
Abstract
Cardiovascular disease (CVD) remains the leading cause of death and morbidity in our society. One of the major risk factors for CVD is hypercholesterolemia. Hypercholesterolemia in children can be caused by a hereditary disorder or can be secondary to other diseases or drugs. In order to prevent CVD later in life, children with hypercholesterolemia should be identified and treated as early as possible. Currently, several different screening strategies have been developed, using either universal screening or case finding to search for children at risk. Once those children are identified, the first step in treatment is lifestyle adjustment. If cholesterol levels remain elevated, the drugs of first choice are statins. Other pharmacological options are ezetimibe or bile acid sequestrants. These agents have all proven to be safe and effective in lowering low-density lipoprotein cholesterol levels and improving surrogate markers of CVD. However, there is a need for long-term follow-up studies to answer the question as to whether it is safe to initiate treatment at a young age to prevent CVD later in life.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Hypercholesterolemia and Cardiovascular Risk
Cardiovascular disease (CVD) is the most prominent cause of morbidity and mortality in our society. A key risk factor for the development of atherosclerotic cardiovascular disease is the presence of hypercholesterolemia. Although CVD is generally not manifest until adulthood, the process of atherosclerosis initiates early in life. This was first documented during the Korean War, when autopsy studies demonstrated direct evidence of coronary atherosclerosis in soldiers with a mean age of 22 years [1]. Subsequent pathological studies have shown that the severity of atherosclerotic lesions in children and young adults increases with the number of pre-existing cardiovascular risk factors, such as hypercholesterolemia, smoking, and a high body mass index (BMI) [2, 3]. Indirect evidence for the development of early atherosclerosis was found in non-invasive imaging studies in children with familial hypercholesterolemia (FH). Impaired flow-mediated dilatation (FMD) of the brachial artery and increased intimal medial thickness (IMT) of the carotid artery illustrated that functional and morphological changes in the vessel wall anatomy are already present in these young individuals [4, 5]. Altogether, these findings have led to the hypothesis that early diagnosis and treatment of hypercholesterolemia in childhood is required to reduce the burden of CVD later in life.
This review aims to provide an overview of the most important causes of hypercholesterolemia in children. Furthermore, different screening strategies to identify children at risk and several treatment options are discussed.
2 Hypercholesterolemia in Childhood
2.1 Autosomal-Dominant Hypercholesterolemia
Autosomal-dominant hypercholesterolemia (ADH) is characterized by severely increased low-density lipoprotein cholesterol (LDL-C) levels and premature coronary artery disease (in men aged <55 years and in women aged <60 years). In the vast majority, ADH is due to mutations in the LDL receptor (LDLR) gene. The associated impairment in the function of these receptors results in reduced clearance of LDL particles from the circulation and elevation of plasma LDL-C [6]. In addition to LDLR defects, a similar phenotype can be caused by a number of mutations in the apolipoprotein B (apoB) gene and gain of function mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene [7, 8].
The prevalence of heterozygous ADH has been reported as being 1 in 500; however, recent data from our group have revealed that the prevalence of ADH in The Netherlands is close to 1:200. Other populations with much higher numbers are the French Canadian (1:270), Lebanese (1:171), and South African populations (1:70) [9, 10]. Individuals with two defective alleles, either homozygous or compound heterozygous (two different mutations on each allele) are rare, occurring in 1:1,000,000. In The Netherlands, we found almost 50 homozygous or compound heterozygous individuals.
The clinical presentation of heterozygous ADH is characterized by two- to three-fold elevations in plasma LDL-C levels, a family history positive for CVD, and physical symptoms of cholesterol deposits in the skin [6]. Historically, left untreated, the cumulative risk of CVD in patients with heterozygous ADH was greater than 50 % in men by the age of 50 years and at least 30 % in women by the age of 60 years [11]. There has been a decline in CVD event rates in recent years, in part due to lifestyle changes and treatment of other CVD risk factors, such as hypertension and diabetes. Homozygous individuals can experience serious cardiovascular events as early as in childhood, but mostly in their twenties [6, 12].
2.2 Familial Combined Hyperlipidemia
Familial combined hyperlipidemia (FCH) is a commonly occurring hereditary lipid disorder, affecting 1–5 % of the general population. It is characterized by increased total cholesterol and/or triglycerides, or both [10]. FCH is a complex disease, and the phenotype is the consequence of interactions between multiple susceptibility genes and the environment. Even within families, the phenotype shows high intra- and inter-person variability [13]. Furthermore, clinical features of FCH frequently overlap with features of diabetes mellitus type 2 and metabolic syndrome; therefore, the diagnosis is often missed [14]. In clinical practice, individuals at risk can be identified by increased apoB and triglyceride levels in combination with a family history of premature CVD [15]. Most subjects with FCH have elevated apoB levels, decreased high-density lipoprotein cholesterol (HDL-C) levels, and the presence of small dense LDL-C [13, 16]. Although clinical expression of FCH was thought to be delayed until adulthood, in some children with parental FCH, hyperlipidemia or hyperapolipoprotein B is present [17].
2.3 Autosomal-Recessive Hypercholesterolemia
Autosomal-recessive hypercholesterolemia (ARH) is a rare monogenic lipid disorder, mostly found in individuals of Sardinian or Middle Eastern origin. Mutations in ARH cause failure of normal internalization of the LDL receptor in some cell types. This results in accumulation of the LDL receptor protein on the cell surface and inadequate clearance of LDL-C from the plasma. The clinical features of ARH resemble those of homozygous ADH, with large bulky xanthomas. Lipid levels are generally more variable and less severe, and respond better to lipid-lowering therapy [18].
2.4 Sitosterolemia
Sitosterolemia, also known as phytosterolemia, is a very rare autosomal-recessive inherited disorder. It is characterized by increased plasma concentrations of plant sterols, such as sitosterol and campesterol, with a chemical structure that resembles that of cholesterol. Sitosterolemia results from mutations in either ABCG5 or ABCG8 genes, encoding for ABC transporter proteins, expressed at the apical membrane of intestinal mucosa cells and hepatocytes. Defects in transporters impair plant sterol excretion from enterocytes and hepatocytes, resulting in accumulation of plant sterols in the plasma and tissues. Patient characteristics include premature atherosclerosis, tendon xanthomas, and occasionally abnormal liver function tests, hemolysis, or thrombocytopenia [19].
2.5 Secondary Hypercholesterolemia
Secondary hypercholesterolemia includes lipid abnormalities due to chronic diseases or drugs. Of the secondary lipid disorders, obesity is of particular concern. As the prevalence of obesity is rapidly increasing, so does the prevalence of associated co-morbidities, especially the metabolic syndrome [20]. The metabolic syndrome is a constellation of risk factors (including abdominal obesity, dyslipidemia, glucose intolerance, and hypertension) for developing CVD and diabetes type 2. Dyslipidemia in obesity and metabolic syndrome is characterized by decreased levels of HDL-C and elevated levels of triglycerides and non-HDL-C.
3 Screening for Hypercholesterolemia
3.1 Screening Strategies
Screening for lipid disorders in children is based on the rationale that early identification and control of pediatric dyslipidemia will reduce the risk and severity of CVD in adulthood. Two different screening approaches are selective screening, aimed at searching specifically for ADH, and universal screening, aimed at identifying any dyslipidemia in childhood.
Most European and the Australasian pediatric guidelines advocate the use of selective screening [21, 22]. This implies that once ADH has been diagnosed or suspected in one parent, screening for ADH should be performed in their children. Selective screening appears to be a cost-effective approach to identify children at risk. However, the use of a self-reported parental history might not be useful to identify children with hypercholesterolemia [23]. In addition, most parents are nowadays treated with lipid-lowering therapy, which has lowered the rate of premature CVD.
In The Netherlands, a national cascade screening program has been used to identify patients at risk of ADH. The index patient is first diagnosed through clinical criteria, followed by a DNA test to confirm the pathological mutation. Screening for the same mutation is undertaken in first-degree relatives to identify new cases. Subsequently, cascade screening continues to screen more distant relatives by using the inheritance pattern across the pedigree. This program has proven to be highly effective in identifying patients with ADH, has reduced the age at which the disease is diagnosed, and has significantly increased the proportion of patients using cholesterol-lowering therapy [24, 25]. However, follow-up of children diagnosed with ADH merits further attention, since only a majority of the children, but not all, have been seen by medical providers [26]. Although cascade screening has been very successful in The Netherlands, this strategy may not be cost effective in larger countries where families are small and geographically dispersed.
In 2011, the executive report and summary of the Expert Panel Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents were published. These guidelines, sponsored by the National Heart, Lung, and Blood Institute (NHLBI) and endorsed by the American Academy of Pediatrics (AAP), comprise several aspects of cardiovascular health in childhood and adolescence, and recommend combining the two approaches of selective and universal screening. Universal screening of all children is based on the concept of identifying the greatest number of individuals with any dyslipidemia. Screening of lipid levels is therefore recommended at two different time points in childhood [27]. Although a majority of children with dyslipidemia might be identified, universal screening has also generated some controversy. In 2007, the US Preventive Services Task Force stated that the evidence is insufficient to recommend for or against population-wide screening for lipid disorders in childhood [28]. Evaluation of any screening program involves weighing benefits against harm and costs. Until today, no long-term studies have evaluated the effects of screening children and adolescents on adult lipid levels and disease outcome. Furthermore, the costs of the universal screening program have not been analyzed by the NHLBI [29]. Serious concerns have also been raised about false positives (children with dyslipidemia who will not develop any premature coronary heart disease) and the possibility that those children will be treated for the rest of their lives with statins, with no appreciable benefit [30, 31].
The NHLBI guidelines also advocate selective screening for dyslipidemia in children with other CVD risk factors. It is well known that the extent of childhood atherosclerosis increases as the number of CVD risk factors increases. For this reason, children with one or more known atherosclerotic risk factors (hypertension, elevated BMI, significant tobacco smoke exposure) or with an underlying primary disease associated with increased risk of CVD should be screened for dyslipidemia. According to the NHBLI guidelines, this selective screening should be performed from the age of 2 years onward [27].
Taking all of this information into account, we would recommend selective screening to identify children with ADH.
Although selective screening programs might fail to diagnose a fair number of ADH cases, this exceeds the potential harm of overdiagnosis and overtreatment caused by universal screening. A novel approach might be to incorporate ADH into the newborn screening program. All newborns could be screened for a mutation in one of the three genes causing ADH. The results of the test could be sent to the family physician for adequate follow-up and referral to a specialized lipid clinic at the age of 8 years. Furthermore, this information could be used to perform ‘reverse’ cascade screening by testing the parents for ADH.
3.2 Screening Tools
Once children are selected for screening, lipid levels are measured. In selective screening, a fasting lipid sample should be obtained with total cholesterol (TC) and triglycerides (TG) in order to calculate LDL-C, according to the Friedewald equation. Many laboratories nowadays determine LDL-C levels directly, without using the Friedewald equation, which makes fasting less important.
In children without any known risk factor for CVD, non HDL-C testing can be used. If the non-HDL-C screening test is abnormal, the NHLBI guidelines recommend that fasting lipid profiles should be measured at least twice at intervals of between 2 weeks and 3 months. If the initial test is a fasting lipid profile, this should similarly be repeated at least once and averaged to determine the need for treatment [27].
3.3 Age at Screening
Total cholesterol levels remain quite constant from the age of 2 years up to puberty, where levels tend to decrease because of rapid growth and sexual maturation [32, 33]. A recent meta-analysis showed that cholesterol levels discriminate best between children with and without FH at the ages of 1–9 years [32]. Considering treatment for children with hypercholesterolemia, dietary interventions are not recommended before the age of 2 years, and pharmacological treatment should not initiated before the age of 8 years. Therefore, screening for childhood hypercholesterolemia is advisable between the age of 2 years and the start of puberty.
The NHLBI guidelines recommend universal screening at two different time points, the first screening between the ages of 9 and 11 years and the second screening between the ages of 17 and 21 years.
4 Non-pharmacological Treatment
The management of children with hypercholesterolemia begins with lifestyle interventions that focus on cholesterol lowering and reducing other cardiovascular risk factors. These interventions include dietary changes, increased physical activity, and eliminating tobacco smoking. For optimal compliance, the whole family should be involved.
The Cardiovascular Health Integrated Lifestyle Diet (CHILD-1) is the recommended first diet for all children at elevated cardiovascular risk from the age of 2 years [27]. It recommends low saturated fat (<10 %) and low cholesterol (<300 mg/day) intake, with sufficient calories to maintain normal growth and development. Sugar and salt intake should be minimized as well. Dietary fat intake is not restricted in children younger than 2 years, and for infants, breastfeeding is recommended. In children with identified hypercholesterolemia, a more aggressive dietary approach (CHILD-2) is necessary [27]. This diet limits the saturated fat intake to less than 7 % of total daily calories and cholesterol intake to less than 200 mg/day. These guidelines should be followed by the entire family. Few data are available on the effectiveness of these diets in children, but they appear to improve cholesterol levels. More importantly, they do not interfere with normal growth and development [34, 35]. Additional dietary supplementation with plant stanols and sterols, water-soluble fiber, soy protein or omega 3 fatty acids might enhance the reduction of LDL-C. However, most studies in children are small; therefore, more studies are warranted before general recommendations can be made.
Although data in children are limited, increased physical activity and a reduction in sedentary behavior might improve fasting lipid profiles and even lower the risk of CVD [36, 37]. Furthermore, there is strong evidence that increased physical activity has a positive influence on other cardiovascular risk factors, such as blood pressure, BMI, and glucose levels. The NHBLI guidelines recommend moderate to vigorous activity for 1 h/day, with vigorous physical activity on 3 days per week for children aged 5 years and older. Total screen time, including television and video games, should be restricted to 2 h or less per day [27].
Smoking is strongly associated with an increased risk of CVD and should therefore be strongly discouraged. It is also associated with an adverse effect on serum lipids, increased inflammation, and vascular dysfunction [38].
5 Pharmacological Treatment
Dietary interventions and increased physical activity are rarely sufficient in children with hereditary hypercholesterolemia. If cholesterol levels are not significantly reduced after 6 months of a suitable lifestyle, pharmacological treatment should be considered (Table 1). NHLBI guidelines recommend initiation of statin therapy for children from the age of 10 years on the following conditions:
-
if LDL-C is ≥190 mg/dL;
-
or if LDL-C remains at 160–189 mg/dL after 6 months of diet in the presence of a family history of early heart disease in a first-degree relative, or at least one high-level risk factor/condition, or at least two moderate risk factors/conditions;
-
or if LDL-C is ≥130–159 mg/dL with at least two high-level risk factors/conditions or at least one high-level risk factor/condition together with two moderate-level risk factors/conditions.
In children aged 8 and 9 years, statin treatment might be considered if LDL-C is persistently ≥190 mg/dL after 6 months of diet, together with a positive family history of premature CVD, or other additional risk factors (Table 2) [27]. Decisions regarding the need for medical therapy should be based on the average of results from at least two fasted lipid profiles obtained at least 2 weeks but no more than 3 months apart.
When initiation of statin therapy is required, children should be referred to either a lipid clinic or a pediatrician who is used to treating hypercholesterolemia. The children, as well as the parents, should be adequately informed of the background, consequences, and treatment of their condition. Several lipid clinics in The Netherlands have started specialized outpatient clinics for FH, where at the same time parents are seen by the lipidologist and children are seen by the pediatrician. Afterwards, the whole family visits the dietician. In this way, all affected family members are screened once a year.
5.1 Statins
Statins are currently the pharmacological agents of first choice in children with hypercholesterolemia (Table 3). Statins work by inhibiting the enzyme hydroxymethylglutaryl coenzyme A reductase (HMG CoA reductase) needed for endogenous production of cholesterol. The reduction of the intracellular cholesterol pool triggers upregulation of the LDL receptors on the cell surface. This upregulation of LDL receptors can increase the clearance of LDL particles. In adults, statins have proven effective in reducing both LDL-C levels and the incidence of coronary and other vascular events [39, 40]. Several pediatric studies have shown statins to be equally effective as in adults, well tolerated, and safe with regard to adverse events, growth, or sexual development [41]. Vascular endothelial function and IMT of the carotid artery, both markers of early atherosclerosis, were also improved in children with FH treated with statins, compared with untreated peers [4, 5].
Several different statins are currently available for use in childhood. The US Food and Drug Administration (FDA) has given approval for the use of lovastatin, simvastatin, atorvastatin and rosuvastatin in children from the age of 10 years onward and for pravastatin 20 mg from the age of 8 years onward. A randomized, placebo-controlled trial of pitavastatin is currently being conducted and has enrolled children from the age of 6 years onward. Adverse events are infrequent in childhood but can include muscle cramps, gastrointestinal complaints, an increase in liver transaminase levels, and rhabdomyolysis. Rhabdomyolysis is extremely rare but may be induced by drug interactions, especially with concomitant use of cyclosporine, erythromycin, or gemfibrozil. Furthermore, the use of statins should be interrupted during pregnancy because of fear of possible fetal harm, which has been described in several case series [42]. A recent systematic review suggested that statins are unlikely to be teratogenic [43]. However, large trials assessing the transplacental passage, benefits, and safety of statins during pregnancy are lacking.
Before initiating treatment with statins, a baseline fasting lipid panel, creatine kinase (CK), and liver transaminases should be measured. The treatment goal is an LDL-C level less than 130 mg/dL. Cholesterol levels should be rechecked after 4 weeks and, if necessary, the medication dose can be doubled and laboratory work should be repeated again after 4 weeks. If target LDL-C levels are still not achieved, the dose may be further increased, or another agent may be added [27]. In the first year of treatment, the fasted lipid panel and liver transaminases should be monitored every 3–4 months, together with growth, sexual maturation, and development.
At initiation of statin treatment, the child and family should be advised to report any adverse muscle-related complaints. CK and transaminase levels should be monitored periodically after the introduction of statins [27]. If CK levels exceed ten times the upper limit of normal or if liver transaminase levels exceed three times the normal levels, treatment should be withheld temporarily. However, CK levels can be increased in children who are actively participating in contact sports, so elevated CK levels should always be taken within the clinical context, before ascribing it to statin use. Ideally, increased physical activity should be avoided for 1–2 days prior to testing.
All taken together, several clinical trials in children with hypercholesterolemia have shown that statins are remarkably safe, well tolerated, and effective in lowering cholesterol levels. These were, however, short-term studies. In the longest follow-up study of 4.5 years, Rodenburg et al. reported that early initiation of statin therapy in children with FH delayed the progression of carotid IMT. There were no serious adverse events reported during follow-up, and normal growth and sexual maturation were not affected [44]. Long-term studies are needed to establish whether early initiation of statin therapy in children is justified in order to prevent cardiovascular events.
5.2 Bile Acid Sequestrants
In the past, bile acid sequestrants (BAS) were considered the only suitable drugs for children, because they act in the intestinal lumen and are not systemically absorbed [45]. These agents bind bile acids in the intestine, resulting in interruption of the reabsorption of bile acids. As a consequence, the formation of bile acids from intrahepatic cholesterol is increased, thereby reducing the intracellular cholesterol concentration. This triggers upregulation of LDL receptors and increased LDL-C clearance. Serum LDL-C can be lowered by 10–20 % by BAS. A major problem of classical BAS is the poor long-term adherence and tolerability, due to gastro-intestinal upset and gritty texture [45].
Recently, a novel second-generation BAS, colesevelam, was evaluated in children [46, 47]. Because of its greater affinity for bile salts, it can be used in a lower dosage, is associated with less unpleasant side effects, and therefore achieves better adherence to treatment. Besides tablets, colesevelam has recently become available as powder for oral suspension and may be administered as monotherapy or in combination with statins. At dosages of 3.75 g once daily or 1.875 g twice daily, it is approved by the FDA for the treatment of pediatric patients aged 10–17 years with heterozygous FH.
5.3 Ezetimibe
Ezetimibe lowers cholesterol by preventing intestinal absorption of dietary and biliary cholesterol. Its mechanism of action may involve Niemann-Pick C1 like 1 (NPCL1L1) protein, a protein involved in cholesterol absorption at the brush border of the intestine. As a result of lower LDL-C levels, LDL receptor expression is upregulated and LDL-C clearance from plasma is increased. In adults, ezetimibe monotherapy reduces LDL-C levels by approximately 17 %, but ezetimibe is mostly used in combination with statin therapy [48, 49].
A small number of short-term studies have investigated the efficacy and tolerability of ezetimibe in children with hypercholesterolemia. Either alone or in addition to simvastatin, it appears to effectively lower LDL-C in young patients, without any significant side effects [50, 51]. However, the clinical benefits of either ezetimibe monotherapy or combining ezetimibe with statin therapy in terms of clinical outcome remain to be proven. In patients who are statin intolerant and in patients with sitosterolemia, ezetimibe is the agent of first choice. Ezetimibe 10 mg is registered by the FDA for pediatric use from the age of 10 years onward.
5.4 Other Agents
Fibric acid derivates increase HDL cholesterol and decrease triglyceride levels. Although there are limited pediatric data on the use of fibrates, they may be useful in children with elevated triglycerides and an associated risk of pancreatitis [27]. In general, fibrates are well tolerated, and their adverse effects are similar to those of statins.
The mechanism of action of nicotinic acid, a water-soluble B complex vitamin, is complex and not fully understood, but results in lowering of both LDL-C and triglyceride concentrations and an increase in HDL-C. Compliance with niacin is poor because of its frequent adverse effects, such as flushing, headache, and rash. Because of those adverse effects, niacin is not recommended for routine pediatric use.
5.5 Novel Agents
A recent study in The Netherlands in 1,249 adult heterozygous FH patients found that although the vast majority used lipid-lowering therapy, only 21 % of them reached LDL-C levels of <2.5 mmol/L. The study suggested that only a small proportion of patients with FH reached LDL-C targets recommended by current guidelines, mainly because they were not treated with the highest dosages of statins, they were statin intolerant, or their levels were too high to be controlled with the current available therapy [52]. This indicates the urge for new treatment options to improve LDL reduction.
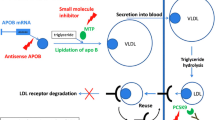
Several novel promising therapeutic strategies for LDL-C lowering have been developed and are currently being investigated in adults. PSCK9 monoclonal antibodies reduce levels of PCSK9, a protease that leads to degradation of LDL receptors and thus increases LDL-C levels. Published phase II data have shown that these monoclonal antibodies, used as monotherapy or added to statins, are effective and well tolerated at 3 months [53, 54]. Larger and much longer phase III trials are now in progress to assess the long-term tolerability, safety, and impact on cardiovascular disease events of these very promising LDL-C–lowering compounds.
A second group of novel lipid-lowering agents are cholesterol ester transport protein (CETP) inhibitors. CETP mediates the process of transfer of cholesteryl esters from HDL particles to apoB-containing particles. Inhibition of CETP activity results in an increase in HDL-C and a decrease in LDL-C. Although a clinical trial with torcetrapib was terminated early because of potential off-target effects on blood pressure, it did not rule out the cardioprotective effect of CETP inhibition [12]. Therefore, two other CETP inhibitors, anacetrapib and evacetrapib, are currently being evaluated in phase III clinical trials. Both agents have shown beneficial effects by increasing HDL-C and decreasing LDL-C concentrations, but long-term outcome trials are eagerly awaited.
Apolipoprotein B is essential for the production of very low-density lipoprotein cholesterol (VLDL-C; the precursor of LDL-C) and for the clearance of cholesterol. Treatment with mipomersen, an oligonucleotide antisense inhibitor directed against apoB messenger RNA, resulted in an additional LDL-C reduction of 25–36 % in patients with homozygous FH or severe heterozygous FH who were already receiving high doses of lipid-lowering therapy [55, 56]. Reported adverse events were injection site reactions and an increase in the fat content of the liver. The FDA has approved mipomersen as an orphan drug for use in patients with homozygous FH.
Microsomal triglyceride transfer protein (MTP) plays an important role in the formation of apoB-containing lipoproteins in hepatocytes and intestinal enterocytes. Lomitapide is an oral MTP inhibitor and has demonstrated substantial LDL-C reductions in patients with homozygous FH treated with aggressive lipid-lowering therapy [57]. Gastrointestinal symptoms and elevations in transaminases were the most commonly reported adverse events. Recently, lomitapide has been approved by both the FDA and EMA as an orphan drug for use in homozygous FH patients.
5.6 LDL Apheresis
Generally, in patients with homozygous ADH or severe heterozygous ADH, statins have only modest effects on plasma levels of LDL-C. In these individuals, LDL apheresis (LDL-A) has proven to be highly effective in removing LDL-C. LDL particles are selectively removed from the circulation by binding to the dextran sulfate or polyacrylamide membrane of this filter. By this technique, LDL-C levels can be effectively reduced by more than 60 % immediately after apheresis [58]. This reduction, however, is temporary, with a rapid rebound of LDL-C over the days following treatment, and the treatment should therefore be performed once weekly [58].
Data on LDL-A in children are limited. Several case reports and case series of children aged 6–17 years treated with LDL-A have shown that this treatment is remarkably safe, and those children who have been treated the longest have shown normal growth and development [59–61].
6 Conclusions
Altogether, in recent decades, we have gained much more insight in the role of hypercholesterolemia in the development of atherosclerosis and subsequent cardiovascular disease. Different screening strategies are used to identify children with hypercholesterolemia who are at risk of premature cardiovascular events. Treatment of hypercholesterolemia starts at a young age with adjustment of lifestyle, adopted by the whole family. If cholesterol levels remain increased, pharmacological treatment, preferably with statins, should be considered. Statins have proven to be safe and effective in short-term studies. The main question that remains is whether long-term statin therapy is indeed justified in order to prevent cardiovascular events later in life. Follow-up of individuals who have initiated statin therapy in childhood will help us to find those answers.
References
McNamara JJ, Molot MA, Stremple JF, Cutting RT. Coronary artery disease in combat casualties in Vietnam. JAMA. 1971;17(216):1185–7.
Berenson G, Srinivasan S. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. N Engl J Med. 1998;338:1650–6.
Relationship of atherosclerosis in young men to serum lipoprotein cholesterol concentrations and smoking. A preliminary report from the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) Research Group. JAMA. 1990;264:3018–24.
Wiegman A, de Groot E, Hutten BA, Rodenburg J, Gort J, Bakker HD, et al. Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia. Lancet. 2004;31(363):369–70.
De Jongh S, Lilien MR, op’t Roodt J, Stroes ESG, Bakker HD, Kastelein JJP. Early statin therapy restores endothelial function in children with familial hypercholesterolemia. J Am Coll Cardiol. 2002;40:2117–21.
Goldstein JL, Hobbs HH, Brown MS. Part 12: Lipids chapter 120: familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. pp. 2863–913.
Innerarity TL, Weisgraber KH, Arnold KAYS, Mahley RW, Kraussf RM, Vega GL, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci USA. 1987;84:6919–23.
Abifadel M, Rabès J-P, Devillers M, Munnich A, Erlich D, Junien C, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat. 2009;30:520–9.
Liyanage KE, Burnett JR, Hooper AJ, van Bockxmeer FM. Familial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distribution. Crit Rev Clin Lab Sci. 2011;48:1–18.
Goldstein JL, Schrott HG, Bierman EL. Hyperlipidemia in coronary heart disease and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–68.
Stone NJ, Levy RI, Fredrickson DS, Verter J. Coronary artery disease in 116 kindred with familial type II hyperlipoproteinemia. Circulation. 1974;1(49):476–88.
Macchiaiolo M, Gagliardi MG, Toscano A, Guccione P, Bartuli A. Homozygous familial hypercholesterolaemia. Lancet. 2012;379:1330.
Veerkamp MJ. Diagnosis of familial combined hyperlipidemia based on lipid phenotype expression in 32 families: results of a 5-year follow-up study. Arterioscler Thromb Vasc Biol. 2002;1(22):274–82.
Ayyobi AF, Brunzell JD. Lipoprotein distribution in the metabolic syndrome, type 2 diabetes mellitus, and familial combined hyperlipidemia. Am J Cardiol. 2003;92:27–33.
Gaddi A, Cicero a FG, Odoo FO, Poli AA, Paoletti R. Practical guidelines for familial combined hyperlipidemia diagnosis: an up-date. Vasc Health Risk Manag. 2007;3:877–86.
De Graaf J, Stalenhoef AF. Defects of lipoprotein metabolism in familial combined hyperlipidaemia. Curr Opin Lipidol. 1998;9:189–96.
Li S, Chen W, Srinivasan SR, Xu J, Berenson GS. Relation of childhood obesity/cardiometabolic phenotypes to adult cardiometabolic profile: the Bogalusa Heart Study. Am J Epidemiol. 2012;1(176 Suppl):S142–9.
Soutar AK, Naoumova RP, Traub LM. Genetics, clinical phenotype, and molecular cell biology of autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2003;1(23):1963–70.
Lütjohann D, von Bergmann K. Phytosterolaemia: diagnosis, characterization and therapeutical approaches. Ann Med. 1997;29:181–4.
Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;3(350):2362–74.
Kusters DM, de Beaufort C, Widhalm K, Guardamagna O, Bratina N, Ose L, et al. Paediatric screening for hypercholesterolaemia in Europe. Arch Dis Child. 2012;97:272–6.
Watts GF, Sullivan DR, Poplawski N, van Bockxmeer F, Hamilton-Craig I, Clifton PM, et al. Familial hypercholesterolaemia: a model of care for Australasia. Atheroscler Suppl. 2011;12:221–63.
Griffin TC, Christoffel KK, Binns HJ, McGuire PA. Family history evaluation as a predictive screen for childhood hypercholesterolemia, Pediatric Practice Research Group. Pediatrics. 1989;84:365–73.
Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in The Netherlands. Lancet. 2001;357:165–8.
Huijgen R, Kindt I, Verhoeven SBJ, Sijbrands EJG, Vissers MN, Kastelein JJP, et al. Two years after molecular diagnosis of familial hypercholesterolemia: majority on cholesterol-lowering treatment but a minority reaches treatment goal. PLoS One. 2010;5:e9220.
Avis HJ, Kusters DM, Vissers MN, Huijgen R, Janssen TH, Wiegman A, et al. Follow-up of children diagnosed with familial hypercholesterolemia in a national genetic screening program. J Pediatr. 2012;161:99–103.
Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128(Suppl):S213–56.
Haney EM, Huffman LH, Bougatsos C, Freeman M, Steiner RD, Nelson HD. Screening and treatment for lipid disorders in children and adolescents: systematic evidence review for the US Preventive Services Task Force. Pediatrics. 2007;120:e189–214.
Schroeder AR, Redberg RF. Cholesterol screening and management in children and young adults should start early—no! Clin Cardiol. 2012;35:665–8.
Gillman MW, Daniels SR. Is universal pediatric lipid screening justified? JAMA. 2012;307:259–60.
Psaty BM, Rivara FP. Universal screening and drug treatment of dyslipidemia in children and adolescents. JAMA. 2012;18(307):257–8.
Wald DS, Bestwick JP, Wald NJ. Child-parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis. BMJ. 2007;22(335):599.
Friedman LA, Morrison J a, Daniels SR, McCarthy WF, Sprecher DL. Sensitivity and specificity of pediatric lipid determinations for adult lipid status: findings from the Princeton Lipid Research Clinics Prevalence Program Follow-up Study. Pediatrics. 2006;118:165–72.
Obarzanek E, Kimm SYS, Barton BA, Van Horn L, Kwiterovich Jr PO, Simons-Morton DG, et al. Long-term safety and efficacy of a cholesterol-lowering diet in children with elevated low-density lipoprotein cholesterol: seven-year results of the Dietary Intervention Study in Children (DISC). Pediatrics. 2001;107:256–64.
Niinikoski H, Lagström H, Jokinen E, Siltala M, Rönnemaa T, Viikari J, et al. Impact of repeated dietary counseling between infancy and 14 years of age on dietary intakes and serum lipids and lipoproteins: the STRIP study. Circulation. 2007;28(116):1032–40.
Pahkala K, Heinonen OJ, Simell O, Viikari JS, Rönnemaa T, Niinikoski H, et al. Association of physical activity with vascular endothelial function and intima-media thickness. Circulation. 2011;124:1956–63.
Pahkala K, Heinonen OJ, Lagström H, Hakala P, Hakanen M, Hernelahti M, et al. Clustered metabolic risk and leisure-time physical activity in adolescents: effect of dose? Br J Sports Med. 2012;46:131–7.
Erhardt L. Cigarette smoking: an undertreated risk factor for cardiovascular disease. Atherosclerosis. 2009;205:23–32.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;8(366):1267–78.
Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DCG, Liem AH, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423.
Avis HJ, Vissers MN, Stein EA, Wijburg FA, Trip MD, Kastelein JJP, et al. A systematic review and meta-analysis of statin therapy in children with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2007;27:1803–10.
Lecarpentier E, Morel O, Fournier T, Elefant E, Chavatte-Palmer P, Tsatsaris V. Statins and pregnancy: between supposed risks and theoretical benefits. Drugs. 2012;16(72):773–88.
Kusters DM, Lahsinoui HH, van de Post JAM, Wiegman A, Wijburg FA, Kastelein JJP, et al. Statin use during pregnancy: a systematic review and meta-analysis. Expert Rev Cardiovasc Ther. 2012;10:363–78.
Rodenburg J, Vissers MN, Wiegman A, van Trotsenburg a SP, van der Graaf A, de Groot E, et al. Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation. 2007;116:664–8.
West RJ, Lloyd JK, Leonard JV. Long-term follow-up of children with familial hypercholesterolaemia treated with cholestyramine. Lancet. 1980;25(2):873–5.
Perry CM. Colesevelam: in pediatric patients with heterozygous familial hypercholesterolemia. Paediatr Drugs. 2010;1(12):133–40.
Stein EA, Marais AD, Szamosi T, Raal FJ, Schurr D, Urbina EM, et al. Colesevelam hydrochloride: efficacy and safety in pediatric subjects with heterozygous familial hypercholesterolemia. J Pediatr. 2010;156:231–6.e1–3.
Dujovne CA, Ettinger MP, McNeer JF, Lipka LJ, LeBeaut AP, Suresh R, et al. Efficacy and safety of a potent new selective cholesterol absorption inhibitor, ezetimibe, in patients with primary hypercholesterolemia. Am J Cardiol. 2002;15(90):1092–7.
Knopp R. Effects of ezetimibe, a new cholesterol absorption inhibitor, on plasma lipids in patients with primary hypercholesterolemia. Eur Heart J. 2003;24:729–41.
Clauss S, Wai K-M, Kavey R-EW, Kuehl K. Ezetimibe treatment of pediatric patients with hypercholesterolemia. J Pediatr. 2009;154:869–72.
Van der Graaf A, Cuffie-Jackson C, Vissers MN, Trip MD, Gagné C, Shi G, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;21(52):1421–9.
Pijlman AH, Huijgen R, Verhagen SN, Imholz BPM, Liem AH, Kastelein JJP, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis. 2010;209:189–94.
Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36.
Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–900.
Raal FJ, Santos RD, Blom DJ, Marais AD, Charng M-J, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
McGowan MP, Tardif J-C, Ceska R, Burgess LJ, Soran H, Gouni-Berthold I, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One. 2012;7:e49006.
Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Thompson GR. Recommendations for the use of LDL apheresis. Atherosclerosis. 2008;198:247–55.
Stefanutti C, Julius U. Lipoprotein apheresis: state of the art and novelties. Atheroscler Suppl. 2013;14:19–27.
Palcoux J-B, Atassi-Dumont M, Lefevre P, Hequet O, Schlienger J-L, Brignon P, et al. Low-density lipoprotein apheresis in children with familial hypercholesterolemia: follow-up to 21 years. Ther Apher Dial. 2008;12:195–201.
Græsdal A, Bogsrud MP, Holven KB, Nenseter MS, Narverud I, Langslet G, et al. Apheresis in homozygous familial hypercholesterolemia: the results of a follow-up of all Norwegian patients with homozygous familial hypercholesterolemia. J Clin Lipidol. 2012;6:331–9.
Conflicts of interest
No external funding was used in the preparation of this manuscript. JJPK is a consultant to, and has received honoraria from, Amgen, Sanofi Aventis, Regeneron, Eli Lilly, Genzyme, Isis Pharmaceuticals, Aegerion, Omthera, AstraZeneca, Pfizer, MSD, and Atheronova. MJAMB, BAH, and AW have no conflicts that might be relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Braamskamp, M.J.A.M., Hutten, B.A., Wiegman, A. et al. Management of Hypercholesterolemia in Children. Pediatr Drugs 16, 105–114 (2014). https://doi.org/10.1007/s40272-013-0060-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-013-0060-2