
Abstract
In 2012, the US National Institute of Mental Health launched three clinical trial contracts under a new FAST initiative. The overall goal for these contracts (Fast-Fail Trials) was to focus early-stage trials, testing novel pharmacologic agents that target the central nervous system, on pharmacologic-based designs to objectively identify doses that produce central nervous system effects. The three contracts targeted different psychiatric populations: psychotic (FAST-PS), mood and anxiety (FAST-MAS), and autism spectrum disorders (FAST-AS). The FAST initiative was a first attempt for the National Institute of Mental Health to adapt an experimental medicine approach to its clinical trial portfolio. As the Fast-Fail trials implemented this new approach for the field, we present the rationale for each trial, design considerations, results, and how each one contributed new knowledge to the field of psychopharmacology; important lessons for pharma and biotech. Under the FAST initiative, the National Institute of Mental Health assembled research teams with a broad range of expertise, who developed and validated the outcome measures and study protocol, and conducted multi-site clinical trials, testing candidate compounds. In the FAST-PS contract, the team validated an imaging-based pharmacodynamic biomarker of the effect of ketamine in the brain that could be utilized in subsequent clinical trials. The initial FAST-AS study was an important first step in the design of early-stage target-engagement trials in autism spectrum disorder, suggesting that a resting electroencephalogram can be used as a pharmacodynamic measure in future studies. The FAST-MAS study showed that blocking the kappa-opioid receptor significantly affects functional magnetic resonance imaging ventral striatal activation in the monetary incentive delay task in anticipation of gain. Together, the outcomes of the FAST-FAIL trials demonstrated the importance of rigorously designed and informative central nervous system trials, including the value of pharmacodynamic measures in early-stage trials. Use of these measures furthered our knowledge about the relationship between specific molecular mechanisms, brain effects, and therapeutic effects in patients with mental illnesses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The FAST-FAIL initiative is a program developed by the US National Institute of Mental Health to support early-stage pharmacodynamic trials of candidate compounds provided by industry that have failed in late-stage central nervous system (CNS) trials. The FAST-Fail initiative is intended to determine if dosing can achieve CNS functional effects. |
Conducting early-stage trials requires teams with a broad range of expertise, including clinical trialists and pharma researchers with legacy information on the candidate drugs, to cooperatively develop study protocols and evaluate progress and outcomes. |
There is a need for rigorously designed and informative early-stage CNS trials, to determine dose ranges and evidence of brain effects that would support future larger scale clinical trials. |
It is highly important to build pharmacodynamic target-based measures into early-stage trials, using CNS functional techniques such as functional magnetic resonance imaging and electroencephalograms, to adequately establish target engagement. |
1 Background
Over the past decade, most large pharmaceutical companies have closed or drastically reduced the size of their central nervous system (CNS) divisions, especially those that were focused on developing compounds for psychiatric disorders. These actions reflect the large number of failures in successfully developing drugs in this field when going beyond mechanisms already known to work. The low probability of success in bringing drugs with new mechanisms to the market means it does not make sense from a financial viewpoint to continue to invest given current financial models. It has been argued by leaders in the field that new incentives are required to mitigate the risks of novel psychiatric drug development [1].
The decrease of large pharma investment in psychiatric drug development was alarming to the community as pharma provides a majority of the drugs that go through regulatory approval to patients and provides the knowledge for developing potent and selective compounds with desirable pharmacokinetic (PK) properties. Because of pharma’s earlier investments, when decisions were made to reduce future investment there were already candidate compounds designed to affect molecular targets in the brain hypothesized to have potential for psychiatric disorders with good safety margins, but which were shelved and/or out-licensed to small ‘virtual’ companies with limited resources. In an attempt to mitigate the consequences of large pharma withdrawing from exploring mechanisms that had not been validated but were of high theoretical interest, the US National Institute of Mental Health (NIMH) awarded three contracts, coined ‘Fast-Fail’, to academic institutions to perform early-stage pharmacodynamic (PD) trials to test candidate drugs with novel mechanisms that industry had advanced to the stage of human studies to determine dose ranges and evidence of brain effects that would support future larger scale clinical trials.
Knowledge of doses that produce a specific brain effect allows one to conclude in a clinical study whether there is a relationship between an action of the drug in the brain and clinical effect. Ideally, this process allows for the testing not only of a specific compound but of the molecular mechanism being pursued. The hope is to achieve more rapid validation or rejection of the mechanistic hypothesis clinically. Such an approach entails requiring a ‘proof-of-molecular-mechanism’ study prior to undertaking a traditional ‘proof-of-efficacy’ study in patients based on extrapolations of the doses shown to produce brain effects in animal studies. The principle behind this approach was to find compounds that had the characteristics required to test a specific mechanism with results that would apply to any other compound with the same mechanism.
Some of the potential candidate drugs had previously been tested in phase II or III pharma efficacy trials for psychiatric indications with negative results. However, the clinical studies had been performed in the absence of either definitive receptor occupancy (RO) or PD measures of the compounds’ brain effects, leaving the negative results uninterpretable with regard to target validation. The NIMH contracts were initiated to better define the dose(s) of selected drug candidates that would produce the targeted mechanism-related effect on CNS function (a PD measure), to allow for much better interpretation of study results. In addition to the scientific goals, the contracts were designed to: provide results more rapidly than the traditional timelines of academic clinical research (hence the contract name ‘Fast-Fail’); provide a collaborative team environment where pharma scientists, NIMH staff, academic and industry consultants along with the academic contractors work together on compound selection and trial designs; and provide resources to the academic sites to ensure registration quality data (in the event the results show promise for further development of the compound).
The scientific questions raised under each FAST trial were distinct and the approaches undertaken to answer those questions were important to present to the field, not just about a particular compound’s viability per se as adequate for testing a mechanistic hypothesis, but also to demonstrate how one could initially test additional compounds in humans for a variety of CNS indications. The contracts focused on specific symptoms/indications. As such, the NIMH awarded three FAST contracts: Fast-Fail Trials in Autism Spectrum Disorders (FAST-AS), awarded to the University of California at Los Angeles (Contract No. HHSN271201200005I, principal investigator: James McCracken); Fast-Fail Trials in Mood and Anxiety Spectrum Disorders (FAST-MAS), awarded to Duke University (Contract No. HHSN271201200006I, principal investigator: Andrew Krystal); and Fast-Fail Trials in Psychotic Spectrum Disorders (FAST-PS), awarded to the Research Foundation for Mental Hygiene (Contract No. HHSN271201200007I, principal investigator: Jeffrey Lieberman).
The goal of this review is to highlight each contract’s goals and outcomes, and the progress made to better understanding the study compounds’ effects on the CNS and viability for subsequent studies. Use of CNS PD measures in human trials is a fairly new approach to ‘go/no go’ decision making in moving compounds forward to late-stage trials. It is important to continue refinement of CNS functional methods such as electroencephalograms (EEGs) and functional magnetic resonance imaging (fMRI) to enable clinical researchers to begin to probe subject-level brain data, which is critical in validating PD measures. The results from these contracts define current capabilities of these measures, as well as their limits, and are described below.
2 Approach
As part of the FAST program, the NIMH established an overarching FAST Committee, which included members with experience from industry, academia, regulatory affairs, and staff from the NIMH and the National Institutes of Health (NIH). The committee’s charge was to evaluate CNS molecular targets of interest for each contract, then rank order CNS compounds for those targets that at a minimum had been tested in first-in-human studies and established initial human safety, for further clinical testing. Criteria for target selection are listed in Fig. 1 and include: (1) a specific and testable hypothesis; (2) a positron emission tomography (PET) ligand to evaluate RO; (3) brain-functional target-engagement measures; (4) a target-selective, CNS-penetrant, Investigational New Drug-ready compound; and (5) consider Research Domain Criteria principles [2] where appropriate. The criteria were considered a gold standard with which to strive for, but not every target of interest met these criteria, as specific PET ligands were not available for the receptor agonists selected. Therefore, it was imperative that in the absence of direct demonstration of target engagement with a PET ligand, a functional measure that could be reasonably inferred to reflect target engagement was available. Once compounds were selected, agreements were established with pharma, and additional consultants were brought into the contract teams who had historical knowledge about the compound to be tested. Given the broad spectrum of patients that fall under the mood and anxiety disorder spectrum as well as the autism spectrum, the committee also considered methods to stratify subjects within each spectrum into more homogeneous subgroups that might be better treatment candidates for the compound.

Criteria for target selection under the National Institute of Mental Health Fast-Fail program. CNS central nervous system, EEG electroencephalogram, fMRI functional magnetic resonance imaging, IND Investigational New Drug, MRS magnetic resonance spectroscopy, PET positron emission tomography, RDoC Research Domain Criteria
2.1 FAST-MAS
The study conducted under the FAST-MAS contract was one of the few, and arguably the only study, with a CNS target choice that met the full gold standard criteria for target selection. The FAST-MAS study focused on a core symptom domain within the broad spectrum of mood and anxiety—anhedonia, the inability to experience pleasure—and introduced measures of associated rewards circuitry as PD measures to incorporate into early-stage trials. From the perspective of taking Research Domain Criteria principles into account, anhedonia falls under a range of Research Domain Criteria constructs (“Reward Responsiveness”, “Reward Learning”, and “Reward Valuation”) [2, 3]. Animal studies had implicated κ-opioid receptor (KOR) antagonism in affecting reward-related brain circuitry (the ventral striatum) to improve reward-associated function and/or reverse anhedonic behaviors [3,4,5,6,7,8,9,10,11]. The FAST Committee and the contract team prioritized the focus on the anhedonia domain and related brain circuit function measures because a KOR antagonist, JNJ-67953964 (formerly known as LY2456302 and CERC-501), met the other criteria for target selection based on existing PET target-engagement data, evidence of human safety, and compound availability.[12,13,14,15]. Zheng and colleagues had established near saturation of RO for a 10-mg dose of JNJ-67953964, using the specific KOR PET tracer [11C]PKAB (LY2879788) [15]. Therefore, all of the criteria in Fig. 1 for compound selection were judged to have been met for the KOR antagonist.
The selected PD measure judged to reflect at least part of what is subsumed under the concept of anhedonia was a task-based fMRI measure: a monetary incentive delay (MID) task effects on ventral striatal activation [16]. As there are several components of this paradigm, an a priori primary outcome measure was specified—mean fMRI ventral striatal activation in the MID task in anticipation of gain testing JNJ-67953964 compared with placebo. We emphasize this pre-specification of the primary outcome because as it turned out the specification of a different measure from the paradigm would have yielded even more impressive findings. In the absence of prior experience with use of the fMRI paradigm in the context of looking for a specific PD effect in humans, an educated guess needed to be made. One of the themes that has emerged from our experience with the FAST program is that the current state of functional brain measures available to be used in humans may need refinement to be optimal for signal detection of drug effects.
The FAST-MAS (ClinicalTrials.gov Identifier: NCT02218736) was a six-site, 8-week, double-blind, placebo-controlled, randomized trial in patients with anhedonia, as measured by the Snaith-Hamilton Pleasure Scale (SHAPS; a 14-item instrument) [17] score of 20 or higher, and a Diagnostic and Statistical Manual of Mental Disorders, 5th Edition mood or anxiety disorder. Eighty-nine subjects (mean age, 39.5 years; 62.9% female) were randomized, 45 (64.4% female) to the JNJ-67953964 group and 44 (61.4% female) to the placebo group. Of these participants, 68 individuals completed the study, 33 in the JNJ-67953964 group and 35 in the placebo group. The primary outcome of this proof-of-mechanism study was striatal activation to reward-predicting cues, as assessed with fMRI in conjunction with the MID task. Further descriptions of procedures, outcome measures, and analyses are provided in the Krystal et al. publications (2018 and 2020) [12, 18]. In the FAST-MAS study, Krystal and colleagues found significant effects for the mean and maximum fMRI ventral striatal activation in the MID task in anticipation of gain contrasted with non-response efficacy trials with JNJ-67953964 compared with placebo (Fig. 2) [18]. The drug effect size, however, was almost twice as large according to a secondary outcome measure, ventral striatal activation to anticipation of loss (as opposed to gain). What the FAST-MAS findings exemplify is the manner in which questioning how a compound affects brain function can reveal insights that go beyond documentation of brain effects and lead us to rethink how we conceptualize the brain processes reward paradigms. It is worth noting that the SHAPS, a traditional behavioral measure of altered hedonic response, was included as a secondary outcome measure and did reveal a drug effect although not as robust as with the fMRI measures.

JNJ-67953964 effect on reward anticipation in FAST-MAS study. a Location of ventral striatal region of interest based on the Harvard–Oxford Subcortical Atlas. b Mean baseline-adjusted functional magnetic resonance imaging signal intensity in the ventral striatum during reward anticipation in the monetary incentive delay task after 8 weeks of treatment with JNJ-67953964 and placebo. Error bars represent 95% confidence intervals. *p < 0.01
A more mundane but operationally important aspect of this study was the attention to detail required to establish confidence that data across sites using a functional brain imaging measure could be pooled. This entailed a degree of standardization of both task administration and fMRI paradigms (summarized in Krystal et al.) [12] that required time and resources that go beyond what has been typical in traditional investigator-initiated studies. What was required for standardization is a common theme across all three contracts. Given the goal to generate data that would inform ‘go/no-go’ decisions on future studies with a compound, one requires a high level of confidence in the validity (and likely replicability) of the results.
Overall, from contract award to the end of the study, it took 4.5 years. The start-up phase was almost 2 years, owing to multiple factors contributing to the extended start-up period, including multiple institutional review boards, independent Data and Safety Monitoring Board, challenges with site calibrations, contract administrations, and subcontract awards. The time from the start of enrollment to the end of the study was 2.5 years. The lessons learned have allowed the NIMH to more realistically anticipate the numerous challenges involved in early-stage clinical trials and multi-site imaging studies, including regulatory aspects, safety monitoring, and data management, all critical components for successful drug development clinical trials.
Subsequent to the FAST-MAS study, we understand that there has been a rise in companies pursuing the development of treatments for anhedonia, and potentially exploring anhedonia as an indication, with the US Food and Drug Administration. This narrowed focus on a symptom within the broad definition of depression in part is being pursued because FAST-MAS legitimized anhedonia as a target. In one example, Takeda, directly following the precedent set by FAST-MAS, pursued a development path for their compound TAK-041 that included a small phase IIa proof-of-mechanism study, smaller in size than the FAST-MAS study, using the MID task to determine if motivation/reward deficits observed in schizophrenia could be attenuated by the drug (ClinicalTrials.gov Identifier: NCT03319953). The question that remains open is whether the CNS effects on the MID, without some evidence of positive effects on the SHAPS, would have provided the same level of stimulus by companies in pursuing this mechanism. Put another way, would knowing that a compound had an effect on a brain functional circuit take precedence over its lack of effect on a clinical measure at an early stage of development?
2.2 FAST-PS
Under the FAST-PS contract, the team identified a target of interest where not all criteria for selection were met; no PET ligand was available. Yet, various pieces of preclinical and clinical evidence suggested the target was promising and that information, together with a target-selective, CNS-penetrant, Investigational New Drug-ready compound, established a specific and testable hypothesis. The decision was to test the activation of the metabotropic glutamate 2,3 receptor (mGluR2/3), a modulating receptor subtype in the CNS that primarily is located presynaptically on neurons and, when stimulated, can reduce the release of glutamate produced by N-methyl-d-aspartate receptor antagonists. The metabotropic group of glutamate receptors, which act through second messenger systems to regulate neuronal function, stands in contrast to the major ionotropic glutamate types, N-methyl-d-aspartate and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate receptors, that open ion channels directly. Drugs acting on glutamatergic CNS ionotropic receptors are associated with immediate effects and used in anesthesia and epilepsies with, to date, no examples of long-term use in the treatment of major mental illnesses.
Observations and then studies on the ability of N-methyl-d-aspartate receptor antagonists to induce psychosis in humans stimulated a focus on glutamatergic function as a target for treating schizophrenia three decades ago [19, 20]. By the late 1990s, it was appreciated that N-methyl-d-aspartate receptor antagonists induce elevated extracellular glutamate, and mGluR2/3 agonists can act presynaptically to attenuate glutamate release [21]. There was considerable excitement in the field when an mGluR2/3 agonist, pomaglumetad (POMA/LY2140023), was reported to show efficacy in a phase II proof-of-concept study in schizophrenia [22] followed by very disappointing phase III studies [23,24,25]. It was unclear whether these overall negative phase III studies be viewed as ruling out the mechanism in the treatment of schizophrenia as no studies had been conducted to establish that the administered doses did indeed have any effect on brain function. Doses for the POMA studies had been established based on cerebrospinal fluid concentrations, which were assumed, based on preclinical studies, to be sufficient to modify brain function.
Only after clinical studies had been underway for years was a method developed that might be applied to show a PD effect of POMA in the human brain. As already noted, no PET ligand is yet available that can directly measure the degree to which mGluR2/3 compounds bind to the receptor. Instead, rat and then human studies were ultimately performed to provide a functional read-out that most likely reflects mGluR2/3 agonism. Building on earlier microdialysis findings of ketamine-induced glutamate release, an associated blood-oxygen-level-dependent (BOLD) fMRI signal increase was demonstrated in rats. This imaging measure provided a PD biomarker of the effects of ketamine that could be investigated in humans who were found to show analogous fMRI increases to those seen in rats [26].
Subsequently, the ability to block or reduce the BOLD response to ketamine was used to determine doses of mGluR group II (mGluR2, mGluR3) prodrugs that produced a CNS functional signal [27]. This study utilized an acute randomized, double-blind, placebo-controlled, PK/PD dose–response design in healthy volunteers, testing POMA, the drug used in clinical trials, and LY2979165, an investigational compound characterized as a selective orthosteric agonist of mGluR2 receptors for the degree of inhibition of the BOLD response to ketamine. Drug candidates were dosed up to a maximum tolerated dose and administered as a single dose 4–6.5 h before ketamine infusion. Only the highest dose of POMA produced a significant reduction in the ketamine BOLD signal at the group level with a high degree of variability whereby some subjects showed total blockade and others none at all, with no observed relationship to blood concentrations. In contrast, the investigational selective mGluR2 agonist LY2979165 did show a significant relationship between blood concentrations achieved at the highest tolerated dose and the degree of inhibition of the BOLD response, suggesting a more consistent blood-to-brain relationship than is the case with POMA. The 80-mg dose of POMA, administered twice a day (BID) used in the phase III studies, was not tested in the ketamine reversal study but the 40-mg dose, used BID in the earlier positive phase II study, was used. There was no evidence of any effect of a single 40-mg dose on the BOLD response. Moreover, the absence of a clear relationship between blood concentration and decreased BOLD response after the 160-mg dose raises the possibility of highly variable penetration of POMA into the brain given that the hypothesized PK/PD relationship between the selective mGluR2 agonist and inhibition of the BOLD signal was observed. Notably, for both compounds, these were maximally tolerated doses determined on the basis of gastrointestinal side effects, which, as will become apparent from what follows, is relevant to the issue of what might be required to administer doses that would achieve consistent brain effects. Taken in its totality, the Mehta et al. study provided a firm foundation on which to build other studies to answer questions about whether doses of POMA could be found that would produce more consistent inhibition of the BOLD response following a BID dosing schedule that had been adopted for clinical efficacy studies [27].
Given this background, a two-part FAST-PS multi-site study designed to compare 40 mg (the phase II positive study dose) and 160 mg (twice the phase III dose that failed) BID for 10 days was undertaken to address the question of whether high enough doses of POMA had been given in any of the efficacy studies to achieve inhibition of the ketamine-induced BOLD effect. Additionally, the possibility was explored that proton magnetic resonance spectroscopy (MRS) measures of glutamate following ketamine infusion would be sufficiently robust to more directly test effects of POMA on stimulated glutamate.
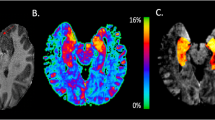
The first part of the overall FAST-PS study consisted of a three-site, randomized, placebo-controlled study (ClinicalTrials.gov Identifier: NCT02134951) of imaging methods to see which best reproducibly detected a signal judged to reflect glutamate release in healthy subjects, using intravenous administration of low-dose ketamine or placebo (2:1 ratio) [28]. Sixty-five subjects (mean age, 31.1 years; 63% male) were randomized, with 53 individuals completing the full study protocol (34 receiving ketamine and 19 receiving placebo). The study provided a side-by-side comparison of three potential imaging approaches for the detection of ketamine-induced alterations in brain function as a model for schizophrenia, comparing feasibility and relative strengths and weaknesses of these imaging-based approaches. Methods included two passive measures: pharmacoBOLD fMRI to measure the metabolic increase associated with elevated glutamate, and MRS of glutamate plus glutamine to measure glutamate directly. A task-related fMRI measure (the Relational and Item Specific Encoding task; RISE) was also employed to target dorsolateral prefrontal cortex and hippocampus, since previously the measure showed reduced brain activation in these regions in subjects with schizophrenia [29]. Further description of the procedures and analyses are provided in Javitt et al. [28] A decision was made in this study to almost double an originally suggested number of subjects to have sufficient power to compare the different biomarkers. Upon ketamine infusion, only the pharmacoBOLD method showed a very large effect (Fig. 3), while MRS of glutamate plus glutamine observed only a moderate increase vs placebo in the first 15 min following ketamine infusion, which returned to levels observed on placebo by 30 min; no changes were observed with the task-related measure [28]. In the absence of a robust MRS-determined increase in the glutamate plus glutamine peak that was highly correlated with the BOLD response, other effects of ketamine beyond affecting glutamate release may be important contributors to the BOLD response. Whatever the mechanistic basis of the response, the question remained whether the reduction in the ketamine BOLD response by POMA seen in rats could be translated into humans. This multi-site study was important not just for recruitment feasibility, but it provided results to better determine how different measures performed in detecting a drug effect and how they could be standardized and reproduced across separate clinical sites to enable wider scale use of specific CNS PD measures in future trials. Importantly, for the design of the study in which POMA was administered (Fig. 3), it was noted that not every subject responded to intravenous ketamine, as observed with a pharmacoBOLD response, although their plasma ketamine and norketamine concentrations were similar to those in subjects who did show an increase in the BOLD signal. While it was unclear why these response differences existed in subjects, in subsequent studies using inhibition of ketamine BOLD responses to assess effects of POMA, the investigators prescreened each subject to evaluate their BOLD response to ketamine; those subjects with an absent baseline BOLD response were excluded [30]. This initial methods development study also allowed for powering of the actual POMA study using fMRI. The important more general methodologic lesson learned is that for functional measures involving responses, it is important to be sure that subjects show the expected response before trying to modify it. This is not a problem when conducting PET RO or tissue exposure studies following drug administration as one is directly measuring either a pharmacologic interaction or concentration and not a response that might suffer from floor or ceiling effects depending on the state of the individual at the time.

Biomarker validation in FAST-PS study. a Ketamine evoked changes in the functional magnetic imaging bold oxygen level-dependent (BOLD) response, ketamine vs placebo. Dotted circle denotes subjects who did not respond to ketamine. b Dorsal anterior cingulate cortex (ACC) region of interest (ROI) for BOLD response (cross-sectional comparison) and magnetic resonance spectroscopy data. c Magnetic resonance spectroscopy results of ketamine vs placebo. Cho choline, Cr creatine, Glx glutamate plus glutamine, NAA N-acetyl aspartate
Under the FAST-PS contract, once the criteria were established for selecting subjects with an adequate baseline BOLD response to ketamine, a four-site, randomized, placebo-controlled, double-blind clinical trial was performed to test the effects of POMA (ClinicalTrials.gov Identifier: NCT02919774; [30]) after repeat BID doses of 40 mg (i.e., low dose of 80 mg/day, used in the positive phase II trial) and 160 mg (i.e., high dose of 320 mg, twice the highest dose explored in any of the phase III trials). For this study, 95 healthy volunteers (aged 18–55 years) were randomized to POMA (low dose, high dose) or placebo (1:1:1 ratio) for 10 days; 81 individuals completed the study protocol and data from 76 subjects (49% male) were included in the efficacy analysis. The primary outcome was ketamine-induced changes in pharmacoBOLD in the dorsal anterior cingulate cortex. Further description of the procedures and analyses are provided in Kantrowitz et al. [30] Consistent with the first phase of the FAST-PS study, more than 10% of subjects did not show a clear positive BOLD response after intravenous ketamine and hence were excluded from the POMA administration phase. Additionally, consistent with prior observations on gastrointestinal side effects, the 160-mg BID dose produced marked nausea and some vomiting in some subjects requiring building in a flexible dose titration schedule to achieve the 160-mg BID dose within a week. After 10 days of dosing, consistent with observations after an acute dose (Mehta et al.), [27] repeat doses of 40 mg BID of POMA had no effect on the ketamine-induced BOLD response. In contrast to the previously reported modest BOLD signal reducing effects of an acute 160-mg POMA dose (Mehta et al.), [27] following repeat dosing of 160 mg BID, no reduction in the ketamine-induced BOLD response was observed. Thus, to the extent that a reduction in the BOLD response is a valid means of detecting functional engagement of mGluR2 receptors, the lack of efficacy of POMA in the doses employed could be simply a function of too low a dose. Put another way, as we have no evidence that doses employed produce consistent functional changes in the brain reflective of mGluR2/3 agonism, the negative phase III trials do not rule out the possibility that this molecular mechanism has potential for the treatment of schizophrenia [27].
In addition to the obvious major takeaway that the potential of mGluR2/3 agonism remains to be tested, other important goals were achieved with regard to putting in place what is needed to rule in or out a molecular mechanism’s range of brain and ultimately clinical effects. By taking information from previous single-site biomarker studies that were designed to be proxy measures intended to detect changes in CNS glutamate levels, the team was able to establish conditions for applying across multiple sites and determining the feasibility, reliability, and limits to incorporating them into clinical trials. An important aspect was to compare different methods for detecting, in this case, effects of ketamine on the brain showing that the functional (BOLD fMRI) response was much more robust than the biochemical (MRS) response. Such a comparison of different types of read-outs allows for the refinement of sample sizes depending on whether one is seeking any PD measure of a drug’s effects vs looking for a very specific effect such as changes in a single neurotransmitter (e.g., glutamate). As already demonstrated, non-trivial refinements of paradigms to assess drug effects in the brain were achieved such as ascertainment of baseline functional response and, as elaborated in the detailed report, the method of administration of an agent. Thus, in the study to assess whether POMA reduced the ketamine response, [30] a bolus of ketamine was employed based on the finding that peak BOLD responses occurred within 5 min of the bolus phase falling off during the infusion phase in the FAST multi-site comparison study of fMRI BOLD and MRS responses [28].
2.3 FAST-AS
The FAST-AS contract study (ClinicalTrials.gov Identifier: NCT01966679) was designed to focus on treatment of social dysfunction, a core deficit in autism spectrum disorder (ASD), which lacks any pharmacologic treatment. A commentary has been published that discusses more thoroughly the experience of working with the contract team to make the decisions of compound and target selection, protocol design, and the resources needed to establish and manage the trial, specifically emphasizing the need to provide industry-type resources to academic sites, when testing candidate compounds with an intent of including the data in a regulatory package [31].
The overall intent was to eventually test a compound in pediatric populations, and therefore the design had to consider this future intent. Less information is currently known about possible pathological underpinnings in ASD, compared with psychosis and the mood and anxiety disorder spectrums. Although limited with regard to sample size, post-mortem studies of subjects with ASD have revealed evidence of complex alterations of the gamma aminobutyric acid (GABA) system [32]. Moreover, in vivo studies that followed the post-mortem reports provide evidence of lower GABA levels in frontal, auditory, and motor cortices by MRS [33] as well as lower occipital GABA/glutamate ratios in ASD [34]. The decision was therefore made to see if there were novel methods of perturbing GABA that might go beyond the perceived limitations of marketed benzodiazepines.
The GABA subtype A (GABA-A) receptor complex can be made up of different subunits (alpha 1, 2, 3, or 5), the composition of which affects receptor function as well as behavioral effects as assessed preclinically. Benzodiazepines are traditional GABA-A receptor agonists with nonselective alpha-subunit binding affinities that have been marketed since the 1960s and used to treat anxiety, seizures, sleep disturbance, and muscle spasms. Although they have been used clinically in patients as young as 6 months old, sedation, cognitive deficits, and drug dependence are side effects that initially limited our interest in testing currently marketed benzodiazepines under this contract. Given the issues limiting further exploration of traditional benzodiazepines in pediatric and other populations, pharma companies have been pursuing the development of selective GABA-A receptor alpha2,3 positive allosteric modulators, based on preclinical evidence that the alpha2 and alpha3 subtypes produce anxiolytic effects without the side effects seen with benzodiazepines [35, 36]. Potent effects on the alpha1 subunit are believed to be most responsible for sedative effects. One of the selective compounds, AZD7325, was available for testing. Two double-blind placebo-controlled trials testing AZD7325 efficacy in adults with generalized anxiety disorder were previously completed; one trial included lorazepam as a positive control [37]. Both trials produced negative primary outcomes to AZD7325, but it remained unclear whether dosing was adequate, leading to a need for a CNS PD trial to be performed before further efficacy trials should be pursued. As in the FAST-PS contract, this agonist compound also lacked RO data for those specific receptor subtypes—no GABA-A receptor alpha2,3 PET ligands have successfully been generated to date—although RO curves for AZD7325 had been generated using a radiolabeled benzodiazepine (11C-flumazenil) and the data were used to infer RO of AZD7325 [38]. The PET RO study observed high occupancy (> 70%) of the GABA-A receptors with AZD7325 doses above 5 mg; maximum apparent occupancy was reached at AZD7325 doses of 20 mg and above. AstraZeneca then used a 10-mg AZD7325 acute dose to further evaluate potential CNS PD to assess sedation, cognition, and resting EEGs in healthy subjects, compared to lorazepam and placebo [39]. The benzodiazepine lorazepam was used as a positive comparator because of its well-established effects on resting EEG, sedation, and cognition. The AZD7325 EEG PD effects in that study included: decrease in delta and theta activity in the frontal-central area, which was distinct from lorazepam that demonstrated increases in delta, beta, and gamma activity, and decreases in theta and alpha activity. The use of EEG as a PD measure was particularly interesting, as the measure is routinely used in ASD research, including in pediatric studies.
Using both the RO and PD data, the FAST-AS contract team designed an initial trial. Given that ASD is a very broad diagnostic category, the researchers first piloted whether it was possible to use an EEG to identify a subset of ASD subjects with EEG function distinct from healthy controls. They established a normative EEG database of 38 healthy control subjects and compared the data with EEG data of 12 subjects with ASD; they identified three EEG parameters that distinguished the ASD subjects from the healthy controls: resting theta power, beta power during FACES task, and beta coherence. Using a cut-off value of 0.6, this measure’s performance metrics were 80% sensitivity, 70% specificity, and an area under the curve of 0.85 [40]. Granted that the subject numbers used to establish this measure were small, there were no obvious clinical differences between those with this EEG pattern and those subjects who had patterns similar to healthy controls. This EEG measure was then used in the AZD7325 clinical trial as an additional stratification criterion, which resulted in an exclusion rate of 17%. The primary outcome selected for this double-blind placebo-controlled trial was the PD EEG spectral power measure established in the previous Chen et al. study, [39] along with safety and tolerance, using an acute resting EEG PD paradigm testing 5 mg of AZD7325 or placebo. Thirty-eight adult subjects, (74% male) aged 18–35 years, diagnosed with ASD as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, and the Autism Diagnostic Observation Schedule, [41] with an IQ estimate of > 80 and Clinical Global Impression–Severity scale of 4 or greater, were included in the trial. Following the acute dosing phase, subchronic exposure was implemented using increasing flexible doses (BID) over 6 weeks up to 15 mg BID, with weekly clinical assessments and EEG measures at 4 and 6 weeks. The active phase of the trial took 1 year to complete. Acute PD EEG shifts were observed with 5 mg of AZD7325, including a decrease in delta power, as observed in the Chen et al. study [39] using a higher 10-mg dose (Fig. 4) [42]. Yet, in the FAST-AS trial, theta power increased rather than decreased. It is unclear whether these differences were due to differences in dosing, EEG device, analytical approach, or the patient population, but these results do suggest 5 mg was sufficient to induce a PD response.

Pharmacodynamic response to a 5-mg acute (single) dose of AZD7325 in FAST-AS study. Topographical locations and pre- vs post-dose effects of AZD7325 vs placebo on resting electroencephalogram spectral power by frequency band in subjects with autism spectrum disorder. Relative electroencephalogram spectral power means and electrode clusters: a Delta power in the parietal midline (PZ), within subject F = 4.3*, interaction F = 3.4+; b Theta power posterior (POZ), within subject F = 4.7*, interaction F = 4.2*; c alpha power frontal; central (FZ) within-subject F = 4.3*, interaction F = 6.9**; d Beta power central midline (CZ), within subject F = 8.6**, interaction F = 4.8*. Interaction represents interaction term of group (AZD7325, placebo) by time (pre- vs post-dose); *p < 0.05; **p < 0.01; +p < 0.10. Error bars represent standard error of the mean
Inspection of the topographical locations of the spectral power pre/post-EEG differences in Fig. 4 raises issues about the criteria by which one selects a measure or a combination of measures as the most informative with regard to a drug effect. Drug/placebo differences are reported for theta and beta spectral power at many more locations that extend beyond a single tight cluster vs the very restricted clusters revealing a difference in alpha and/or delta power. As the results from Chen et al. [37] are not presented in a manner to know whether the delta decrease was limited to a small PZ cluster as observed in the ASD subjects, given the complexity of EEG data and the many methods in which it can be analyzed, a far more standardized approach to reporting all relevant pieces of data is needed to compare results across studies. In the absence of such standardization of study design, acquisitional paradigms, and analytic pipelines, it is impossible, as noted above, to know whether differences in aspects of drug effects across studies reflect differences in the responses of individuals or differences in methods.
Extending the dosing to 4 and then 6 weeks using a flexible dosing schedule indicated a shifting of the primary theta EEG endpoint to increased power. However, blood concentrations of AZD7325 at both the 4- and 6-week time points revealed that although a significant number of subjects assigned the compound (in a double-blinded manner) had reported taking the compound, they had low concentrations (based on PK data in AstraZeneca’s investigator’s brochure) or nondetectable amounts measured in the plasma, indicating that medication adherence was a potential issue and therefore the subchronic results were less informative. It also revealed the importance of periodic plasma sampling to assess adherence, rather than solely relying on subjective diaries.
Even with the limits of the study, this initial FAST-AS study performed in adults was an important first step in the design of early-stage target-engagement trials in ASD. It suggested that a resting EEG could be used as a PD measure, and it highlighted the need to build a dose range into initial acute dosing trials AND a positive control (when possible), in this case, a benzodiazepine. With those changes to study design, one could evaluate whether the different EEG systems used in FAST-AS vs the Chen et al. study [39] produced the same or different benzodiazepine PD effects. If different, then the somewhat disparate FAST-AS PD results might be explained by the different EEG systems. Adding dose ranges would provide more confidence in the PD measure—that certain shifts in power would be consistent in doses that alter CNS function, presumably at the site of action of the drug. The data from FAST-AS are available in the National Institute of Mental Health Data Archive (https://nda.nih.gov/) for other researchers to analyze. Future studies with GABA receptor alpha2,3 compounds could also begin to pursue pediatric ASD testing, using a pediatric PK/PD bridging approach, with design considerations based on the results from FAST-AS and future design consideration. Recently, Baergic Bio entered into an exclusive licensing agreement with AstraZeneca to advance the clinical development of AZD7325 (now called BAER-101) in select CNS disorders [43]. These types of transitions in intellectual property occurred with all three compounds tested in the FAST contracts. Moreover, licensing or selling compounds to other companies could result in a loss of NIMH access to them (one of the difficulties in testing shelved candidate drugs). For FAST-MAS and FAST-PS, the studies were ongoing and the new companies honored the original agreements (e.g., clinical supply agreement). Given this situation, the NIMH is reaching out to various companies that own selective GABA-A receptor alpha2,3 positive allosteric modulators that are ready for human testing, to continue these studies. The National Advisory Mental Health Council has also established a workgroup that is considering optimizing strategies and approaches for testing novel interventions.
3 Conclusions
Under these Fast-Fail contracts, the NIMH was able to create a team environment whereby experts from various affiliations were able to work together to develop PD measures and implement them in candidate compound trials. While improvements in CNS functional techniques such as fMRI and EEGs continue to be developed, there are still limits to their capabilities, when using subject-level data to make large financial decisions about whether to continue a drug program. The decisions, in terms of PD approaches and methods, populations to test, dosing, and trial design, required a group effort where clinical trialists, pharma researchers with legacy information on the candidate drugs, and other consultants were able to cooperatively develop the plan and then evaluate progress and outcomes together. It would not have been feasible to accomplish these goals under an investigator-initiated grant. Each project taught us the value of building PD measures into early-stage trials, and the limits of each measure. For these types of trials to have the greatest impact, one needs to design the trial such that a negative result ‘definitively’ rules that compound out from further testing under those conditions. Put another way, the goal is to have sufficient confidence in the validity and interpretation of the trial results to be able to make a ‘no go’ decision with a low likelihood of making a type 2 error. Level of certainty depends, of course, on the robustness of the methods utilized, which are still evolving in terms of detecting effects of agents on brain function.
Thus, there is often a tension between moving quickly in terms of study design and one’s openness to adjusting protocols to incorporate emerging information on the utilization of a biomarker. We presented the importance of excluding those subjects who did not show a response to ketamine infusion in a prespecified region of interest, in the FAST-PS trial, as an example. Unlike the situation in which an orthosteric antagonist can definitively be ruled in or out as occupying a receptor when an appropriate PET ligand is available, other functional CNS pharmacodynamics remain evolving methods. Our experience teaches us that it is useful to have researchers publish, or at least make available upon request, all of their findings (positive and negative) and present the data at a subject level, rather than population-based differences. Only with such information can one properly power future studies and anticipate potential issues with variation, especially when carrying out multi-site trials.
We emphasize these issues because we are operating from the perspective of the importance of building the link between a specific molecular mechanism and physiologic and therapeutic effects in humans. Beyond finding a means of incorporating methodologic advances, there are important operational lessons learned given that most precedents for incorporation of target-engagement studies in early drug development come from pharmaceutical companies that utilize industrialized processes different from those available under NIH funding mechanisms. Even those, however, can evolve and when trial contracts were awarded in 2012, start-up phases involved unanticipated complexities including the use of NIH contracting mechanisms, establishing sites while depending on primary contractor subcontracting mechanisms, and evolving NIMH oversite structures. Once those were in place, trials were conducted (clinical data collection) in 1–3 years. Given the normal NIH grant cycles of 5 years, with study designs not focused on prespecifying outcomes, this FAST FAIL model was successful through establishment of concrete outcomes over this period of time. It also provided evidence that the NIMH could fund and oversee these trials, while not having the resources of industry.
References
Choi DW, Armitage R, Brady LS, Coetzee T, Fisher W, Hyman S, et al. Medicines for the mind: policy-based “pull” incentives for creating breakthrough CNS drugs. Neuron. 2014;84(3):554–63. https://doi.org/10.1016/j.neuron.2014.10.027.
Insel TR, Cuthbert BN, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research Domain Criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):750–1.
Carlezon WA Jr, Krystal AD. Kappa-opioid antagonists for psychiatric disorders: from bench to clinical trials. Depress Anxiety. 2016;33(10):895–906. https://doi.org/10.1002/da.22500.
Carlezon WA, Béguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, et al. Depressive-like effects of the κ-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316(1):440–7. https://doi.org/10.1124/jpet.105.092304.
Bruijnzeel AW. kappa-Opioid receptor signaling and brain reward function. Brain Res Rev. 2009;62(1):127–46. https://doi.org/10.1016/j.brainresrev.2009.09.008.
Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA. Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology. 2012;62(1):167–76. https://doi.org/10.1016/j.neuropharm.2011.06.014.
Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology. 2010;210(2):241–52. https://doi.org/10.1007/s00213-010-1836-5.
Maisonneuve IM, Archer S, Glick SD. U50,488, a κ opioid receptor agonist, attenuates cocaine-induced increases in extracellular dopamine in the nucleus accumbens of rats. Neurosci Lett. 1994;181(1):57–60. https://doi.org/10.1016/0304-3940(94)90559-2.
Muschamp JW, Van't Veer A, Parsegian A, Gallo MS, Chen M, Neve RL, et al. Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci. 2011;31(8):3095–103. https://doi.org/10.1523/JNEUROSCI.5973-10.2011.
Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon WA Jr. The kappa-opioid agonist U69,593 blocks cocaine-induced enhancement of brain stimulation reward. Biol Psychiatry. 2008;64(11):982–8. https://doi.org/10.1016/j.biopsych.2008.05.029.
Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology. 2010;210(2):121–35. https://doi.org/10.1007/s00213-010-1825-8.
Krystal AD, Pizzagalli DA, Mathew SJ, Sanacora G, Keefe R, Song A, et al. The first implementation of the NIMH FAST-FAIL approach to psychiatric drug development. Nat Rev Drug Discov. 2018;18:82–4. https://doi.org/10.1038/nrd.2018.222.
Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, et al. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology. 2014;77:131–44. https://doi.org/10.1016/j.neuropharm.2013.09.021.
Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, et al. Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J Clin Pharmacol. 2014;54(9):968–78. https://doi.org/10.1002/jcph.286.
Zheng M-Q, Nabulsi N, Kim SJ, Tomasi G, Lin S-F, Mitch C, et al. Synthesis and evaluation of 11C-LY2795050 as a κ-opioid receptor antagonist radiotracer for PET imaging. J Nucl Med. 2013;54(3):455–63. https://doi.org/10.2967/jnumed.112.109512.
Knutson B, Bhanji JP, Cooney RE, Atlas LY, Gotlib IH. Neural responses to monetary incentives in major depression. Biol Psychiatry. 2008;63(7):686–92. https://doi.org/10.1016/j.biopsych.2007.07.023.
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167(1):99–103. https://doi.org/10.1192/bjp.167.1.99.
Krystal AD, Pizzagalli DA, Smoski M, Mathew SJ, Nurnberger J Jr, Lisanby SH, et al. A randomized proof-of-mechanism trial applying the 'fast-fail' approach to evaluating kappa-opioid antagonism as a treatment for anhedonia. Nat Med. 2020;26(5):760–8. https://doi.org/10.1038/s41591-020-0806-7.
Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301–8. https://doi.org/10.1176/ajp.148.10.1301.
Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199–21414. https://doi.org/10.1001/archpsyc.1994.03950030035004.
Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349–52. https://doi.org/10.1126/science.281.5381.1349.
Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13(9):1102–7. https://doi.org/10.1038/nm1632.
Adams DH, Zhang L, Millen BA, Kinon BJ, Gomez JC. Pomaglumetad methionil (LY2140023 monohydrate) and aripiprazole in patients with schizophrenia: a phase 3, multicenter, double-blind comparison. Schizophr Res Treatment. 2014;2014:758212. https://doi.org/10.1155/2014/758212.
Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S, et al. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J Clin Psychopharmacol. 2011;31(3):349–55. https://doi.org/10.1097/JCP.0b013e318218dcd5.
Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry. 2015;78(11):754–62. https://doi.org/10.1016/j.biopsych.2015.03.016.
De Simoni S, Schwarz AJ, O'Daly OG, Marquand AF, Brittain C, Gonzales C, et al. Test-retest reliability of the BOLD pharmacological MRI response to ketamine in healthy volunteers. Neuroimage. 2013;64:75–90. https://doi.org/10.1016/j.neuroimage.2012.09.037.
Mehta MA, Schmechtig A, Kotoula V, McColm J, Jackson K, Brittain C, et al. Group II metabotropic glutamate receptor agonist prodrugs LY2979165 and LY2140023 attenuate the functional imaging response to ketamine in healthy subjects. Psychopharmacology. 2018;235(7):1875–86. https://doi.org/10.1007/s00213-018-4877-9.
Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, et al. Utility of imaging-based biomarkers for glutamate-targeted drug development in psychotic disorders: a randomized clinical trial. JAMA Psychiatry. 2018;75(1):11–9. https://doi.org/10.1001/jamapsychiatry.2017.3572.
Ragland JD, Ranganath C, Harms MP, Barch DM, Gold JM, Layher E, et al. Functional and neuroanatomic specificity of episodic memory dysfunction in schizophrenia: a functional magnetic resonance imaging study of the relational and item-specific encoding task. JAMA Psychiatry. 2015;72(9):909–16. https://doi.org/10.1001/jamapsychiatry.2015.0276.
Kantrowitz JT, Grinband J, Goff DC, Lahti AC, Marder SR, Kegeles LS, et al. Clinical efficacy and target engagement of glutamatergic drugs: placebocControlled RCTs of pomaglumetad and TS-134 for reversal of ketamine-induced psychotic symptoms and PharmacoBOLD in healthy volunteers. medRxiv. 2020:2020.03.09.20029827. 10.1101/2020.03.09.20029827.
Grabb MC, Cross AJ, Potter WZ, McCracken JT. Derisking psychiatric drug development: the NIMH's fast fail program, a novel precompetitive model. J Clin Psychopharmacol. 2016;36(5):419–21. https://doi.org/10.1097/JCP.0000000000000536.
Blatt GJ, Fatemi SH. Alterations in GABAergic biomarkers in the autism brain: research findings and clinical implications. Anat Rec (Hoboken). 2011;294(10):1646–52. https://doi.org/10.1002/ar.21252.
Gaetz W, Bloy L, Wang DJ, Port RG, Blaskey L, Levy SE, et al. GABA estimation in the brains of children on the autism spectrum: measurement precision and regional cortical variation. Neuroimage. 2014;86:1–9. https://doi.org/10.1016/j.neuroimage.2013.05.068.
Drenthen GS, Barendse EM, Aldenkamp AP, van Veenendaal TM, Puts NA, Edden RA, et al. Altered neurotransmitter metabolism in adolescents with high-functioning autism. Psychiatry Res Neuroimaging. 2016;256:44–9. https://doi.org/10.1016/j.pscychresns.2016.09.007.
Christian EP, Snyder DH, Song W, Gurley DA, Smolka J, Maier DL, et al. EEG-beta/gamma spectral power elevation in rat: a translatable biomarker elicited by GABA(Aalpha2/3)-positive allosteric modulators at nonsedating anxiolytic doses. J Neurophysiol. 2015;113(1):116–31. https://doi.org/10.1152/jn.00539.2013.
Nickolls SA, Gurrell R, van Amerongen G, Kammonen J, Cao L, Brown AR, et al. Pharmacology in translation: the preclinical and early clinical profile of the novel alpha2/3 functionally selective GABAA receptor positive allosteric modulator PF-06372865. Br J Pharmacol. 2018;175(4):708–25. https://doi.org/10.1111/bph.14119.
Chen X, van Gerven J, Cohen A, Jacobs G. Human pharmacology of positive GABA-A subtype-selective receptor modulators for the treatment of anxiety. Acta Pharmacol Sin. 2019;40(5):571–82. https://doi.org/10.1038/s41401-018-0185-5.
Jucaite A, Cselenyi Z, Lappalainen J, McCarthy DJ, Lee CM, Nyberg S, et al. GABAA receptor occupancy by subtype selective GABAAalpha2,3 modulators: PET studies in humans. Psychopharmacology. 2017;234(4):707–16. https://doi.org/10.1007/s00213-016-4506-4.
Chen X, Jacobs G, de Kam M, Jaeger J, Lappalainen J, Maruff P, et al. The central nervous system effects of the partial GABA-Aα2,3-selective receptor modulator AZD7325 in comparison with lorazepam in healthy males. Br J Clin Pharmacol. 2014;78(6):1298–314. https://doi.org/10.1111/bcp.12413.
Bentin S, Allison T, Puce A, Perez E, McCarthy G. Electrophysiological studies of face perception in humans. J Cogn Neurosci. 1996;8(6):551–65. https://doi.org/10.1162/jocn.1996.8.6.551.
Lord CR, Dilavore M, Risi PC, Gotham S, Bishop K, Somer L. Autism diagnostic observation schedule (ADOS-2). Modules 1 through 4. 2nd ed. Torrence, California: Western Psychological Services; 2012.
McCracken JT. 12.3 Target engagement of AZD7325 in adults with ASD. J Am Acad Child Adolesc Psychiatry. 2018;57(10):S287. https://doi.org/10.1016/j.jaac.2018.07.683.
Fortress Biotech announces exclusive worldwide license agreement with AstraZeneca and Cincinnati Children’s Hospital Medical Center to develop a novel treatment for select CNS disorders. Baergic Bio, a Fortress partner company, enters into an agreement with AstraZeneca for AZD7325, a novel α2/3-subtype-selective GABA A positive allosteric modulator. Globe Newswire; 2019.
Acknowledgements
The authors thank Andrew Krystal, Jeffrey Lieberman, Sandra Loo, James McCracken, and Diego Pizzagalli for providing figures for the article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to assist with the preparation of this review.
Conflict of interest
Margaret C. Grabb and Mi Hillefors have no conflicts of interest that are directly relevant to the content of this article. William Z. Potter consulted for Otsuka, AgeneBio, Takeda, Lilly, Noven, and Karuna.
Rights and permissions
About this article

Cite this article
Grabb, M.C., Hillefors, M. & Potter, W.Z. The NIMH ‘Fast-Fail Trials’ (FAST) Initiative: Rationale, Promise, and Progress. Pharm Med 34, 233–245 (2020). https://doi.org/10.1007/s40290-020-00343-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40290-020-00343-y