
Abstract
As part of its Single Technology Appraisal process, the National Institute for Health and Care Excellence (NICE) invited the manufacturer of olaparib (AstraZeneca) to submit evidence on the clinical and cost effectiveness of olaparib for the maintenance treatment of BRCA1/2 mutated (BRCAm), platinum-sensitive relapsed (PSR) ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy. The Evidence Review Group (ERG) produced a critical review of the evidence contained within the company’s submission (CS) to NICE. The clinical evidence related to one phase II, double-blind randomised controlled trial that recruited 265 patients with PSR serous ovarian cancer (OC) regardless of BRCAm status. Patients received olaparib 400 mg twice daily (b.i.d.) or matched placebo. In the whole population, the primary endpoint of progression-free survival (PFS) was met (hazard ratio [HR] 0.35; 95 % confidence interval [CI] 0.25–0.49, p < 0.01) for olaparib versus placebo. The BRCAm subgroup analysis (added after the study commenced but 1 month before the primary analysis was undertaken) reported an HR for PFS of 0.18 (95 % CI 0.10–0.31, p < 0.0001) for olaparib versus placebo, though interaction tests appeared inconclusive. Overall survival was not statistically significant in the whole group (HR 0.88; 95 % CI 0.64–1.21; p = 0.44) or the BRCAm subgroup (0.73; 95 % CI 0.45–1.17; p = 0.19), though treatment switching may have confounded results. The exclusion of data from sites allowing crossover resulted in an HR for overall survival (OS) of 0.52 (95 % CI 0.28–0.97, p = 0.039) in the BRCAm group. Health-related quality-of-life measures were not significantly different between groups. All post hoc exploratory outcomes (time to treatment discontinuation/death, time to first subsequent therapy/death, and time to second subsequent therapy/death) were statistically significantly better in the olaparib arm in the whole population and the BRCAm subgroup analyses. Adverse events were more frequent for olaparib but were largely minor or manageable. The company’s semi-Markov model assessed the cost effectiveness of olaparib versus routine surveillance in patients with BRCAm PSR OC from a National Health Service (NHS) and Personal Social Services (PSS) perspective over a lifetime horizon. The model suggests that the incremental cost-effectiveness ratio (ICER) for olaparib versus routine surveillance is expected to be approximately £49,146 per quality-adjusted life-year (QALY) gained. The ERG did not consider the company’s cost-effectiveness estimates to be credible. Additional ERG analyses suggested that the ICER is likely to be more than £92,214 per QALY gained. Additional analyses provided by the company in patients who received three or more lines of chemotherapy suggested a more favourable cost-effectiveness profile for olaparib. The NICE Appraisal Committee recommended olaparib for this subgroup provided the cost of olaparib for people who continue to receive treatment after 15 months will be met by the company.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The supporting clinical evidence for olaparib was a subgroup analysis of a phase II trial, considered to be at moderately high risk of bias overall. The BRCAm subgroup was considered clinically plausible, but interaction tests were inconclusive. Progression-free survival (PFS) was significantly better for patients receiving olaparib (p < 0.0001). Overall survival (OS) was not significantly better (p = 0.19) except in the crossover-adjusted analysis (p = 0.039), though this analysis did not correct for unlicensed treatment with olaparib beyond PFS. |
Additional work undertaken by the Evidence Review Group (ERG) suggested that the incremental cost-effectiveness ratio (ICER) for olaparib versus routine surveillance in BRCAm platinum-sensitive relapsed (PSR) ovarian cancer (OC) patients who have received two or more lines of chemotherapy is likely to be greater than £92,214 per quality-adjusted life-year (QALY) gained. |
The National Institute for Health and Care Excellence (NICE) Appraisal Committee concluded that the most plausible ICERs for olaparib versus routine surveillance in BRCAm PSR OC patients who have received three or more lines chemotherapy were £46,600–46,800 per QALY gained. |
Olaparib was recommended within its marketing authorisation for adults with BRCAm PSR OC that has responded to platinum-based chemotherapy only if they have received three or more courses of platinum-based chemotherapy and the drug cost of olaparib for people who continue to receive treatment after 15 months is met by the company. |
1 Introduction
Health technologies must be shown to represent a clinically effective and cost-effective use of resources in order to be recommended for use within the National Health Service (NHS) in England. The National Institute for Health and Care Excellence (NICE) is an independent organisation responsible for providing national guidance on promoting good health and preventing and treating ill health in priority areas with a significant impact. The NICE Single Technology Appraisal (STA) process usually covers new technologies within a single indication shortly after they have received UK marketing authorisation [1]. Within this process, the company provides NICE with a written submission that summarises the company’s estimates of the clinical effectiveness and cost effectiveness of the technology together with an executable health economic model. The company’s submission (CS) is reviewed by an external organisation independent of NICE, the Evidence Review Group (ERG), which consults with clinical specialists and produces an ERG report. After consideration of the CS, the ERG report and testimony from experts and other stakeholders, the NICE Appraisal Committee formulates preliminary guidance in the form of an Appraisal Consultation Document (ACD), which indicates the committee’s initial recommendations on the use of the technology. Stakeholders are subsequently invited to comment on the submitted evidence and the ACD, after which a subsequent ACD may be produced or a Final Appraisal Determination (FAD) is issued, which is open to appeal. An ACD is not produced when the technology is recommended without restriction; in such instances, the FAD is produced directly. This paper presents a summary of the ERG report [2] and subsequent analyses [3–5] for the STA of olaparib for the maintenance treatment of BRCA1/2 mutated (BRCAm), platinum-sensitive relapsed (PSR) ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy, and the subsequent development of the NICE guidance for the use of this drug in England [6]. Full details of all relevant appraisal documents can be found on the NICE website (https://www.nice.org.uk/guidance/indevelopment/ta381/documents).
2 The Decision Problem
Ovarian cancer (OC) represents a group of tumours that arise from diverse types of tissue contained in the ovary. The most common type of OC arises from epithelial cells on the surface of the ovary and can often spread to any surface within the abdominal cavity, including the fallopian tubes and peritoneal cavity. The symptoms of OC commonly include persistent abdominal distension, early satiety and/or loss of appetite, pelvic or abdominal pain and increased urinary urgency and/or frequency [7]. Approximately 6100 women are diagnosed with OC in England each year [8]. Incidence increases with age, and most cases are diagnosed in older postmenopausal women. Most OCs are sporadic. However, the presence of BRCA mutations account for more than 10 % of all OCs, and carriers of BRCA mutations have an increased lifetime risk of developing breast cancer and OC. In England and Wales, the 5-year survival rate is approximately 46 %; however, prognosis is considerably worse for patients with advanced disease [8]. Approximately 10–15 % of women presenting with advanced disease achieve long-term remission through chemotherapy. However, following initial response to treatment, the majority of patients subsequently relapse.
2.1 Current Treatment
There are currently no licensed therapies for the maintenance treatment of PSR OC. In addition, very few economic analyses of maintenance therapies for PSR OC have been undertaken [9]. Prior to January 2015, bevacizumab was available in England as a maintenance therapy; this is no longer routinely available through the Cancer Drugs Fund (CDF) in the relapsed setting. Current care involves routine surveillance, with further chemotherapy upon relapse. Surveillance typically involves routine outpatient appointments to assess for symptomatic disease progression. Cancer antigen 125 (CA125), a serum tumour marker, may be used to detect relapse in OC, although the benefits of routine measurement are disputed, and its use across England is variable [10]. In people whose disease relapses following initial therapy, NICE recommends the following as options for second- or subsequent-line therapy: (1) paclitaxel in combination with a platinum compound in platinum-sensitive or partially platinum-sensitive disease; (2) pegylated liposomal doxorubicin hydrochloride in partially platinum-sensitive, platinum-resistant or platinum-refractory disease; (3) single-agent paclitaxel in platinum-refractory or platinum-resistant disease; and (4) topotecan in platinum-refractory or platinum-resistant disease for people for whom pegylated liposomal doxorubicin hydrochloride and single-agent paclitaxel are considered inappropriate [11].
Olaparib (Lynparza®) is a potent inhibitor of poly (ADP-ribose) polymerase (PARP)-1, PARP-2 and PARP-3. Olaparib is licensed for the maintenance treatment of adult patients with PSR BRCAm (germline and/or somatic) high-grade serous epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy [12]. The recommended dose of olaparib is 400 mg (eight 50-mg capsules) twice daily (b.i.d.). Treatment should be continued until disease progression; treatment interruptions and dose reductions may be used to manage adverse reactions [12]. As of June 2016, olaparib had not been listed on the British National Formulary (BNF) [13]. The original anticipated NHS list price was £3950.00 per pack (448 capsules) [14]. During the appraisal, a Patient Access Scheme (PAS) was agreed whereby the cost of olaparib for people who continued to receive treatment for more than 18 months would be met by the company. This was subsequently reduced to 15 months, and a price of £3550 per pack was agreed. Unless otherwise stated, all results presented here include the original 18-month PAS and original list price.
To receive olaparib, patients must have confirmation of BRCAm. Current NICE guidelines recommend BRCA testing for women with OC in whom the combined BRCA1/2m carrier probability is 10 % or more [15]. Currently, the use of BRCAm testing across England remains variable.
In November 2014, NICE issued a final scope to appraise the clinical effectiveness and cost effectiveness of olaparib within its licensed indication for the maintenance treatment of BRCA1/2m, PSR ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy [16].
3 Independent Evidence Review Group (ERG) Review
The company (AstraZeneca) provided a submission to NICE on the clinical effectiveness and cost effectiveness of olaparib for the maintenance treatment of BRCA1/2m PSR OC [14]. This submission was critically appraised by the ERG. In addition, the ERG identified areas requiring clarification, for which the company provided additional evidence prior to completion of the ERG report [2, 17].
3.1 Clinical Evidence Submitted by the Company
The CS included an unpublished systematic review of studies in patients with OC of any BRCAm status. The scope of this review was wider than that required by the decision problem. One relevant study was identified for inclusion (Study 19 [18]).
3.1.1 Clinical Trial Design
Population and trial design Study 19 was a pivotal phase II, double-blind randomised controlled trial. The study recruited 265 patients aged ≥18 years with a histological diagnosis of recurrent high-grade (grade 2 or 3) serous OC (including primary peritoneal or fallopian tube cancer) that was platinum-sensitive (progression >6 months) as determined by response to the most recent round of chemotherapy and at least one previous round (not necessarily sequential rounds), and regardless of BRCAm status. Patients who had received previous PARP inhibitor therapy were excluded. Patients had to have an Eastern Co-operative Oncology Group (ECOG) performance status ≤2, a life expectancy of at least 16 weeks and a CA125 measurement below the upper limit of normal, or if above, not significantly rising over time.
Intervention and comparator Patients were randomised by an interactive voice response system (IVRS) to olaparib 400 mg b.i.d. or matched placebo. Interruptions and dose reductions were permitted to address toxicity or adverse events (AEs), but re-treatment was not allowed. Continuation of treatment was permitted for patients who were still benefitting. Some concomitant medications were allowed, and patients in the placebo arm could crossover to receive a PARP inhibitor after the study endpoint was reached.
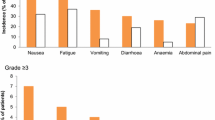
Outcomes The primary outcome was progression-free survival (PFS) as assessed by the Response Evaluation Criteria in Solid Tumours (RECIST) criteria or death. Pre-specified secondary outcomes relevant to the scope included overall survival (OS), AEs and health-related quality of life (HRQoL) by the Trial Outcome Index (TOI), the Functional Assessment of Cancer Therapy–Ovarian (FACT-O) and the FACT/National Comprehensive Cancer Network Ovarian Symptom Index (FOSI). Additional post hoc exploratory analyses were reported for the safety population, including time to treatment discontinuation/death (TTD/D), time to first subsequent therapy/death (TFST/D) and time to second subsequent therapy/death (TSST/D) (Fig. 1). Only TFST/D was listed in the NICE scope, with TSST/D presented as a proxy for the second PFS period (PFS2). AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 3.0 [19].

Outcome measurement in Study 19
Analysis plan The pivotal data for this assessment was a subgroup analysis of BRCAm patients from Study 19 [14, 20]. The testing of all patients for BRCAm status, a subgroup analysis of PFS in BRCAm patients, and a global interaction test were added to the statistical plan approximately 1 month before the PFS data cut-off (DCO) point was reached (June 2010). This replaced the subgroup analysis of patients who had homologous-recombination-deficient (HRD) tumours (of which BRCA mutations are a subset), as an HRD test was not developed in time. Additional analyses of all other clinical endpoints in this subgroup were added to the analysis plan after the DCO in consultation with the European Medicines Agency (EMA). Changes were also made to the timing of OS analyses after the DCO. In the whole population analysis, OS was analysed at two main points: (1) at the same time as the PFS analysis and (2) at an interim point when the data were 58 % mature.
3.1.2 Clinical Study Results
Patient characteristics The most notable imbalances in patient characteristics between treatment arms related to objective response to the most recent platinum-based chemotherapy and, to a lesser extent, in ECOG performance status. Adjustments for imbalances were applied in Cox proportional hazards model analyses in the full analysis set (FAS); whether adjustment was applied to the BRCAm subgroup was unclear.
PFS: In the whole population analysis, the primary study endpoint was met, with a hazard ratio (HR) for PFS of 0.35 (95 % confidence interval [CI] 0.25–0.49, p < 0.01) for olaparib versus placebo. Median PFS was 8.4 months for olaparib versus 4.8 months for placebo (95 % CI not reported [NR]). The BRCAm subgroup analysis reported an HR for PFS of 0.18 (95 % CI 0.10–0.31, p < 0.0001) for olaparib versus placebo; median PFS was 11.2 months for olaparib (95 % CI 8.3 to ‘not calculable’) versus 4.3 months for placebo (95 % CI 3.0–5.4). A treatment–subgroup interaction test was not presented within the CS but was reported in the Clinical Study Report and the European Public Assessment Report (EPAR); each reports a significant interaction for BRCAm (p = 0.030 or p = 0.025, respectively) when considered alone but a non-significant interaction (p = 0.15647 or p = 0.142, respectively) when a global test adding treatment interaction terms for all non-treatment covariates was performed [21, 22].
OS Within the whole population, OS was not significantly different between groups at either analysis point. The HR for death was 0.94 (95 % CI 0.63–1.39; p = 0.75) for olaparib versus placebo (median OS 29.7 vs. 29.9 months, respectively; 95 % CI NR) at the June 2010 DCO [17]. At 58 % OS data maturity (November 2012), the HR for death was 0.88 (95 % CI 0.64–1.21, p = 0.44) for olaparib versus placebo, with a median survival of 29.8 months (95 % CI 27.2–35.7) in the olaparib arm versus 27.8 months (95 % CI 24.4–34.0) in the placebo arm [21, 22]. For the BRCAm subgroup, OS was reported only at the November 2012 DCO (52 % maturity); the HR for death was 0.73 (95 % CI 0.45–1.17, p = 0.19) for olaparib versus placebo. Median OS was 34.9 months in the olaparib group and 31.9 months in the placebo group. A crossover analysis within the BRCAm group in which sites allowing placebo group crossover to PARP inhibitors reported a significant OS difference (HR 0.52, 95 % CI 0.28–0.97, nominal p = 0.039) [23]. No correction was applied for patients in the olaparib arm who continued to receive olaparib beyond disease progression.
HRQoL Study 19 reported “no significant difference in improvement rates or time to worsening of TOI, FOSI or Total FACT-O” and concluded that HRQoL was not negatively impacted during therapy [24].
Other outcomes All post hoc exploratory outcomes (TTD/D, TFST/D and TSST/D) were statistically significantly better for the olaparib group, for both the whole population and the BRCAm subgroup. In the whole population, the HR for TTD/D was 0.39 (95 % CI 0.30–0.51) for olaparib versus placebo and 0.36 (95 % CI 0.24–0.53) for olaparib versus placebo in the BRCAm subgroup. In the whole population, the HR for TFST/D was 0.41 (95 % CI 0.31–0.54) for olaparib versus placebo and 0.33 (95 % CI 0.22–0.50) for olaparib versus placebo in the BRCAm subgroup. In the whole population, the HR for TSST/D was 0.54 (95 % CI 0.41–0.72) for olaparib versus placebo and 0.44 (95 % CI 0.29–0.67) for olaparib versus placebo in the BRCAm subgroup.
AEs occurred more often in the olaparib group but were largely minor and manageable with dose reductions or interruptions. More patients receiving olaparib experienced severe AEs such as fatigue, anaemia and neutropenia than those receiving placebo. Serious AEs occurred in 21.6 % of olaparib patients versus 9.7 % of placebo patients. These included anaemia, small bowel obstruction, dyspnoea and gastritis.
3.2 Critique of Clinical Effectiveness Evidence and Interpretation
3.2.1 Critique of Systematic Review
The company adapted the systematic review to bring it into line with the NICE scope. Clarifications provided by the company suggest that the review was well conducted. The ERG concluded that all relevant evidence had been identified.
3.2.2 Critique of Clinical Evidence
Study 19 had several limitations, both methodologically and with respect to its relevance to the decision problem.
Population The ERG and their clinical advisors considered the inclusion criteria for the FAS broadly appropriate [12]. Two ERG advisors thought the criteria requiring stable CA125 status to be reasonable and considered that CA125 status would likely be used in clinical practice as this biomarker is used to monitor response to chemotherapy. Baseline imbalances were considered likely to be due to problems with the IVRS, which led to mis-stratification of patients. Although these were corrected using Cox model analyses for the FAS, it remained unclear whether all BRACm analyses were also adjusted.
The BRCAm subgroup was considered clinically relevant, but the study used both germline (blood test) and tumour (tissue sample test) BRCAm testing to select patients. Tumour testing is not routinely performed in England, and it is unclear whether this will be possible on a large scale. Consequently, this may potentially lead to problems regarding generalisability.
Intervention The intervention was considered largely appropriate, with the exception of the continuation of olaparib beyond progression (which is not in accordance with the licence), and the assessment of progression (halting treatment in most cases) using RECIST rather than CA125 (which generally indicates progression before RECIST). These factors are likely to mean that treatment was administered in the trial for longer than would be the case in usual clinical practice in England.
Comparator The ERG concluded that the comparator reflected clinical practice. Data on differences in concomitant treatments (e.g. ascites drainage, pain relief) between groups were not presented.
Outcomes The primary outcome was considered appropriate, though it was noted that PFS is a proxy for OS, and that OS is the most relevant outcome. The ERG argued that, as Study 19 was being used as pivotal evidence, it should conform to EMA guidelines for phase III trials [25], which state that PFS should be supported by a trend toward OS benefit or outcomes such as PFS2 or time to next-line therapy. In Study 19, TFST/D and TSST/D were considered by the ERG to be suitable supporting endpoints instead of PFS2, despite not being listed in the NICE scope. However, the clinical advisors were concerned that practice in the countries included in Study 19 may be to commence subsequent therapy earlier than in England, thus truncating these outcomes. Furthermore, these outcomes were added to the study plan after PFS data had been collected, hence they are at high risk of bias.
Conversely, continuation of treatment beyond PFS and the use of RECIST criteria rather than CA125 means that TTD/D and PFS may be longer than would be expected in clinical practice. Other outcomes such as TFST/D, TSST/D, OS and AEs may have been affected by the increased dose allowed in the trial, unblinding of study participants, and by placebo group crossover. Generalisability was therefore a concern to the ERG.
AE measurement was largely adequate, despite a lack of clarity about the methods of elicitation. The choice of HRQoL measures appeared appropriate, though a preference-based measure was not used and measurement was only performed during the treatment phase of the trial.
Study design The multiple changes to the statistical analysis plan, particularly the timing of OS measurement and the addition of the BRCAm subgroup analyses, were a matter of concern as they were performed post hoc. The ERG’s clinical advisors thought the company’s rationale for selecting the BRCAm subgroup was clinically plausible, although interaction tests were inconclusive. Based on published quality-assessment criteria [26], the ERG scored the study as low risk for four domains (allocation concealment, imbalances in dropouts between groups, outcome reporting bias and analysis methods), but high risk for randomisation (due to problems with the IVRS) and balance between groups in prognostic factors at baseline, and unclear risk for blinding as some patients were unblinded under an emergency protocol.
The ERG concluded the study results were associated with considerable uncertainty in relation to their accuracy and generalisability because these biases and relevance issues may operate in unknown directions and to unknown extents and because of the small sample size of the study and subgroup analyses. To compound these issues further, the history of changes to the study protocol and the post hoc definition of the BRCAm subgroup and inconclusive interaction tests means that the hypothesis that olaparib has superior efficacy in BRCAm patients compared with other patients had not been robustly tested or proved. The ERG noted that a phase III trial of olaparib in BRCAm OC patients was ongoing (clinicaltrials.gov identifier: NCT01874353) and would provide the required confirmation of the study’s results. The lack of conclusive evidence to support an OS advantage for olaparib does not detract from the benefits inherent to a postponement of PFS but does make it difficult to conclude whether or not olaparib confers a survival benefit.
3.3 Cost-Effectiveness Evidence Submitted by the Company
The company submitted a de novo health economic model to assess the cost effectiveness of olaparib versus routine surveillance in patients with BRCAm PSR OC. The company’s economic analysis comprised two related evaluations:
-
(i)
The base-case economic evaluation of olaparib maintenance treatment versus routine surveillance in patients with BRCAm PSR OC. This excluded the costs of BRCAm testing and considered costs and benefits relating to the index BRCAm OC patient.
-
(ii)
A broader economic evaluation that also accounted for (1) the costs of BRCAm testing in BRCAm PSR OC patients and (2) the costs and benefits of expanding BRCAm testing to family members of relapsed BRCAm OC patients undergoing BRCAm testing as a prerequisite in consideration of olaparib as a potential treatment option. This analysis considers costs and benefits relating to the index BRCAm OC patient and family members.
The company’s base-case analysis adopted a semi-Markov approach and evaluated costs and health outcomes from an NHS and Personal Social Services (PSS) perspective over a lifetime horizon (15 years) discounted at a rate of 3.5 % per year. The company’s model (Fig. 2) includes five health states: (1) progression-free (on maintenance treatment), (2) progression-free (discontinued maintenance treatment), (3) first subsequent chemotherapy (on treatment or discontinued), (4) second subsequent chemotherapy (on treatment or discontinued) and (5) dead. Transitions between progressive states are modelled using parametric survivor functions fitted to time-to-event data together with fixed estimates of the proportion of these progression events that are deaths. Clinical input parameters were estimated using data from the Study 19 BRCAm subgroup [20]. For the progression-free states, health utilities were mapped from the FACT-O to the EuroQol five dimensions questionnaire (EQ-5D) [27]; utilities for subsequent states were sourced from a previous NICE submission [28]. Resource use estimates were based on Study 19 [20], previous appraisals [29], clinical guidelines [15], literature [30–32] and assumptions. Unit costs were derived from NHS Reference Costs 2013–14 [33], the Personal Social Services Research Unit (PSSRU) [34], the NHS Commercial Medicines Unit (CMU) [35] and the BNF [13]. The additional costs and benefits of BRCAm testing within the secondary analysis were taken from a cost-effectiveness report published as part of the NICE familial breast cancer guideline [15]. The CS argues that olaparib satisfies NICE’s criteria for life-extending therapies at the end of life (EoL) [36].

Company’s model structure
The probabilistic version of the company’s model suggests that olaparib is expected to produce an additional 0.90 quality-adjusted life-years (QALYs) at an additional cost of £72,232 compared with routine surveillance; this corresponds to an incremental cost-effectiveness ratio (ICER) for olaparib versus routine surveillance of approximately £49,146 per QALY gained. The deterministic model yielded a similar ICER of £49,826 per QALY gained. Assuming willingness-to-pay thresholds of £30,000 and £50,000 per QALY gained, the probability that olaparib produces more net benefit than routine surveillance is approximately 0.02 and 0.52, respectively. The company’s secondary analysis, which was based on five family pedigrees, suggested a lower average deterministic ICER for BRCAm testing plus olaparib versus routine surveillance without BRCAm testing of £39,343 per QALY gained.
3.3.1 Critique of Cost-Effectiveness Evidence and Interpretation
The ERG critically appraised the company’s economic analysis and double-programmed the company’s model. No significant programming errors were found. However, the ERG had concerns regarding the model structure and the evidence used to inform the model’s parameters.
3.3.1.1 Choice of Model Structure and Use of Outcomes Data from Study 19 BRCAm Subgroup
The company’s model assumes that all patients who survive their first subsequent therapy event (the ‘progression-free’ period) subsequently receive a first subsequent chemotherapy and that all patients who survive the second subsequent therapy event subsequently receive a second course of chemotherapy. However, for some patients with advanced disease, chemotherapy may offer limited benefit, and patients may instead receive supportive care. Furthermore, the model structurally limits the number of lines of subsequent chemotherapy to a maximum of two, yet more than 36 % of patients within the Study 19 BRCAm subgroup received three or more subsequent lines of therapy [14]. The ERG’s main concerns surrounded the outcomes data included in the model and the range of evidence that had been excluded from it. The model is based on the TFST/D from randomisation and TSST/D from first subsequent therapy and survival within those states, with olaparib conferring a clinical benefit in delaying the time to first and second subsequent therapy and, as a consequence, delaying time to death. The modelled ‘progression-free’ interval does not relate to the PFS endpoint but is instead defined by TFST/D. PFS data were not used in the model. Both TFST/D and TSST/D were post hoc exploratory outcomes and may have been influenced by subjective decisions regarding future chemotherapy use, eligibility for treatment and loss of blinding within Study 19. The ERG also had concerns that the observed OS data from Study 19 were not directly used in the company’s model. Instead, the model applies the risk of death as (1) a fixed proportion of time-dependent progression events upon leaving the progression-free and subsequent therapy states and (2) a treatment-independent time-to-event curve for all patients from entry into the second subsequent therapy state. Mortality is therefore captured as a conditional event for patients reaching different health states rather than by fitting survivor functions to the Kaplan–Meier OS data.
The CS argued that their model structure better represented the benefits of maintenance treatments and the treatment pathway following relapse compared with a simple partitioned survival approach [14]. The ERG argued that the best model is that which (1) represents clinical reality and (2) makes the best use of the evidence available. Excluding PFS, compounding multiple assumptions regarding mortality risks associated with specific health states within and between treatment groups and limiting the treatment pathway to two lines of chemotherapy does not satisfy both of these criteria.
3.3.1.2 Potential Confounding of Endpoints Used in the Company’s Model
The model attempts to deal with placebo group crossover by assuming that the time from first subsequent therapy to second subsequent therapy or death, the probability that a second subsequent therapy event is death, and the time from second subsequent therapy to death are independent of treatment. The company provided analyses in which placebo group OS was adjusted for treatment switching by (1) excluding sites allowing placebo group crossover and (2) using a Rank Preserving Structural Failure Time Model (RPSFTM) [14, 17]. Kaplan–Meier curves produced using these methods each suggested an apparent OS benefit for olaparib versus placebo but indicated little difference between the groups by around 3 years post-randomisation. As OS was not directly included as a model input, the impact of using these crossover-adjusted data on the cost effectiveness of olaparib could not be assessed using the company’s model. No attempt was made to correct for confounding due to the continuation of olaparib beyond progression.
3.3.1.3 Concerns Regarding the Methods for Modelling of Time-to-Event Outcomes
According to the CS [14], the process for survival modelling was based on Latimer et al. [37]. However, the inclusion of baseline characteristics as covariates in the model-fitting process was neither justified nor explained, model discrimination did not appear to have included judgements about the plausibility of extrapolations, assumptions of proportional hazards appeared inappropriate, and sensitivity analyses using alternative survivor functions were not presented for outcomes except TFST/D.
3.3.1.4 Discordance Between Model Predictions and Observed Data from Study 19
Model-predicted OS did not provide a good fit to the observed data, irrespective of whether crossover was adjusted for. A comparison of the modelled and empirical OS curves indicated the following:
-
The crossover-site-excluded (CSE) and RPSFTM-adjusted OS Kaplan–Meier curves were similar.
-
Despite adjustment, the gap between the olaparib and placebo curves appears to close, or nearly close, at around 3 years post-randomisation irrespective of the crossover method applied.
-
OS is reasonably predicted for olaparib for the first 2 years post-randomisation but is subsequently overestimated.
-
The model does not provide a good fit to the empirical placebo group data irrespective of the method of crossover adjustment.
-
Whilst the empirical OS data, both with and without crossover adjustment, suggest that the curves for olaparib and placebo intersect, or nearly intersect, at around 3 years post-randomisation, this is not reflected in the model-predicted OS. Rather, it is around this timepoint within the model whereby the company’s model predicts the greatest difference between the groups.
These apparent biases in model-predicted OS are likely to be symptomatic of poorly fitting parametric models, inappropriate assumptions regarding proportional hazards, assumptions regarding the proportion of events that are deaths and the equivalence of time-to-event outcomes between groups following the first progression event. Overall, the ERG did not have confidence in the model results.
3.3.1.5 Concerns Regarding the Company’s Secondary Analysis
The secondary analysis compared BRCAm testing plus olaparib against no BRCAm testing and routine surveillance. However, the comparison that should have been made is BRCAm testing plus olaparib versus BRCAm testing plus routine surveillance; this was absent from the CS. Consequently, much of the apparent benefit of using olaparib suggested by the analysis was conflated with the benefits of BRCAm testing.
3.4 Additional Work Undertaken by the ERG
3.4.1 ERG Exploratory Analysis Methods
The ERG replicated the individual patient data (IPD) from the Study 19 BRCAm subgroup using methods reported by Guyot et al. [38] and fitted multiple candidate survivor functions to (1) TTD/D, (2) TFST/D, (3) RPSFTM-adjusted OS and (4) CSE-adjusted OS. The analyses focussed on addressing two questions: (1) “What is the expected incremental OS gain for olaparib versus routine surveillance?” and (2) “What is the expected incremental QALY gain for olaparib versus routine surveillance?”. With respect to the first question, the ERG used a restricted means approach to estimate the area under the curve (AUC) using the ERG-fitted parametric models of crossover-adjusted OS for olaparib versus placebo. With respect to the second question, the ERG developed a partitioned survival model in which parametric curves were fitted directly to the crossover-adjusted OS data. Uncertainty was explored across 108 combinations of candidate parametric functions (see the Appendix in the Electronic Supplementary Material [ESM]).
3.4.2 Restricted Mean Survival
The most optimistic estimate of undiscounted incremental survival benefit for olaparib versus routine surveillance produced by the ERG’s restricted means analysis was 0.68 life-years; this is considerably lower than the 1.36 additional life-years predicted by the company’s model.
3.4.3 Partitioned Survival Model
The ERG’s partitioned survival model suggests that the most optimistic discounted incremental QALY gain for olaparib versus routine surveillance is approximately 0.52 QALYs (see Appendix in the ESM). This is markedly lower than the company’s modelled estimate of 0.90 QALYs. Assuming that the company’s estimated incremental costs of olaparib are reasonable, this implies that the ICER for olaparib versus routine surveillance is likely to be in excess of £92,214 per QALY gained, but may be considerably higher. Undiscounted OS in the placebo group was consistently greater than 2 years irrespective of the selected survivor function.
3.5 Conclusion of the ERG Report
The ERG considered the evidence for olaparib for the maintenance treatment of BRCA1/2m PSR OC to be relatively weak and at relatively high risk of bias. The ERG did not consider the company’s ICERs to be credible. Additional work undertaken by the ERG suggested that the ICER for olaparib versus routine surveillance is likely to be greater than £92,214 per QALY gained. On the basis of the ERG’s exploratory analyses and the company’s model-predicted OS for the routine surveillance group (approximately 30 months), olaparib does not appear to satisfy NICE’s EoL criteria.
4 Key Methodological Issues
Study 19 was subject to several methodological issues. The hypothesis that olaparib has superior efficacy in BRCAm patients compared with other patients had not, in the ERG’s view, been robustly tested or proved, and no phase III trial was available to confirm results. Whilst the HR for PFS suggested a considerable treatment effect, administration of olaparib was not in accordance with its licence or with clinical practice in England, and outcomes were at risk of internal and external bias. The immaturity of OS data made it difficult to conclude whether PFS advantages would translate into improved survival. The ERG considered that the company’s model did not handle competing risks of events or treatment crossover in an unbiased manner. The model appears to over-predict OS for olaparib and under-predict OS for routine surveillance. Direct modelling of crossover-adjusted OS data from Study 19 indicated a markedly smaller incremental survival gain compared with the company’s modelled predictions. Consequently, the ICER for olaparib is likely to be considerably higher than that suggested by the company’s model.
5 NICE Guidance
The Appraisal Committee reviewed the data available on the clinical and cost effectiveness of olaparib, having considered evidence on the nature of recurrent OC and the value placed on the benefits of olaparib by people with the condition, those who represent them, and clinical experts. It also took into account the effective use of NHS resources. The first ACD (published June 2015) did not recommend olaparib for the treatment of BRCAm PSR OC [23]. The committee noted that substantial disagreement between the results from Study 19 and the model predictions undermined confidence in the company’s model, that the model over-predicted the survival gains associated with olaparib, and that olaparib did not satisfy NICE’s EoL criteria. The committee considered that the company’s secondary analysis did not produce a valid cost-effectiveness estimate.
Following the first ACD, the company submitted additional analyses, including further survival modelling using CSE-adjusted OS data [39]. The company’s ACD response also included clinical evidence suggesting a greater benefit for olaparib in patients who had received three or more courses of platinum-based chemotherapy; however, this was not accompanied by any formal economic analysis in this subgroup. Despite being based on similar data, the company’s new survival models for the overall BRCAm population did not reflect those produced in the ERG’s exploratory analyses; in one example, the company’s OS estimate was almost double that estimated by the ERG. The ERG was concerned that the company’s new survival models had been implemented incorrectly [4]. At the second ACD, the committee was minded not to recommend olaparib for patients who have had three or more courses of platinum-based chemotherapy; within this subgroup, the committee requested from the company a ‘robust’ estimate of the cost effectiveness of olaparib, taking account of the cost of somatic testing and the committee’s concerns about its previous models. The company subsequently produced additional analyses for this subgroup, including the lower price for olaparib and a reduction in the number of cycles from which olaparib would be provided free of charge (15 rather than 18 cycles) [40]. The ERG remained concerned that the company’s new modelled OS predictions in the third- and subsequent-line subgroup still did not reflect the observed Study 19 OS data [5]. However, the committee concluded that, within this subgroup, the most plausible ICER was approximately £46,600–46,800 per QALY gained and that there was sufficient evidence to suggest that olaparib satisfied NICE’s EoL criteria [41].
In December 2015, NICE published its FAD, which stated that “olaparib is recommended within its marketing authorisation as an option for treating adults with PSR ovarian, fallopian tube or peritoneal cancer who have BRCA1 or BRCA2 mutations and whose disease has responded to platinum-based chemotherapy only if: they have had 3 or more courses of platinum-based chemotherapy, and; the drug cost of olaparib for people who remain on treatment after 15 months will be met by the company” [41].
5.1 Consideration of Clinical and Cost-Effectiveness Issues
This section discusses the key issues considered by the Appraisal Committee. The full list can be found in the FAD [41].
5.1.1 Uncertainty Surrounding Validity of the BRCA1/2 Subgroup
The committee noted that the key clinical-effectiveness evidence was derived the Study 19 BRCAm subgroup. It also heard from the company that most of the trial population had been tested for BRCAm retrospectively. The committee noted comments from the ERG that interaction tests between the BRCAm subgroup and the whole population were inconclusive, hence it was not possible to be certain that the treatment effect was different in the BRCAm subgroup. The committee heard that there is a biologically plausible reason why people with BRCAm disease would benefit more from olaparib than the whole trial population, which could be explained by the relationship between malfunctioning BRCA genes and the development of HRD, and the subsequent effect on DNA repair. The committee concluded that olaparib was clinically effective in the treatment of PSR OC and accepted there is a biologically plausible reason for olaparib being particularly effective in the BRCAm subgroup.
5.1.2 Uncertainty Surrounding the Size of the Treatment Effect Estimates
The committee noted that olaparib was associated with statistically significant improvements in median PFS, TFST/D and TSST/D compared with placebo in the BRCAm subgroup and the whole trial population. The committee concluded that, whilst relevant, TFST/D and TSST/D had been identified post hoc and should be viewed with caution. It also noted that the OS data were immature and may have been confounded by crossover. The committee noted that, without adjustment, the difference between treatment groups in median OS in the BRCAm subgroup was 3 months (not statistically significant); however, excluding crossover sites resulted in a statistically significant difference in median OS of 8.3 months. It concluded that uncertainty remained about the extent to which olaparib increases OS compared with placebo in patients with BRCAm OC.
The committee considered the company’s further evidence relating to BRCAm patients in Study 19 who had received three or more lines of platinum-containing therapy. The committee noted that this subgroup contained fewer patients than the total BRCAm subgroup and that there were imbalances in baseline characteristics; some potentially favoured placebo and others potentially favoured olaparib. Nevertheless, the PFS benefit in this subgroup was 6.9 months (HR 0.11), and the median CSE-adjusted OS benefit was 12.3 months (HR 0.56). The committee noted clinical experts’ comments that a difference of this magnitude had never previously been seen in OC treatment. The committee concluded there was evidence of benefit for olaparib in patients who had received three or more lines of platinum-based chemotherapy.
5.1.3 Uncertainties Relating to the Cost Effectiveness of Olaparib in the BRCAm Subgroup
The committee considered the company’s model structure to be unconventional and very different to those used in previous appraisals. The committee expressed concern that PFS data from Study 19 had not been included, despite this being the primary outcome in Study 19. In addition, OS data had not been directly incorporated into the model. The committee was concerned that intermediate outcomes had been used to make assumptions about longer-term OS and considered that it would have been more conventional to fit a curve directly to the OS data, with adjustment for placebo group crossover. The committee concluded that the company’s model was a novel design that lacked external validity and that the use of sequential intermediate outcomes to model OS relied on numerous assumptions that may not all be reasonable. It also noted that graphical plots of survival probabilities from the model showed that the difference between the curves for olaparib and placebo increased at later time points, implying OS benefits for olaparib increase over time. The committee noted that no data were provided to support this and that greater separation of the curves over time would not be expected during treatment for cancer. The committee also noted that the substantial disagreement between the results from Study 19 and the model predictions undermined confidence in the company’s model. The committee concluded that the company’s modelling of benefit for the BRCAm subgroup overestimated the benefit of olaparib and therefore underestimated the ICER for olaparib.
5.1.4 Cost Effectiveness of Olaparib in the Third- and Subsequent-Line Subgroup
The committee considered the additional cost-effectiveness analyses provided by the company following the second ACD, which related to the subgroup of BRCAm patients who had received three or more lines of platinum-based chemotherapy. The committee concluded that the company’s three health-state (partitioned survival) model provided a better basis for decision making than their original model. It noted that the ICERs in this subgroup varied according to the curve used to model OS and, although it considered that on visual inspection the Gompertz curve might be an option, it heard from the company that the log normal and log logistic curves provided the best fit to the data. The committee accepted this was not unreasonable and concluded that the most plausible ICERs were £46,600–46,800 per QALY gained. The committee considered whether the EoL criteria would apply to third- and subsequent-line subgroups. It understood that median CSE-adjusted OS for this subgroup in the placebo arm of Study 19 was 20.6 months. The committee was persuaded that life expectancy was likely to be less than 24 months in people who had received three or more lines of platinum-based chemotherapy, but greater than 24 months in the overall BRCAm population.
6 Appraisal Committee’s Key Conclusion
The committee concluded that, in Study 19, olaparib increased PFS and time to subsequent therapy compared with placebo in the whole trial population and in the BRCAm subgroup. It also concluded that there was uncertainty about whether, and the extent to which, olaparib increases OS compared with placebo. The committee concluded that the ICERs presented by the company for olaparib compared with routine surveillance for the overall population of patients with BRCAm PSR OC were considerably above the range normally considered to be a cost-effective use of NHS resources (£20,000–30,000 per QALY gained). The committee concluded that the EoL criteria did not apply to olaparib when considering the overall BRCAm PSR OC population. For the subgroup of patients with BRCAm PSR who have received three or more previous lines of platinum-based chemotherapy, the committee accepted that the most plausible ICERs were £46,600–46,800 per QALY gained. The committee concluded there was sufficient evidence to suggest that olaparib met the EoL criteria for this subgroup.
References
National Institute for Health and Care Excellence. Guide to the processes of technology appraisal. London: NICE; 2014.
Tappenden P, Harnan S, Ren S, Thokala P, Wong R, Mukuria C, et al. Olaparib for maintenance treatment of BRCA 1 or 2 mutated, relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy: a Single Technology Appraisal. Sheffield: University of Sheffield; 2015.
Tappenden P. Olaparib for maintenance treatment of BRCA 1 or 2 mutated, relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy: a Single Technology Appraisal. Addendum—analysis of olaparib Patient Access Scheme. Sheffield: University of Sheffield; 2015.
Tappenden P, Harnan S, Ren S. Olaparib for maintenance treatment of BRCA 1 or 2 mutated, relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy: a Single Technology Appraisal. Addendum—ERG comments on the company’s response to the ACD. Sheffield: University of Sheffield; 2015.
Tappenden P, Harnan S, Bermejo I. Olaparib for maintenance treatment of BRCA 1 or 2 mutated, relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy: a Single Technology Appraisal. Addendum—ERG critique of company’s response to ACD2. Sheffield: University of Sheffield; 2015.
National Institute for Health and Care Excellence. Olaparib for maintenance treatment of relapsed, platinum-sensitive, BRCA mutation-positive ovarian, fallopian tube and peritoneal cancer after response to second-line or subsequent platinum-based chemotherapy. Technology Appraisal Guidance 381. London: NICE; 2016.
National Institute for Health and Care Excellence. Ovarian cancer: recognition and initial management. Clinical guideline 122. London: NICE; 2011.
Cancer Research UK. Ovarian cancer statistics. 2016. Available from: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/ovarian-cancer. Accessed 01 May 2016.
Poonawalla IB, Parikh RC, Du XL, VonVille HM, Lairson DR. Cost effectiveness of chemotherapeutic agents and targeted biologics in ovarian cancer: a systematic review. Pharmacoecnomics. 2015;33(11):1155–85.
Rustin GJ, van der Burg ME, Griffin CL, Guthrie D, Lamont A, Jayson GC, et al. Early versus delayed treatment of relapsed ovarian cancer (MRC OV05/EORTC 55955): a randomised trial. Lancet. 2010;376(9747):1155–63.
National Institute for Health and Care Excellence. Paclitaxel, pegylated liposomal doxorubicin hydrochloride and topotecan for second-line or subsequent treatment of advanced ovarian cancer. Technology Appraisal Guidance 91. London: NICE; 2005.
European Medicines Agency. Summary of product characteristics—olaparib. London: EMA; 2015.
BMJ Group, RCPCH Publications Ltd and the Royal Pharmaceutical Society of Great Britain. British National Formulary; 2014.
AstraZeneca UK Ltd. Olaparib as a monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed (PSR) BRCA-mutated (germline and/or somatic) high-grade, serous epithelial ovarian, fallopian tube or primary peritoneal cancer who are in response (complete response or partial response) to platinum-based chemotherapy. Single Technology Appraisal. Manufacturer’s submission to the National Institute for Health and Care Excellence. Luton: AstraZeneca; 2015.
National Institute for Health and Care Excellence. Familial breast cancer: classification, care and managing breast cancer and related risks in people with a family history of breast cancer. Clinical Guideline 164. London: NICE; 2013.
National Institute for Health and Care Excellence. Olaparib for maintenance treatment of BRCA 1 or 2 mutated, relapsed, platinum-sensitive ovarian, fallopian tube and peritoneal cancer in people whose relapsed disease has responded to platinum-based chemotherapy—final scope. London: NICE; 2014.
AstraZeneca UK Ltd. Olaparib as a monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed (PSR) BRCA-mutated (germline and/or somatic) high-grade, serous epithelial ovarian, fallopian tube or primary peritoneal cancer who are in response (complete response or partial response) to platinum-based chemotherapy. Single Technology Appraisal. Response to clarification questions. Luton: AstraZeneca; 2015.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–92.
National Cancer Institute. Common Terminology Criteria for Adverse Events v3.0 (CTCAE). Bethesda: NCI; 2006.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61.
AstraZenecca Ltd. Phase II randomised, double blind, multicentre study to assess the efficacy of AZD2281 in the treatment of patients with platinum sensitive serous ovarian cancer following treatment with two or more platinum containing regimens. Macclesfield: AstraZeneca; 2015.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP)—assessment report—olaparib. London: EMA; 2014.
National Institute for Health and Care Excellence. Olaparib for maintenance treatment of relapsed, platinum-sensitive, BRCA mutation-positive ovarian, fallopian tube and peritoneal cancer following response to second-line or subsequent platinum-based chemotherapy. Appraisal Consultation Document. London: NICE; 2015.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Health-related quality of life (HRQoL) during olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC) and a BRCA mutation (BRCAm). In: European Society for Medical Oncology Madrid, Spain, 26–30 September 2014.
European Medicines Agency. Guideline on the evaluation of anticancer medicinal products in man. London: EMA; 2015.
Centre for Reviews and Dissemination. Systematic reviews: CRD’s guidance for undertaking reviews in health care. York: University of York; 2009.
Longworth L, Yang Y, Young T, Mulhern B, Hernandez Alava M, Mukuria C. Use of generic and condition-specific measures of health-related quality of life in NICE decision-making: a systematic review, statistical modelling and survey. Health Technol Assess. 2014;18(9):i-224.
National Institute for Health and Care Excellence. Trabectedin for the treatment of relapsed ovarian cancer. Technology Appraisal Guidance 222. London: NICE; 2011.
National Institute for Health and Care Excellence. Bevacizumab in combination with gemcitabine and carboplatin for treating the first recurrence of platinum-sensitive advanced ovarian cancer. Technology Appraisal Guidance 285. London: NICE; 2013.
Guest J, Ruiz F, Greener M, Trotman I. Palliative care treatment patterns and associated costs of healthcare resource use for specific advanced cancer patients in the UK. Eur J Cancer Care. 2006;15(1):65–73.
Gao W, Ho Y, Verne J, Glickman M, Higginson I, on behalf of the GUIDE Care Project. Changing patterns in place of cancer death in England: a population-based study. PLoS Med 2013;10(3).
Yorkshire Cancer Network Gynaecology Network Group. Epithelial ovarian carcinoma—2nd and subsequent line. Leeds: YCNGNG; 2010.
Department of Health. NHS reference costs 2013–14. London: DH; 2014.
Curtis L. Unit costs of health and social care. Kent: Personal Social Services Research Unit; 2013.
Department of Health Commercial Medicines Unit. Drugs and pharmaceutical electronic market information (eMit). London: DH; 2014.
National Institute for Health and Care Excellence. Guide to the methods of technology appraisal. London: NICE; 2013.
Latimer N. NICE DSU Technical Support Document 14: Survival analysis for economic evaluations alongside clinical trials—extrapolation with patient-level data. Sheffield: University of Sheffield; 2013.
Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan–Meier survival curves. BMC Med Res Methodol 2011;12(9).
AstraZeneca UK Ltd. Olaparib as a monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed (PSR) BRCA-mutated (germline and/or somatic) high-grade, serous epithelial ovarian, fallopian tube or primary peritoneal cancer who are in response (complete response or partial response) to platinum-based chemotherapy. Single Technology Appraisal. Response to ACD. Luton; AstraZeneca; 2015.
AstraZeneca UK Ltd. Olaparib as a monotherapy for the maintenance treatment of adult patients with platinum-sensitive relapsed (PSR) BRCA-mutated (germline and/or somatic) high-grade, serous epithelial ovarian, fallopian tube or primary peritoneal cancer who are in response (complete response or partial response) to platinum-based chemotherapy. Single Technology Appraisal. Response to second ACD. Luton: AstraZeneca; 2015.
National Institute for Health and Care Excellence. Olaparib for maintenance treatment of relapsed, platinum-sensitive, BRCA mutation-positive ovarian, fallopian tube and peritoneal cancer after response to second-line or subsequent platinum-based chemotherapy. Final Appraisal Determination. London: NICE; 2016.
Acknowledgments
This project was funded by the National Institute for Health Research (NIHR) Health Technology Assessment (HTA) Programme (Project No. 14/57/01). See the HTA programme website for further project information (http://www.hta.ac.uk). This summary of the ERG report was compiled after NICE issued the FAD. All authors have commented on the submitted manuscript and have given their approval for the final version to be published. The views expressed in this report are those of the authors and not necessarily those of the NIHR HTA Programme. Any errors are the responsibility of the authors.
Author contributions
Sue Harnan summarised and critiqued the clinical-effectiveness data reported within the CS. Ruth Wong critiqued the company’s search strategy. Shijie Ren critiqued the statistical analyses undertaken by the company. Clara Mukuria advised on the company’s use of health utility mapping. Paul Tappenden and Praveen Thokala critiqued the health economic analysis submitted by the company. Paul Tappenden and Shijie Ren undertook the ERG’s exploratory analyses. Clare Green, Simon Pledge and John Tidy provided clinical advice to the ERG throughout the project. All authors were involved in drafting and commenting on the final report. Paul Tappenden acts as the guarantor of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Paul Tappenden, Sue Harnan, Shijie Ren, Praveen Thokala, Ruth Wong, Clara Mukuria, Clare Green, Simon Pledge and John Tidy declare no financial conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tappenden, P., Harnan, S., Ren, S. et al. Olaparib for Maintenance Treatment of BRCA 1 or 2 Mutated, Relapsed, Platinum-Sensitive Ovarian, Fallopian Tube and Peritoneal Cancer in People Whose Relapsed Disease has Responded to Platinum-Based Chemotherapy: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. PharmacoEconomics 35, 97–109 (2017). https://doi.org/10.1007/s40273-016-0440-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-016-0440-x