaches was considered highly significant for such regimens. On the other hand, because targeted therapies act upon cancer-cell specific genes or proteins, there has been less emphasis on investigating PGx of targeted therapeutics. However, the importance of PGx of targeted therapeutics becomes clear in cases where patients either do not respond, develop resistance, or react adversely to drugs. One example of this phenomenon includes chronic myeloid leukemia (CML) patients with the BCR-ABL1 fusion protein who do not respond to the tyrosine kinase inhibitor (TKI) imatinib and require consideration of additional genetic factors in selecting appropriate drugs. Studying the PGx of targeted therapies is especially important when considering that, in the case of CML, up to one-third of patients treated with imatinib may not demonstrate complete cytogenetic response or become imatinib resistant [5].
1.1 Overview of CML Biology and Treatment
CML is a myeloproliferative disorder characterized by the presence of a reciprocal translocation between chromosomes 9 and 22, also known as the Philadelphia chromosome (Ph). This translocation results in the fusion of Abelson murine leukemia viral oncogene homolog 1 (ABL1) gene from chromosome 9 with the breakpoint cluster region (BCR) gene on chromosome 22, thus forming the BCR-ABL1 fusion gene [6]. As a constitutively active tyrosine kinase, the fusion protein-product of this gene activates a number of oncogenic signaling pathways, including PI3K/AKT/mTOR, RAS/RAF/MEK/ERK, and JAK/STAT [7].
The annual incidence rate of CML is 1–2 cases per 100,000 and accounts for 10% of newly diagnosed cases of leukemia in the USA [8]. The progression of CML is structured by phases, with 85% of patients diagnosed in the benign chronic phase (CP) [9]. Without successful treatment during CP, the disease can progress to the accelerated phase (AP), and to the often-fatal blast crisis (BC) phase. Prognosis varies with the phase of CML, age, and therapeutic response [10].
Prior to the advent of targeted therapy for CP-CML, patients were treated with busulfan and hydroxyurea chemotherapy, producing complete hematological response (CHR: ≤12,000 white cells/μL blood) in 50–80% of patients. Complete cytogenetic remissions (CCyR: no detectable Ph+ cells) were rare [11, 12]. The subsequent introduction of human cytokine interferon-α therapy produced higher rates of complete hematologic and cytogenetic responses [13]. However, the BCR-ABL1 inhibitor imatinib mesylate, which was approved by the US FDA in 2001, outperformed all previous therapeutics achieving CHR in 95%, and CCyR in 76% of the patients in the IRIS trial for CML treatment [14, 15]. Since the introduction of imatinib as frontline therapy for CP-CML, the 5-year survival rates have dramatically increased from 31% in the early 1990s to 66% in the years between 2006 to 2012 [16].
As noted above, about 25% of the patients in the IRIS trial were resistant to imatinib, while an additional 20–25% of the patients achieving CHR eventually developed resistance with increased risk of disease progression [17]. In the 8-year follow-up study of the original IRIS trial, only 55% of the patients remained on imatinib with 16% of withdrawals attributed to unsatisfactory treatment outcome [18]. A common mechanism of resistance to imatinib was point mutations in the kinase domain of BCR-ABL1 oncogene. To overcome this, second-generation TKIs, including dasatinib and nilotinib, were developed with several-fold higher affinity to BCR-ABL1 than imatinib. These drugs significantly improved patient outcomes in imatinib-resistant CML [19, 20]. In addition, the second-generation TKIs were superior in achieving major molecular response (MMR: BCR-ABL1 RNA level ≤0.1% on the International Scale) and CCyR by month 12 in newly diagnosed CP-CML patients as compared to imatinib, and were approved as frontline treatment options [21, 22]. However, only about 50% of the resistant cases can be explained on the basis of BCR-ABL1 point mutations [23]. In addition, a number of patients develop drug intolerance or cytotoxicity, experiencing both physical and mental side effects (physical functioning, role limitations because of physical problems, bodily pain, general health perceptions, vitality, social functioning, role limitations because of emotional problems, and mental health; patient-reported) [24], highlighting the importance of PGx targeted therapies.
In this review, we will discuss the role of patient germline genetics and acquired somatic alterations that may influence response to targeted TKIs. We discuss the role of genetic determinants that influence processes such as enzymatic drug metabolism, cellular influx and efflux, in addition to the role of mutations in genes directly targeted by the TKIs. We further discuss the role of alternative downstream pathways that circumvent the effects of BCR-ABL1 inhibition and could influence response to TKIs.
2 Germline Genetic Determinants of Drug Response
Targeted TKIs for CML are administered orally, and their bioavailability is dependent on gastrointestinal absorption, first-pass metabolism, and cellular influx and efflux. Thus, variations in the genes regulating metabolism and transport can impact pharmacokinetics and pharmacodynamics of TKIs in CML patients. A figure illustrating the enzymes involved in pharmacokinetics and pharmacodynamics of imatinib can be found online at https://www.pharmgkb.org/pathway/PA164713427.
2.1 Drug Metabolism Genes
2.1.1 Cytochrome P450
The metabolism of TKIs primarily occurs in the cytochrome P450 complex. This complex comprises a family of hemoproteins that catalyze reactions relating to drug metabolism, as well as the synthesis of cholesterol, steroids, and other lipids. The cytochrome P450 3A4 (CYP3A4) is the main enzyme responsible for the first-pass metabolism of TKIs, while other enzymes including CYP3A5, CYP2C8, and CYP2D6 are also involved to a lesser extent.
Imatinib is converted by the CYP3A enzymes [25] into its main metabolite N-desmethyl imatinib, which is pharmacologically active but three to four times less cytotoxic than imatinib [26]. The importance of CYP3A enzymes was highlighted in a study that showed CML patients achieving a complete molecular response (CMR; no detectable BCR-ABL1 transcript using qPCR) and demonstrated higher CYP3A4 and CYP3A5 enzymatic activity than patients with partial molecular responses [27]. However, recent pharmacokinetic studies suggest that the predominant role of CYP3A4 may be taken over by CYP2C8 over the course of imatinib treatment, driven by time-dependent auto-inhibition of CYP3A4 activity by imatinib [28]. Thus, the inhibition of CYP3A4 activity over time may elevate the role of other cytochrome P450 enzymes in imatinib metabolism. An example of such compensatory effect comes from a study that examined the influence of the CYP3A5*3 polymorphisms on the clinical outcome and steady-state trough plasma concentration of imatinib in 110 Nigerian CML patients [29]. This study reported that the GG genotype in CYP3A5*3 was associated significantly with higher trough plasma concentrations, but not associated with clinical outcomes. Another study investigated the influence of the CYP2B6 15631G > T polymorphisms on imatinib response in 48 CML patients in Morocco [30]. This study reported that CHR loss was higher in patients carrying the CYP2B6 15631GG/TT genotype when compared with 15631GT, while CCyR was higher in the 15631GG/GT genotype compared with 15631TT. In addition, primary cytogenetic resistance was higher in patients with the 15631GG/TT genotype compared with 15631GT carriers, whereas side effects were more common in patients with the 15631GG genotypes compared with GT/TT carriers.
CYP3A4 is also the primary isoform responsible for the metabolism of nilotinib [31] and dasatinib [32]. Dasatinib functions as a competitive inhibitor of CYP3A4 and CYP2C8 [33, 34], whereas nilotinib has inhibitory activity against CYP3A4, CYP2C8, CYP2C9 and CYP2D6 [35]. While the role of polymorphisms in these genes has not been extensively studied in CML patients receiving dasatinib or nilotinib, several studies have focused on drug-drug interactions with these TKIs, considering most CML patients take TKIs throughout their lives along with other drugs (reviewed in [36]).
These findings provide preliminary evidence that along with CYP3A4 activity, identifying polymorphisms in additional P450 enzymes could help predict imatinib therapeutic response. However, these reported variants may be population-specific and must be profiled in larger CML cohorts to assess clinical relevance.
2.1.2 UGT1A1
Another aspect of TKI metabolism involves glucuronidation of the active metabolite into inactive compounds for elimination. This reaction is catalyzed by the uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) enzyme, which transforms small lipophilic molecules, including TKIs, into water-soluble metabolites for excretion. UGT1A1 also glucuronidates bilirubin in humans, and the (TA)7 polymorphism in the TATAA element of the promoter (UGT1A1*28), downregulating its expression, has been strongly linked with hyperbilirubinemia [37]. A pharmacogenetic analysis of the TA-repeat polymorphism demonstrated an association between the (TA)7/(TA)7 genotype and the risk of hyperbilirubinemia in imatinib-resistant or intolerant CML patients treated with nilotinib [38]. This study also suggested that since UGT1A1 did not glucuronidate nilotinib, potent inhibition of UGT1A1 activity by nilotinib, combined with the (TA)7 polymorphism, was the most probable reason for increased rate of hyperbilirubinemia in CML patients. A separate study investigated the influence of UGT1A1 polymorphisms and nilotinib trough plasma concentrations on nilotinib-induced hyperbilirubinemia in Japanese CML patients [39]. A higher proportion of patients with hyperbilirubinemia carried the UGT1A1 *6/*6 and *6/*28 genotypes when compared to the other genotypes (allele *6 represents UGT1A1 211G > A). Additional case studies of Japanese CML patients reported that in addition to grade 2 hyperbilirubinemia, some patients with the *6/*6 or *6/*28 genotype had other severe toxicities including QT interval prolongation, elevated lipase levels, anemia, and hepatic cyst hemorrhage [40]. These findings indicate the benefit of identifying the patients’ risk for increased hyperbilirubinemia and severe toxicity through prospective UGT1A1 genotyping prior to nilotinib therapy.
Table 1 provides a summary of PGx variants of drug metabolism genes associated with CML targeted drugs.
2.2 Drug Transport Genes
The intracellular concentration of TKIs depends on influx and efflux of the drugs via transmembrane transporter proteins and can determine the efficiency of BCR-ABL1 inhibition. The multidrug resistance ATP-binding cassette (MDR-ABC) family proteins ABCB1 (also known as MDR1 or P-GP) and ABCG2 (also known as BCRP2) are key mediators of active drug efflux. The role of MDR-ABC proteins in multidrug resistance has been widely studied in several cancer types and polymorphisms in these genes serve as predictive markers for drug response [41]. However, the precise mechanism of MDR-ABC transporter-mediated resistance in CML patients is contentious.
Clinical studies have established that ABCB1 expression levels are elevated in advanced stages of CML [42], and that high ABCB1 expression is associated with lower rates of MMR and imatinib resistance [43, 44]. At the molecular level, in vitro studies with the K562 cell line demonstrated that an increase in ABCB1-mediated drug efflux is a potential mechanism of resistance to imatinib [45, 46]. However, the results of in vitro studies with other TKIs are far from unambiguous. One study reported that ABCB1 only induced resistance against dasatinib and not nilotinib, while ABCG2 induced resistance against both drugs [47]. Another study reported that nilotinib resistance was only associated with ABCB1 activity and not ABCG2 [48], while a different study showed that nilotinib resistance was indeed correlated with both ABCB1 and ABCG2 protein expression [49]. A large amount of variation observed across in vitro studies could be attributed to differences in drug concentrations tested, duration of the experiment, and end-point measure of drug efficacy. Nevertheless, consistent clinical correlations observed between MDR-ABC protein activity and imatinib resistance have prompted studies to investigate the role of commonly inherited variants of ABCB1 and ABCG2.
2.2.1 ABCB1
Gurney et al. [50] investigated the association between the three most common ABCB1 variants, 1236T > C, 2677G > T/A and 3435C > T, and imatinib clearance in 8 CML and 14 gastrointestinal stromal tumor (GIST) patients [50]. They reported that the rate of reduction in imatinib clearance was significantly lower in patients with the TT genotype at all three loci. Subsequently, Dulucq et al. [51] evaluated the clinical impact of ABCB1 variants on imatinib response in a cohort of 90 CML patients in France [51]. In this study, patients with the 1236TT or 2677TT/TA genotype achieved significantly higher rates of MMR. However, another study conducted by Deenik et al. [52] with 46 CML patients in the Netherlands found that the 1236CT/TT, 3435TT, and 2677TT genotypes were actually associated with reduced rates of MMR and CMR [52], in agreement with the initial report suggesting a lower reduction in imatinib clearance rate in patients with TT genotypes [50]. In response, Dulucq et al. [53] expanded their analysis to a larger cohort of 557 patients and reported that the 2677 G allele was indeed associated with higher MMR rates [53], now in agreement with results reported by Deenik et al. [52].
Evidently, the conclusions drawn in these reports were affected by the sample size of the patient cohorts. To address this issue, meta-analyses of a number of other trials that evaluated the associations between ABCB1 variants and imatinib response were performed. One meta-analysis covering 1826 patients across 12 studies reported significant associations between 2677 G, 3435 T, and worse response to imatinib in all populations studied, whereas 1236 CC predicted better response specifically in Asian populations [54]. The result in Asian populations was corroborated in a separate meta-analysis, confirming the association between 1236T > C locus with imatinib response in this demographic [55].
At the molecular level, in vitro studies so far do not agree whether these polymorphisms indeed play a role in TKI response, with one study suggesting no role [56], while another reporting substantial and statistically significant influence on the activity of all CML TKIs [57]. Nevertheless, researchers agree that expression levels of ABCB1 are strong predictors of imatinib responsiveness [43, 44] and could be monitored as early response markers in CML patients receiving imatinib [58].
2.2.2 ABCG2
Imatinib, nilotinib, and dasatinib have been shown to be high-affinity substrates of ABCG2, another member of the MDR-ABC family of efflux proteins [47, 59, 60]. In vitro studies with ABCG2 overexpression in K562 cells show elevated resistance to imatinib [61], nilotinib [59], and dasatinib [47]. Interestingly, the third-generation TKI ponatinib was not affected by either ABCB1 or ABCG2 expression at clinically relevant concentrations [47], while all TKIs could inhibit ABCG2 activity at high concentrations [47, 61]. While multiple in vitro studies support the role of ABCG2 expression in mediating TKI resistance, few clinical studies have evaluated the role of ABCG2 expression and variants in CML patients. A study with 188 CML patients in Brazil showed that ABCG2 expression levels were higher in imatinib-resistant patients as well as in patients who did not achieve MMR [62]. Another study with 215 CML patients in Malaysia evaluated two variants: ABCG2 34G > A and 421C > A and found that the ABCG2 diplotype A34A421 was significantly correlated with a better imatinib response [63]. A recent meta-analysis compiled data from 14 studies across 2184 patients and also concluded that the 421A variant was significantly associated with higher MMR and CCyR in CML patients [64]. As with ABCB1, the correlation between ABCG2 expression and TKI response seems to be less contentious, but establishing the overall importance of genetic variants would greatly benefit from studies in larger cohorts.
2.2.3 SLC22A1
The active intracellular uptake of TKIs is controlled by the expression and activity of influx proteins like the solute carrier family 22 member SLC22A1 (also known as organic cation transporters or OCT1). The involvement of OCT1 in the transport of imatinib has been somewhat contentious with in vitro studies showing OCT1 can bind to imatinib, but changes in SLC22A1 expression levels may or may not be associated with imatinib response. A critical discrepancy in several studies was attributed to primers designed to quantify SLC22A1 using qPCR, with several studies using primers that spanned SNPs that affected expression levels [65]. A recent study evaluated the association between SLC22A1 expression and imatinib uptake in multiple cell lines and mouse models and reported that the uptake of imatinib was not dependent on SLC22A1 expression [66]. On the other hand, OCT1 activity seems to be a strong predictor of imatinib response at the clinical level. In one study, the activity of OCT1 was reported to be a strong predictor of MMR in a cohort of 56 CML patients from Australia [67]. Additional studies from the UK with 70 patients [68] and Brazil with 88 patients [69] confirmed that SLC22A1 expression levels were predictive of imatinib response over time.
The role of SLC22A1 genetic variants has also been studied in small cohorts of CML patients, but no clear consensus has emerged about their effect on imatinib sensitivity. A study in 38 Asian CML patients revealed three SLC22A1 polymorphisms (rs3798168, rs628031, and IVS7 + 850C > T) to be significantly associated with imatinib clearance [70]. Another study in 50 CML patients from Italy found SLC22A1 480C > G to be significantly associated with imatinib clearance [71], whereas the 401G/A genotype, occurring in a small fraction of patients, was associated with MMR in a study of 132 CML patients from the UK [72]. However, additional studies have reported no association between SLC22A1 variants and imatinib response [73]. Thus, the rare occurrence of these genotypes necessitates exercising caution in interpreting results from small-sized studies.
2.2.4 Additional Transporters
In addition to the primary efflux and influx transporters discussed above, various other transmembrane transporters that may introduce variability in TKI response have been evaluated. These include the influx transporters solute carrier organic anion transporter family member 1B3 (SLCO1B3), solute carrier organic anion transporter family member 1A2 (SLCO1A2; also known as OATP1A2), solute carrier family 22 member 4 (SLC22A4; also known as OCTN1) and the efflux transporters ATP-binding cassette subfamily A member 3 (ABCA3) and subfamily C member 4 (ABCC4). The main motivation for exploring additional transporters comes from conflicting results about the ability of TKIs to bind these transporters in vitro. For example, one study reported that imatinib failed to bind OCT1, and its expression was correlated with expression of other transporters, thereby potentially acting as a proxy biomarker for these proteins [74].
A study with 34 Japanese CML patients found that the rate of imatinib clearance was significantly higher in patients with the SLCO1B3 334GG, ABCB1 3435CC [75], and SLCO1A2 361GG genotypes [76]. Another study with 118 CML patients in Brazil reported reduced CMR in patients with the ABCA3 4548-91 CC/CA genotypes compared to AA carriers, while SLCO1B3 699GG and 344TT genotypes were more prevalent in patients who responded to imatinib [77]. A study with 189 CML patients from Italy found that the presence of the C allele in the SLC22A4 variant rs1050152 was significantly associated with increased rates of MMR [78].
While it appears that there is surmounting evidence that additional drug influx and efflux genes could determine responsiveness to imatinib, most data were obtained from small, population-specific cohorts, and further clinical and experimental validation would help in supporting their overall influence.
Table 2 provides a summary of PGx variants of drug transport genes associated with CML targeted drugs.
3 Somatic Determinants of Drug Response
One of the most widely studied mechanisms of resistance to imatinib treatment involves point mutations in the tyrosine kinase domain of the BCR-ABL1 fusion oncogene. However, BCR-ABL1 point mutations are not detected in all patients with TKI resistance and may explain only about half of the resistant cases [79, 80]. Recent reports suggest that the activation of alternate oncogenic pathways following TKI treatment may be responsible for acquired resistance in a BCR-ABL1-independent manner. In this section, we discuss the role of BCR-ABL1 mutations and the activation of alternate pathways in determining TKI response in CML patients.
3.1 BCR-ABL1
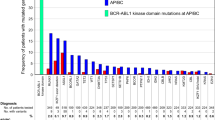
Since the discovery of an association between BCR-ABL1 kinase domain mutations with imatinib resistance [81], more than 90 point mutations that alter TKI’s ability to bind BCR-ABL1 have been identified [80, 82]. As TKIs inhibit BCR-ABL1 activity by binding and inactivating the ATP-binding loop of the ABL1 kinase domain, mutations can confer resistance by altering the conformation of the protein to prevent TKI access, reduce binding affinity, or to weaken hydrogen bonds required for binding or maintain the ATP loop in an active conformation [82].
The first study investigating mechanisms of imatinib resistance identified the T315I mutation arising from a substitution of C to T at base 944 of the ABL1 gene in about half of the 11 imatinib-resistant patients evaluated [83]. The authors confirmed the inability of imatinib to bind the mutated BCR-ABL1 protein using in vitro experiments. Subsequently, mutation screening in imatinib-resistant patients identified several additional ABL kinase domain mutations, including E255 K, E255 V, Y253F, Y253H, F317L, H396P, M351T, along with T315I [84,85,86] that conferred varying degrees of resistance [87]. One study evaluated BCR-ABL1 mutations in 144 CML patients and found that these mutations were strongly associated with resistance in 27 cases [88]. Another study evaluated the mutational profile of 171 patients across all phases of CML who failed imatinib treatment and reported BCR-ABL1 mutations in about 1 in 3 patients [89]. This study also found that the rate of mutations was significantly higher in patients who attained the BC or AP stage.
The inability of imatinib to bind mutated BCR-ABL1 prompted the development of second-generation TKIs: dasatinib, nilotinib, and bosutinib. The second-generation TKIs were able to overcome imatinib resistance arising from point mutations attributable to several-fold higher binding affinities for BCR-ABL1 as compared to imatinib [90]. However, one notable exception was the “gatekeeper” T315I mutation, which conferred resistance to second-generation TKIs as well. Phase II trials with the third-generation TKI ponatinib show promising results, with MMR and CCyR observed in patients regardless of T315I status [91].
Longitudinal monitoring of CML patients resistant to TKIs suggests that these point mutations were likely selected during the course of treatment [92], but their presence in pre-treatment CML-CP patients is not well documented. Consequently, the European LeukemiaNet expert panel recommended against monitoring for BCR-ABL1 mutations prior to treatment or in patients with sustained CCyR. Direct sequencing of BCR-ABL1 would be most beneficial to patients who exhibit primary or secondary resistance to imatinib or second-generation TKIs [93].
3.2 BCR-ABL1 Independent TKI Resistance Mechanisms
Given the significant proportion of patients who are resistant to TKIs without evidence of BCR-ABL1 mutations, studies have shown that pathways that function downstream of BCR-ABL1 could compensate for the loss of BCR-ABL1 activity and may be responsible for acquired resistance to TKI treatment. The main rationale for the existence of such pathways comes from the early investigations that reported the presence of an imatinib-resistant population of CD34+ progenitor stem cells in the blood of patients [94]. These CD34+ CML stem cells were distinct from normal CD34+ cells in their transcriptomic and molecular profile, expressing a number of oncogenic signaling pathways [95]. Furthermore, survival of human CML stem cells was reported to be independent of BCR-ABL1 activity, and imatinib treatment failed to completely clear CD34+ CML stem cells [96]. Investigations of therapeutic response revealed that the second-generation TKIs nilotinib and dasatinib were also ineffective in inducing apoptosis in CD34+ leukemic cell populations [97, 98]. An emerging theme from these studies seems to indicate that the resistance to TKIs may develop in CML patients despite a complete inhibition of BCR-ABL1 activity. In this section, we outline such acquired somatic alterations that may influence drug response in CML patients.
3.2.1 KIT and HIF1A
The KIT proto-oncogene (also known as SCFR, CD117, or c-KIT) encodes a hematopoietic stem cell surface cytokine receptor that is activated in several cancer types and regulates cellular proliferation, differentiation, and migration. CML cells that are Ph+ also show elevated expression of KIT, and certain KIT polymorphisms may be associated with increased WBC count in CML patients [99]. Molecular studies showed that the TKIs imatinib and nilotinib also directly bind to and inhibit c-KIT activity, but the efficiency of inhibition of CD34+ CML cells with elevated KIT can vary between drugs [100]. Moreover, complete inhibition of c-KIT seems to be necessary for inducing apoptosis in CD34+ cells upon inhibition of BCR-ABL1 [100]. These results suggest c-KIT activation could influence therapeutic response to TKIs. While c-KIT activating mutations have been extensively studied in other diseases such as GIST and AML, reports of c-KIT mutations in CML patients are rare and could be the subject of future studies.
One of the targets activated by c-KIT, hypoxia-inducible factor 1-alpha (HIF1A), is a transcription factor that regulates the response to hypoxia. Overexpression of HIF1A is associated with several cancer types, with roles in cancer cell metabolism, survival, and invasion. In the absence of BCR-ABL1 mutations, imatinib-resistant Ph+ CML cell populations were shown to have elevated HIF1A expression [101]. This study suggested that HIF1A activity could help CML progenitor cells to survive in the hypoxic bone marrow environment. These results were further corroborated in a study with primary CD34+ cells isolated from CML patients where the hypoxic environment was shown to protect CML cells despite effective BCR-ABL1 inhibition [102]. Thus, secondary inhibition of HIF1A could enhance the elimination of CML progenitor cells.
3.2.2 Downstream Signaling Pathways
Early investigations into the mechanisms of CML oncogenesis revealed that the PI3 K/AKT/mTOR [103, 104], p38/MAPK [105], and STAT5 [106, 107] signaling pathways are activated downstream of the BCR-ABL1 kinase. Subsequent studies showed that these pathways were not only mediating the effects of BCR-ABL1 activation, their complete inhibition was also necessary in order to control growth and proliferation of CML cells.
In a study with 54 untreated and 62 imatinib-resistant CML patients, it was reported that resistant patients had significantly lower levels of BCR-ABL1, CRKL (CRK- like proto-oncogene), and AKT phosphorylation levels [108], suggesting alternative pathways independent of BCR-ABL1 may be activated in resistant populations. In an in vitro study, multiple Ph+ cell lines that did not carry mutations in the BCR-ABL1 kinase domain, but were resistant to imatinib, nilotinib, and dasatinib were reported to carry mutations for constitutive activation of the PI3 K/AKT pathway [109].The role of downstream signaling was corroborated in another study that reported activation of the MAPK signaling pathway in CD34+ CML cells and found that co-inhibition of both PI3 K/AKT and MAPK signaling resulted in a significantly enhanced elimination of CD34+ CML progenitor cells [110]. Other studies have reported constitutively active MAPK signaling in CML progenitor cells independent of BCR-ABL1, driven by activating mutations in c-Raf [111].
The involvement of JAK/STAT signaling in TKI resistance has also received attention in recent years, with a report suggesting STAT5 expression levels are upregulated in patients following imatinib treatment [112]. Thus, clonal selection of STAT5 expressing cells could be a mechanism of secondary resistance in Ph+ patients who respond to TKIs initially. Multiple recent studies also report that the inhibition of JAK-STAT signaling either via JAK inhibitors [113], or the suppression of STAT5A and STAT5B, significantly enhances responsiveness to imatinib in Ph+ CML cells and to chemotherapy in imatinib-resistant cells [114, 115].
As shown in multiple reports, pathways downstream of BCR-ABL1 are clearly involved in the persistence of CD34+ CML progenitor stem cells. Evidently, insufficient clearance of CML stem cells could result in resistance to TKIs. However, these could be challenging to predict in patients at the time of diagnosis and may require longitudinal genomic profiling of patients to assess the status of oncogenic pathways during the course of treatment.
Table 3 provides an outline of acquired somatic alterations associated with TKI resistance in CML patients.
4 Conclusions
The clinical management of CML has improved dramatically since the introduction of BCR-ABL1-targeting TKIs. However, therapeutic resistance and relapse still affect a significant proportion of patients undergoing TKI treatment. As discussed above, several germline variants influence the efficacy of targeted drugs in CML patients, including variants in genes involved in drug transport and metabolism. In addition, somatic variants in drug target genes, as well as a compensatory activation of downstream signaling pathways, can also influence the efficacy of targeted drugs. Despite continual growth in PGx knowledge of targeted therapeutics, CML patients are not routinely profiled for genetic variants to predict treatment efficacy, in part due to most results being reported in small-scale clinical studies. Thus, to obtain reliable clinical biomarkers, the current PGx interactions must be validated in large population studies. Furthermore, to obtain a better understanding of the role of somatic variants in drug resistance or disease relapse, patients receiving targeted therapies should be monitored over time to observe how their cancer genome changes in response to treatment. With a comprehensive understanding of a patient’s genetic profile, better treatment decisions can be made in the future, which would ultimately lead to improved treatment outcomes and quality of life.
References
Westbrook K, Stearns V. Pharmacogenomics of breast cancer therapy: an update kelly. Pharmacol Ther. 2014;139(1):1–11.
Panczyk M. Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J Gastroenterol. 2014;20(29):9775–827.
Huang RS, Dolan ME. Approaches to the discovery of pharmacogenomic markers in oncology: 2000–2010–2020. Pharmacogenomics. 2010;11(4):471–4.
Huang RS, Ratain MJ. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J Clin. 2009;59(1):42–55.
Bhamidipati PK, et al. Management of imatinib-resistant patients with chronic myeloid leukemia. Ther Adv Hematol. 2013;4(2):103–17.
Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385(9976):1447–59.
Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96(10):3343–56.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29.
Faderl S, et al. Chronic myelogenous leukemia: biology and therapy. Ann Intern Med. 1999;131(3):207–19.
Sillaber C, et al. Chronic myeloid leukemia: pathophysiology, diagnostic parameters, and current treatment concepts. Wiener klinische Wochenschrift. 2003;115(13–14):485–504.
Kantarjian HM, et al. Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. Am J Med. 1987;83(3):445–54.
Kennedy BJ. The evolution of hydroxyurea therapy in chronic myelogenous leukemia. Semin Oncol. 1992;19(3 Suppl 9):21–6.
Kujawski LA, Talpaz M. The role of interferon-alpha in the treatment of chronic myeloid leukemia. Cytokine Growth Factor Rev. 2007;18(5–6):459–71.
Kantarjian H, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346(9):645–52.
O’Brien SG, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004.
American Cancer S. Cancer facts & figures. 2016.
Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8(11):1018–29.
Deininger M, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114(22):1126.
Kantarjian H, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–51.
Talpaz M, et al. Dasatinib in imatinib-resistant philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41.
Saglio G, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–9.
Kantarjian H, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260–70.
Jabbour E, et al. Chronic myeloid leukemia: mechanisms of resistance and treatment. Hematol Oncol Clin N Am. 2011;25(5):981–95.
Efficace F, et al. Health-related quality of life in chronic myeloid leukemia patients receiving long-term therapy with imatinib compared with the general population. Blood. 2011;118(17):4554–60.
Peng B, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–42.
Mlejnek P, et al. Interactions of N-desmethyl imatinib, an active metabolite of imatinib, with P-glycoprotein in human leukemia cells. Ann Hematol. 2011;90(7):837–42.
Gréen H, et al. CYP3A activity influences imatinib response in patients with chronic myeloid leukemia: a pilot study on in vivo CYP3A activity. Eur J Clin Pharmacol. 2010;66(4):383–6.
Filppula AM, et al. Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism: variability in CYP2C8 activity may alter plasma concentrations and response. Drug Metab Dispos. 2012;41(1):50–9.
Adeagbo BA, et al. Influence of CYP3A5*3 and ABCB1 C3435T on clinical outcomes and trough plasma concentrations of imatinib in Nigerians with chronic myeloid leukaemia. J Clin Pharm Ther. 2016;41(5):546–51.
Kassogue Y, et al. Functional polymorphism of CYP2B6 G15631T is associated with hematologic and cytogenetic response in chronic myeloid leukemia patients treated with imatinib. Med Oncol. 2014;31(1):782.
Duckett DR, Cameron MD. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin Drug Metab Toxicol. 2010;6(10):1175–93.
Christopher LJ, et al. Metabolism and disposition of dasatinib after oral administration to humans. Drug Metab Dispos. 2008;36(7):1357–64.
Filppula AM, Neuvonen PJ, Backman JT. In vitro assessment of time-dependent inhibitory effects on CYP2C8 and CYP3A activity by fourteen protein kinase inhibitors. Drug Metab Dispos. 2014;42(7):1202–9.
Li X, et al. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37(6):1242–50.
DeRemer DL, Ustun C, Natarajan K. Nilotinib: a second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clinical Therapeutics. 2008;30(11):1956–75.
Haouala A, et al. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2011;117(8):e75–87.
Bosma PJ, et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333(18):1171–5.
Singer JB, et al. UGT1A1 promoter polymorphism increases risk of nilotinib-induced hyperbilirubinemia. Leukemia. 2007;21(11):2311–5.
Abumiya M, et al. Influence of UGT1A1 6, 27, and 28 polymorphisms on nilotinib-induced hyperbilirubinemia in Japanese patients with chronic myeloid leukemia. Drug Metab Pharmacokinet. 2014;29(6):449–54.
Shibata T, et al. Association between severe toxicity of nilotinib and UGT1A1 polymorphisms in Japanese patients with chronic myelogenous leukemia. Int J Clin Oncol. 2014;19(2):391–6.
Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26(9):2039–54.
Giles FJ, et al. Multidrug resistance protein expression in chronic myeloid leukemia. Cancer. 1999;86(5):805–13.
Eadie LN, et al. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia. 2016;31(1):75–82.
da Cunha Vasconcelos F, et al. Low ABCB1 and high OCT1 levels play a favorable role in the molecular response to imatinib in CML patients in the community clinical practice. Leuk Res. 2016;51:3–10.
Illmer T, et al. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia. 2004;18(3):401–8.
Bouchet S, et al. From in vitro to in vivo: intracellular determination of imatinib and nilotinib may be related with clinical outcome. Leukemia. 2013;27(8):1757–9.
Hegedűs C, et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br J Pharmacol. 2009;158(4):1153–64.
Eadie LN, et al. Degree of kinase inhibition achieved in vitro by imatinib and nilotinib is decreased by high levels of ABCB1 but not ABCG2. Leuk Lymphoma. 2012;54(3):569–78.
Kosztyu P, et al. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem Biol Interact. 2014;219:203–10.
Gurney H, et al. Imatinib disposition and ABCB1 (MDR1, P-Glycoprotein) genotype. Clin Pharmacol Ther. 2007;82(1):33–40.
Dulucq S, et al. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2008;112(5):2024–7.
Deenik W, et al. Polymorphisms in the multidrug resistance gene MDR1 (ABCB1) predict for molecular resistance in patients with newly diagnosed chronic myeloid leukemia receiving high-dose imatinib. Blood. 2010;116(26):6144–5.
Dulucq S, et al. Response: is there really a relationship between multidrug resistance gene (MDR1) polymorphisms and major molecular response to imatinib in chronic myeloid leukemia? Blood. 2010;116(26):6145–6.
Zheng Q, et al. ABCB1 polymorphisms predict imatinib response in chronic myeloid leukemia patients: a systematic review and meta-analysis. Pharmacogenom J. 2014;15(2):127–34.
Zu B, et al. MDR1 gene polymorphisms and imatinib response in chronic myeloid leukemia: a meta-analysis. Pharmacogenomics. 2014;15(5):667–77.
Skoglund K, et al. ABCB1 haplotypes do not influence transport or efficacy of tyrosine kinase inhibitors in vitro. Pharmacogenom Pers Med. 2013;6:63–72.
Dessilly G, et al. Impact of ABCB1 1236C > T-2677G > T-3435C > T polymorphisms on the anti-proliferative activity of imatinib, nilotinib, dasatinib and ponatinib. Sci Rep. 2016;6:29559. doi:10.1038/srep29559.
Némethová V, Rázga F. Overexpression of ABCB1 as prediction marker for CML: how close we are to translation into clinics? Leukemia. 2016;31(1):266–7.
Brendel C, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21(6):1267–75.
Burger H, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104(9):2940–2.
Nakanishi T. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108(2):678–84.
de Lima LT, et al. Reduced ABCG2 and increased SLC22A1 mRNA expression are associated with imatinib response in chronic myeloid leukemia. Med Oncol. 2014;31(3):851.
Au A, et al. Association of genotypes and haplotypes of multi-drug transporter genes ABCB1 and ABCG2 with clinical response to imatinib mesylate in chronic myeloid leukemia patients. Biomed Pharmacother. 2014;68(3):343–9.
Jiang Z-P, et al. Trough concentration and ABCG2 polymorphism are better to predict imatinib response in chronic myeloid leukemia: a meta-analysis. Pharmacogenomics. 2017;18(1):35–56.
Watkins DB, Hughes TP, White DL. OCT1 and imatinib transport in CML: is it clinically relevant? Leukemia. 2015;29(10):1960–9.
Nies AT, et al. Cellular uptake of imatinib into leukemic cells is independent of human organic cation transporter 1 (OCT1). Clin Cancer Res. 2013;20(4):985–94.
White DL, et al. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J Clin Oncol. 2010;28(16):2761–7.
Wang L, et al. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin Pharmacol Ther. 2007;83(2):258–64.
Nardinelli L, et al. Pretherapeutic expression of the hOCT1 gene predicts a complete molecular response to imatinib mesylate in chronic-phase chronic myeloid leukemia. Acta Haematol. 2012;127(4):228–34.
Singh O, et al. SLC22A1-ABCB1 haplotype profiles predict imatinib pharmacokinetics in Asian patients with chronic myeloid leukemia. PLoS One. 2012;7(12):e51771.
Di Paolo A, et al. The c.480C>G polymorphism of hOCT1 influences imatinib clearance in patients affected by chronic myeloid leukemia. Pharmacogenom J. 2014;14(4):328–35.
Bazeos A, et al. hOCT1 transcript levels and single nucleotide polymorphisms as predictive factors for response to imatinib in chronic myeloid leukemia. Leukemia. 2010;24(6):1243–5.
Vine J, et al. Polymorphisms in the human organic cation transporter and the multidrug resistance gene: correlation with imatinib levels and clinical course in patients with chronic myeloid leukemia. Leuk Lymphoma. 2014;55(11):2525–31.
Hu S, et al. Interaction of imatinib with human organic ion carriers. Clinical Cancer Research. 2008;14(10):3141–8.
Yamakawa Y, et al. Association of genetic polymorphisms in the influx transporter SLCO1B3 and the efflux transporter ABCB1 with imatinib pharmacokinetics in patients with chronic myeloid leukemia. Ther Drug Monit. 2011;33(2):244–50.
Yamakawa Y, et al. Pharmacokinetic impact of SLCO1A2 polymorphisms on imatinib disposition in patients with chronic myeloid leukemia. Clin Pharmacol Ther. 2011;90(1):157–63.
de Lima LT, et al. Relationship between SLCO1B3 and ABCA3 polymorphisms and imatinib response in chronic myeloid leukemia patients. Hematology. 2014;20(3):137–42.
Angelini S, et al. Association between imatinib transporters and metabolizing enzymes genotype and response in newly diagnosed chronic myeloid leukemia patients receiving imatinib therapy. Haematologica. 2012;98(2):193–200.
Khorashad JS, et al. The presence of a BCR-ABL mutant allele in CML does not always explain clinical resistance to imatinib. Leukemia. 2006;20(4):658–63.
Soverini S, et al. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12(24):7374–9.
Shah NP, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–25.
O’Hare T, Eide CA, Deininger MWN. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242–9.
Gorre ME. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–80.
Hochhaus A, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16(11):2190–6.
von Bubnoff N, et al. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359(9305):487–91.
Branford S. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99(9):3472–5.
Corbin AS. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood. 2003;101(11):4611–4.
Branford S. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102(1):276–83.
Jabbour E, et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20(10):1767–73.
O’Hare T. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500–5.
Cortes JE, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–96.
Roche-Lestienne C. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100(3):1014–8.
Soverini S, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208–15.
Graham SM. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–25.
Diaz-Blanco E, et al. Molecular signature of CD34+ hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia. 2007;21(3):494–504.
Corbin AS, et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Investig. 2011;121(1):396–409.
Jorgensen HG, et al. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9):4016–9.
Copland M. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–9.
Inokuchi K, et al. Abnormality of c-kit oncoprotein in certain patients with chronic myelogenous leukemia—potential clinical significance. Leukemia. 2002;16(2):170–7.
Belloc F, et al. The stem cell factor–c-KIT pathway must be inhibited to enable apoptosis induced by BCR–ABL inhibitors in chronic myelogenous leukemia cells. Leukemia. 2009;23(4):679–85.
Zhao F, et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1α-induced metabolic reprograming. Oncogene. 2010;29(20):2962–72.
Ng KP, et al. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood. 2014;123(21):3316–26.
Tang X. Role of phosphatidylinositol 3-kinase and specific protein kinase B isoforms in the suppression of apoptosis mediated by the abelson protein-tyrosine kinase. J Biol Chem. 2000;275(17):13142–8.
Skorski T. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16(20):6151–61.
Parmar S, et al. Role of the p38 mitogen-activated protein kinase pathway in the generation of the effects of imatinib mesylate (STI571) in BCR-ABL-expressing cells. J Biol Chem. 2004;279(24):25345–52.
Nieborowska-Skorska M, et al. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med. 1999;189(8):1229–42.
Klejman A. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002;21(21):5766–74.
Jilani I, et al. Phosphorylation levels of BCR-ABL, CrkL, AKT and STAT5 in imatinib-resistant chronic myeloid leukemia cells implicate alternative pathway usage as a survival strategy. Leuk Res. 2008;32(4):643–9.
Quentmeier H, et al. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4(1):6.
Chu S. BCR/ABL kinase inhibition by imatinib mesylate enhances MAP kinase activity in chronic myelogenous leukemia CD34+ cells. Blood. 2004;103(8):3167–74.
Hentschel J. BCR-ABL- and Ras-independent activation of Raf as a novel mechanism of Imatinib resistance in CML. Int J Oncol. 2011;39(3):585–91.
Warsch W, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117(12):3409–20.
Okabe S, et al. Combination of the ABL kinase inhibitor imatinib with the Janus kinase 2 inhibitor TG101348 for targeting residual BCR-ABL-positive cells. J Hematol Oncol. 2014;7(1):37.
Kaymaz BT, et al. Suppression of STAT5A and STAT5B chronic myeloid leukemia cells via siRNA and antisense-oligonucleotide applications with the induction of apoptosis. Am J Blood Res. 2013;3(1):58–70.
Kosova B, et al. Suppression of STAT5A increases chemotherapeutic sensitivity in imatinib-resistant and imatinib-sensitive K562 cells. Leuk Lymphoma. 2010;51(10):1895–901.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
RSH received support from the Avon Foundation research grant, NIH/NIGMS grant K08GM089941, NIH/NCI grant R21 CA139278, NIH/NIGMS grant UO1GM61393, Circle of Service Foundation Early Career Investigator Award, University of Chicago Support Grant (#P30 CA14599), Breast Cancer SPORE Career Development Award (CA125183), the National Center for Advancing Translational Sciences of the NIH (UL1RR024999), University of Chicago CTSA core subsidy grant, and a Conquer Cancer Foundation of ASCO Translational Research Professorship Award In Memory of Merrill J. Egorin, MD (awarded to Dr. MJ Ratain).
Conflicts of interest
The authors (AN, JW and RSH) declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Nath, A., Wang, J. & Stephanie Huang, R. Pharmacogenetics and Pharmacogenomics of Targeted Therapeutics in Chronic Myeloid Leukemia. Mol Diagn Ther 21, 621–631 (2017). https://doi.org/10.1007/s40291-017-0292-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-017-0292-x