
Abstract
Inflammatory bowel disease, including Crohn’s disease and ulcerative colitis, comprises multiple complex immune-mediated disorders. Early diagnosis and prompt disease control may prevent long-term complications and hospitalization. The therapeutic options have expanded in the last two decades, with the development of biologics and small molecules targeting specific pathways implicated in inflammatory bowel disease pathogenesis. The interleukin (IL)-23/Th-17 axis is one such example. Targeting IL-12/23 is effective for the treatment of both moderate-to-severe Crohn’s disease and ulcerative colitis, and ustekinumab (an IL-12/23p40 antagonist) is approved for both indications. In patients with psoriasis, improved clinical outcomes were observed with agents that more selectively targeted IL-23 (IL-23p19 antagonists) compared with those that target both IL-12 and IL-23. Many specific IL-23p19 antagonists are currently being investigated in Crohn’s disease and ulcerative colitis, and risankizumab has been recently approved for moderate-to-severely active Crohn’s disease. In this review, we summarize the mechanisms of action and the evidence from clinical trials supporting the efficacy and safety of IL-23p19 antagonists for the treatment of inflammatory bowel disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Targeting interleukin-23 (IL-23) has been shown to be associated with significant therapeutic benefits in clinical trials of patients with inflammatory bowel disease. |
Ustekinumab (an IL-12/23 antagonist) is approved for the treatment of moderate-to-severe Crohn’s disease and ulcerative colitis, and risankizumab (a selective IL-23p19 antagonist) is approved for the treatment of moderate-to-severe Crohn’s disease. |
Additional agents selectively targeting IL-23p19 are currently in clinical development. |
1 Introduction
Inflammatory bowel disease (IBD) is a complex immune-mediated disease with diverse manifestations. Crohn’s disease (CD) and ulcerative colitis (UC) are subcategories of IBD that are phenotypically different yet share similar pathogenesis and management algorithms. Long-term medical therapy to modulate dysregulated immune responses and surgery are frequently required, resulting in a substantial burden to patients [1, 2] and the healthcare system [3, 4]. Approximately 7 million people globally have IBD, and there has been a substantial increase in IBD prevalence since the 1990s, particularly in recently industrialized countries [5, 6].
Despite the availability of effective pharmacotherapy, clinical remission rates during maintenance therapy for IBD generally do not exceed 50% [7, 8]. Since their introduction over 20 years ago, biologics targeting tumor necrosis factor (TNF) have transformed IBD care and are mainstays of therapy in this class. However, approximately one-third of patients will experience a primary nonresponse to induction therapy with TNF antagonists, and half of patients with an initial response may lose response over time [9–11]. Evolution of immunological mechanisms in response to biologic therapy has been documented, and can lead to therapeutic resistance [12]. This is an important clinical problem that highlights the need for new therapies. In the last two decades, additional biologics and small molecules with novel mechanisms of action, including those targeting the interleukin (IL)-12/23 cytokines, have been developed.
A strong association between the development of CD and IL-23 receptor (IL-23R) gene polymorphisms on chromosome 1p31 was first described in 2006 in a pivotal genome wide association (GWAS) study [13]. This study also identified rare IL-23R gene variants with reduced IL-23 expression that were protective for the development of CD, thus underscoring the importance of IL-23 in the pathogenesis of IBD. IL-23 is a key activator of pathogenic Th17 cells that is implicated in many chronic autoimmune conditions, such as psoriasis and IBD. Ustekinumab, a monoclonal antibody targeting the p40 subunit common to both IL-12 and IL-23 (IL-12/23p40, discussed in greater detail below), was the first agent in this class approved for treatment of CD and UC [14, 15]. Antibodies selectively targeting a subunit unique to IL-23 (IL-23p19) have also been developed and approved for the treatment of psoriasis (risankizumab, guselkumab, tildrakizumab), psoriatic arthritis (risankizumab and guselkumab), and most recently for the treatment of CD (risankizumab). Additional agents targeting IL-23p19 (e.g., mirikizumab, brazikumab, and guselkumab) are currently in phase 3 of clinical development for the treatment of both CD and UC. Recently, application for marketing authorization of mirikizumab for the treatment of adults with moderate-to-severe UC has been submitted to regulatory authorities. Although there are no completed head-to-head studies in IBD that compare the two classes of IL-23 antagonists, agents that selectively target IL-23p19 were associated with substantially greater efficacy for the treatment of psoriasis compared with IL-12/23p40 blockers [16]. In this review, we summarize the role of IL-23 in the immunopathogenesis of IBD as well as data from placebo-controlled trials on the efficacy and safety of IL-23p19 antagonists in CD and UC (Table 1).
2 Role of the IL-23 Axis in the Immunopathogenesis of IBD
2.1 Structure and Physiological Function of IL-12 and IL-23 and Their Receptors
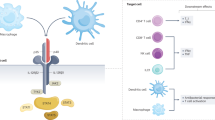
IL-12 and IL-23 are structurally similar, heterodimeric IL-12 family cytokines with diverse yet overlapping functions. IL-12 was first identified in 1989 as a soluble factor that stimulated natural killer cells and was later found to play a key role in innate immunity against bacterial pathogens [29]. Structurally, IL-12 consists of two disulfide-bound subunits, p35 and p40, that transmit signals via binding to the IL-12 receptor (IL-12R)β1 and IL-12Rβ2 subunits, respectively [30]. IL-12 is secreted predominantly by antigen presenting cells, dendritic cells, and macrophages (Fig. 1) [31,32,33]. Interactions between p40 and p19, a protein originally described in the early 2000s as having no intrinsic cytokine activity, was subsequently shown to stimulate memory T-cell proliferation and activation of the signal transducer and activator of transcription 4 (STAT4)/Janus kinase 2 (JAK2) pathway. Consequently, this led to designating it as IL-23 [34]. The IL-23 receptor is also a heterodimeric protein consisting of IL-12Rβ1 and IL-23R subunits. The IL-12Rβ1 subunit is common to both the IL-12R and IL-23R, whereas the IL-23R subunit binds specifically to the p19 subunit of IL-23. Macrophages and dendritic cells are the major sources of IL-23 [35].

A Production of IL-12 and IL-23 and receptor binding. Interleukin (IL)-23 and IL-12 are heterodimeric cytokines produced by macrophages, neutrophils, and dendritic cells in response to microbial and nonmicrobial stimuli, and which share a common subunit. The common IL-12/23p40 subunit combines with either IL-12p35 or IL-23p19 to form IL-12 and IL-23, respectively. Binding of IL-12 and IL-23 to their respective receptors on target cells, induces a chain of events leading to a number of downstream effects. The IL-23 receptor comprises IL-12Rβ1 and IL-23R chains whereas the IL-12 receptor comprises IL-12Rβ1 and IL-12Rβ2 chains. IL-23 receptor binding leads to phosphorylation of Janus kinase-2 (JAK-2) and tyrosine kinase-2 (TYK-2) and subsequent phosphorylation of (predominantly) signal transducer and activator of transcription (STAT)-3. Similarly, IL-12 receptor binding activates STAT4. Phosphorylation of STATs leads to their dimerization and subsequent translocation to the nucleus where they regulate gene transcription. B Downstream cellular effect and cytokine expression. IL-23 stimulates a variety of cells including TH17 T cells, type 3 innate lymphoid cells (ILC3), gamma delta T cells, and natural killer (NK) T cells to produce IL-17 cytokines, IL-22, interferon gamma (IFN-γ), and granulocyte monocyte-colony stimulating factor (GM-CSF). IL-12 stimulates naïve CD4+ T cells and induces their differentiation towards the T helper 1 (Th1) phenotype and subsequent production of IFN-γ. IL-12 induces IFNγ and tumor necrosis factor (TNF) release by CD8+ T cells, NK cells, and type 1 ILC.
Dimerization of the IL-12R upon binding of IL-12 leads to activation of the tyrosine kinases (TYK) JAK2 and TYK2, which in turn phosphorylate STAT4. Once phosphorylated, cytoplasmic STAT4 homodimerizes and translocates to the nucleus, where it promotes transcription of target genes [36]. IL-12 production occurs in response to microbial products such as lipopolysaccharide that signal through the Toll-like receptor-4. IL-12 stimulates Th1 CD4 cells and NK cells through activation of the key transcription factor T-bet, which results in the production of interferon-γ (IFN-γ) [37]. IFN-γ in turn enhances antigen presentation by inducing the expression of major histocompatibility complex molecules and activates cells to produce cytolytic molecules, such as perforin and granzyme that are important for clearance of intracellular bacteria. Accordingly, IL-12 acts as a bridge between the innate and adaptive immune systems and plays a crucial role in mucosal defense. This concept is supported by the increased susceptibility to infections such as Salmonella and mycobacterium observed in patients with genetic deficiency of IL-12 [38].
IL-23 mediates T-cell activation and antibacterial responses at the mucosal level [39] and is crucial for T-cell-dependent immune responses orchestrated by Th17-mediated proliferation and stimulation of memory T cells [34] Similar to IL-12, receptor-binding of IL-23 activates JAK2 and TYK2; however, signaling occurs predominantly through STAT3 [40]. Phosphorylated STAT3 activates RORγt, a master transcription factor responsible for transcription of IL-17 genes in Th17 CD4+ T cells [41]. This process results in production of IL-17, IL-17F, TNF-α, IL-6, granulocyte monocyte colony stimulating factor, and IL-22. While IL-23 cannot induce the differentiation of Th17 cells, as naïve T cells lack IL-23 receptors, it is a crucial factor for maintenance of Th17 cell proliferation [42]. IL-6 and TGF-β are the key cytokines that drive Th17 differentiation through activation of RORγt in naïve T-cells [43]. IL-23 receptors are also expressed by other IL-23 target cells including NK cells, NKT cells, gamma delta T cells, innate lymphoid cells, and Th17 T cells.
2.2 Role of IL-12 and IL-23 in the Pathogenesis of IBD
Although IL-12 and IL-23 are constitutively expressed in the healthy human intestine (particularly in the terminal ileum) [44] increased levels of both cytokines are observed in the presence of intestinal infections and in inflammatory states [44,45,46]. Early experiments demonstrated that blockade of IL-12 with either IL-12p40 neutralizing antibodies or disruption of the IL-12p40 gene (IL-12B) protected against experimentally induced autoimmune and inflammatory conditions [47,48,49,50,50]. In subsequent experiments, mice with IL-23p19 and IL-12p40 deficiency were protected against development of both experimental autoimmune encephalomyelitis and colitis, whereas mice with IL-12p35 deficiency were not; suggesting that IL-23, rather than IL-12, was fundamental in driving chronic inflammation [49, 51,52,53]. The functional role of IL-23 in intestinal inflammation is complex and includes mediation of both innate and T-cell-mediated responses. Elevated local concentrations of both IL-23 and IL-17 in the intestinal tissue of animal colitis models support their involvement in inflammation [51, 54]. Many single nucleotide polymorphisms in the IL-23 pathway conferring susceptibility to IBD have been identified in GWAS in the past 2 decades, and further support the importance of IL-23 and IL-17 in the pathogenesis of IBD [55]. IL-23 may also play a role in maintenance of intestinal epithelial integrity and promotion of mucosal healing via stimulation of IL-17 and IL-22 production. A study by Maxwell et al. demonstrated a differential role of IL-17 and IL-23 in intestinal inflammation. Impaired mucosal barrier function was observed with inhibition of IL-17 in a multidrug resistance-1a-ablated (Abcb1a−/−) mouse model of colitis [56]. This may partially explain why treatment with the IL-17 antagonists secukinumab and brodalumab led to worsening CD, rather than improvement [57, 58]. Finally, additional evidence suggests a pleiotropic effect of IL-23 via induction of IL-22 expression by innate lymphoid cells (ILC), a cytokine crucial for STAT3 activation in intestinal epithelial cells that has been associated with mucosal healing [56, 59, 60].
3 Efficacy of Biologics Targeting IL-23 in Moderate-to-Severe Crohn’s Disease
3.1 Brazikumab
The safety and efficacy of brazikumab (MEDI2070), a monoclonal antibody targeting IL-23p19, was evaluated in a phase 2, double-blind, randomized controlled trial (RCT) [18]. The primary endpoint was clinical response (100 point decrease in the CDAI score or CDAI < 150) at week 8. A statistically significant proportion of patients treated with brazikumab achieved a clinical response at week 8 compared with patients treated with placebo (49.2% versus 26.7%, p = 0.01); however, this response was not sustained at week 12 (37.3 versus 28.6, p = 0.29). Although a higher proportion of patients treated with brazikumab achieved clinical remission (CDAI < 150) at week 8 compared with those treated with placebo, the difference between the groups was not statistically significant (Table 1). At week 24, 53.8% (28/52) of patients who continued to receive open-label brazikumab following induction treatment with brazikumab achieved a clinical response compared with 57.7% (30/52) of patients who were treated with placebo during the induction phase. In this study, higher concentrations of IL-22 at baseline were associated with a greater likelihood of achieving response to treatment with brazikumab. A phase 2b study of brazikumab (NCT02574637) was terminated by the sponsor after recruiting 29 patients for reasons unrelated to safety and efficacy.
3.2 Mirikizumab
In the phase 2, double-blind, SERENITY trial, 191 patients with moderate-to-severe CD were randomized (2:1:1:2) to intravenous (IV) treatment with placebo, 200, 600, or 1000 mg mirikizumab q4w [19]. The primary outcome was endoscopic response (50% reduction in the SES-CD from baseline) at week 12. Patients who received mirikizumab and achieved ≥ 1 point improvement in the SES-CD at week 12 were re-randomized to continue their initial intravenous (IV) induction dose or receive 300 mg mirikizumab subcutaneous (SC) q4w. All patients in the maintenance phase received both IV and SC dosing between weeks 12 and 52 in a double-dummy design to maintain blinding. At week 12, the primary outcome (endoscopic response) and important secondary outcomes such as endoscopic remission (SES-CD < 4 for ileocolonic disease, SES-CD < 2 for isolated ileal disease, and no SES-CD subscore > 1), patient-reported outcome (PRO)-defined remission (average daily AP ≤ 1 and average daily SF ≤ 2.5), and clinical remission (CDAI < 150) were statistically superior to placebo for the two highest doses of mirikizumab (600 and 1000 mg) but not for the 200 mg dose (Table 1). Although a consistent dose-response relationship was observed for endoscopic outcomes (response and remission), higher rates for clinical outcomes (PRO remission and CDAI remission) were observed with the 600 mg dose relative to those observed with the 1000 mg dose. In the maintenance phase, endoscopic response rates were 58.5% (24/41) and 58.7% (27/46) and endoscopic remission rates were 19.5% (8/41) and 32.6% (15/46) at week 52 in the IV maintenance and SC maintenance cohorts, respectively. No consistent dose-response relationship was observed for outcomes at week 52. A substudy that assessed histological outcomes demonstrated superior histological response [absence of neutrophils in the lamina propria, epithelial damage, erosions, and ulceration; or a ≥ 50% decrease in the Robarts Histopathology Index (RHI) score; or Global Histologic Disease Activity Score (GHAS)] rates with all doses of mirikizumab compared with placebo (200 mg: 53.6%, p < 0.1; 600 mg: 50%, p < 0.05; 1000 mg: 66%, p < 0.01; and placebo: 29.2%) at week 12 [61]. However, only the 1000 mg mirikizumab dose was statistically superior to placebo in inducing histological remission [GHAS score epithelial damage: normal; infiltration of polymorphonuclear cells in lamina propria: normal; polymorphonuclear cells in epithelium: absent; and erosions or ulcerations: no; or RHI ≤ 3 and sum of RHI items 2–4 (lamina propria neutrophils, neutrophils in epithelium, and erosions or ulcerations) equal to 0]. At week 52, 51.8% and 22.3% of patients with active histological disease at study entry achieved histological response and remission, respectively. With the exception of two patients, all patients in histological remission were also in deep histological remission (GHAS or RHI score = 0) [61].
3.3 Guselkumab
GALAXI-1 was a phase 2, double-blind, RCT that evaluated the efficacy and safety of guselkumab in 309 adult patients with moderate-to-severe CD [20]. Patients were randomized to treatment with IV guselkumab (200 mg, 600 mg, 1200 mg) or placebo (1:1:1:1) q4w. Patients were also randomized to IV treatment with 6 mg/kg ustekinumab at week 0 followed by SC treatment with 90 mg ustekinumab at week 8 as a reference arm. Approximately 50% of patients had a prior inadequate response or intolerance to biologics prior to enrollment. The primary endpoint was change in CDAI score from baseline at week 12. Statistically significant decreases in the CDAI score from baseline were observed in all guselkumab treatment groups compared with placebo (Table 1), although no apparent dose-response relationship was observed for this endpoint. The proportion of patients who achieved important secondary endpoints was also statistically greater in the pooled guselkumab treatment groups compared with placebo, and included the outcomes of clinical remission (CDAI < 150, 53% versus 16.4%; p < 0.05), endoscopic response (50% decrease in SES-CD from baseline or SES-CD ≤ 2, 35.7% versus 11.5%, p < 0.05), clinical response (100-point reduction from baseline in CDAI score or CDAI score < 150, 65.9% versus 24.6%; p < 0.05), PRO-2 remission (AP ≤ 1 and SF ≤ 3 and no worsening from baseline, 42.7% versus 16.4%; p < 0.05), and clinical biomarker response (≥ 50% reduction from baseline in C-reactive protein (CRP) or fecal calprotectin concentration, 47% versus 6.6%; p < 0.05). In the subgroup of patients with inadequate response or intolerance to prior biologic therapy, 47.5% in the combined guselkumab dose group and 10.0% in the placebo group achieved clinical remission at week 12. Patients with a response at week 12 were transitioned to open-label SC dosing (placebo responders to placebo SC q4w; guselkumab 200 mg IV to 100 mg SC q8w; 600 mg IV to 200 mg SC q4w; 1200 mg IV to 200 mg SC q4w; and ustekinumab to 90 mg SC q8w) for a total of 48 weeks. No apparent dose-response was observed; clinical remission rates at week 48 ranged from 57.4% to 73.0% among patients treated with guselkumab, and clinical response rates ranged from 67.2% to 84.1% [62]. Phase 3 clinical trials evaluating the efficacy of guselkumab in patients with moderate-to-severe CD [GRAVITI (NCT05197049)] and perianal CD [FUZION CD (NCT05347095)] are currently ongoing.
3.4 Risankizumab
In a phase 2, double-blind RCT, 121 adult patients with moderate-to-severe CD were randomized in a 1:1:1 ratio to receive treatment with IV risankizumab (200 mg or 600 mg) or placebo at weeks 0, 4, and 8 [17]. A total of 93% (113/121) of patients had previous exposure to at least one anti-TNF agent at baseline of the study. The proportion of patients achieving the primary outcome of clinical remission (CDAI < 150) at week 12 was significantly higher in the combined risankizumab treatment groups compared with the placebo group (Table 1). The observed differences in clinical remission rates between patients treated with risankizumab and placebo were higher with the 600 mg dose (36.6%, Δ = 20.9%, p = 0.02) than the 200 mg dose (24.4%, Δ = 9.0%, p = 0.31). For important secondary outcomes including clinical response (CDAI < 150 or a reduction in the CDAI score ≥ 100 points from baseline), endoscopic response (> 50% reduction in CDEIS score from baseline), and endoscopic remission (CDEIS ≤ 4 or CDEIS ≤ 2 for patients with initial isolated ileitis) at week 12, risankizumab 600 mg was statistically superior to placebo, whereas risankizumab 200 mg was superior to placebo only in achieving endoscopic remission.
The phase 3 induction (ADVANCE and MOTIVATE) and maintenance (FORTIFY) trials provided additional evidence supporting the efficacy of risankizumab for the treatment of moderate-to-severe CD [22, 23]. These studies were the first to include both a clinical outcome and an endoscopic outcome based upon central assessment as co-primary endpoints. The ADVANCE trial (n = 931) included patients who had failed conventional or biological therapy, whereas the MOTIVATE trial (n = 618) included only patients who had failed biologic therapy. [22] Eligible patients were randomized (2:2:1 in ADVANCE, 1:1:1 in MOTIVATE) to induction treatment with 600 mg or 1200 mg IV risankizumab, or placebo. The co-primary endpoint was clinical remission (CDAI < 150 or average daily SF subscore ≤ 2 and average daily AP subscore ≤ 1 and both not worse than baseline) and endoscopic response (> 50% decrease in SES-CD from baseline) at week 12. A total of 30% and 53% of patients in the ADVANCE and MOTIVATE trials, respectively, had failed two or more biologics, and approximately 20% of patients in both trials had previously received ustekinumab. A statistically significant proportion of patients in both risankizumab arms achieved the co-primary endpoint of clinical remission and endoscopic response at week 12 in both the ADVANCE and MOTIVATE trials. A numerically greater proportion of biologic-naïve patients achieved clinical remission compared with patients with prior exposure to biologics. Patients who achieved clinical response (≥ 30% decrease in average daily SF subscore and/or ≥ 30% decrease in average daily AP subscore, both not worse than baseline) at week 12 were re-randomized (1:1:1) to maintenance treatment with SC risankizumab (180 mg or 360 mg q8w) or placebo in the FORTIFY trial [23]. The co-primary outcomes were clinical remission and endoscopic response (as defined above). Patients randomized to treatment with risankizumab achieved statistically superior rates of clinical remission and endoscopic response at week 52 (Table 1). Similarly, a significantly higher proportion of patients treated with 180 and 360 mg risankizumab achieved important secondary endpoints such as clinical remission, endoscopic remission, and deep remission at week 52 compared with placebo. Patients who did not achieve clinical response at week 12 in the ADVANCE and MOTIVATE trials were also eligible to receive an additional 12 weeks of risankizumab therapy (180 mg or 360 mg SC) in the FORTIFY trial. Of the patients who received the 180 and 360 mg dose, 53.3% (16/30) and 66.7% (22/33) achieved clinical remission, respectively [63].
4 Efficacy of Biologics Targeting IL-23 in Moderate-to-Severe Ulcerative Colitis
4.1 Mirikizumab
A phase 2 study assessed the efficacy and safety of mirikizumab in patients with moderate-to-severe UC. A total of 249 patients were randomized 1:1:1:1 to IV treatment with 50 mg exposure-based (EB) dosing, 200 mg EB dosing, 600 mg fixed dosing of mirikizumab, or placebo at weeks 0, 4, and 8 in a phase 2 study [24]. Patients who achieved a clinical response (a decrease in the 9-point Mayo Clinic score, including ≥ 2 points and ≥ 35% from baseline, with either a ≥ 1 point decrease in the RB subscore or a RB subscore 0 or 1) to mirikizumab at week 12 were re-randomized to SC maintenance treatment with 200 mg mirikizumab every 4 or 12 weeks, while placebo responders received treatment with SC placebo q4w. In the first phase of the study, only the 200 mg mirikizumab dose was statistically superior to placebo for induction of clinical remission (22.6% versus 4.8%, p < 0.01) at week 12. However, all mirikizumab doses were statistically superior to placebo for induction of clinical response at week 12. The 50 and 200 mg mirikizumab doses were also statistically superior to placebo for induction of endoscopic response at week 12. Among patients who were naïve to treatment with biologics, only mirikizumab 200 mg was superior to placebo for induction of clinical remission and response, whereas both the 50 and 200 mg doses were superior to placebo for induction of endoscopic improvement. Mirikizumab was not statistically superior to placebo for induction of clinical remission and endoscopic improvement at any dose in patients with prior exposure to biologics, although the 200 and 600 mg doses were statistically superior to placebo for induction of clinical response. At week 52, 53.7% (22/47) and 39.7% (17/46) of patients who responded to mirikizumab at week 12 and were treated with 200 mg mirikizumab every 4 and 12 weeks, respectively, achieved clinical remission [64]. Treatment of patients who failed to respond to initial IV induction doses of mirikizumab at week 12 with an additional 12 weeks of IV mirikizumab at doses of 600 or 1000 mg (after a protocol amendment) resulted in a clinical response in 50.0% (10/20) of patients who received treatment with an additional 12 weeks of 600 mg mirikizumab, and 43.8% (28/64) of those who received treatment with 1000 mg mirikizumab. Among patients who responded to extended dosing, 68.8% maintained a clinical response for up to 52 weeks with SC maintenance mirikizumab 200 mg q4w.
A subsequent phase 3 induction study (LUCENT 1) randomized 1281 adult patients with moderate-to-severe UC who had inadequate response, loss of response, or intolerance to corticosteroids, immunosuppressants, biologic therapies, or tofacitinib in a 3:1 ratio to treatment with 300 mg IV mirikizumab or placebo at 0, 4, and 8 weeks [25]. This study was designed as a single induction trial with a low alpha error of 0.00125. The primary outcome was clinical remission (as defined above) at week 12. A significantly greater proportion of patients treated with mirikizumab achieved clinical remission at week 12 compared with placebo (24.2% versus 13.3%; p < 0.001). Moreover, a significantly higher proportion of patients receiving mirikizumab achieved clinical response at week 12 compared with placebo (63.5% versus 42.2%; p < 0.00001). Patients who were biologic naïve (70.1% versus 50.3%; p < 0.001) were more likely to achieve clinical response than those who received prior biologic therapy (54.6% versus 29.7%; p < 0.001) when compared with placebo [65]. All key secondary endpoints, including the average reduction in bowel urgency severity, were significantly superior in the mirikizumab treatment group compared with placebo (p < 0.001), as was the reduction in CRP concentration from baseline (p < 0.001) at week 12. A significantly greater proportion of patients achieved histological remission (Geboes score ≤ 2B.0, 29.3% versus 15.6%; p < 0.001) and histological improvement (Geboes score ≤ 3.1, 39.2% versus 20.7%; p < 0.001) in the mirikizumab treatment group compared with placebo at week 12 [66].
Patients who demonstrated a clinical response to treatment with mirikizumab at week 12 (n = 544) in the LUCENT-1 induction trial were re-randomized in a 2:1 ratio to double-blind SC maintenance treatment with either 200 mg mirikizumab (n = 365) or placebo (n = 179) q4w up to week 52 in the LUCENT-2 trial (NCT03524092). At week 52, 49.9% of patients randomized to treatment with mirikizumab achieved the primary endpoint of clinical remission compared with 25.1% of patients treated with placebo (p < 0.001) [26]. At week 52, a statistically greater proportion of patients receiving mirikizumab maintenance treatment achieved SF remission (SF = 0, or SF = 1 with a ≥ 1 point decrease from induction baseline) (75.1% versus 44.7%, p < 0.001) and RB remission (RB = 0) (79.7% versus 49.7%, p < 0.001) compared with placebo [67]. Histological remission and improvement at week 52 occurred in 54.8% and 48.5% of patients treated with mirikizumab, respectively, compared with 24.6% and 25.7% of patients treated with placebo (p < 0.001 for both comparisons with placebo) [66].
4.2 Guselkumab
The phase 2b randomized, double-blind, placebo-controlled QUASAR study was conducted to evaluate guselkumab as an induction treatment for 313 patients with moderate-to-severe UC [27]. Patients were randomized (1:1:1) to treatment with IV guselkumab (200 or 400 mg) or placebo at weeks 0, 4, and 8. The primary outcome was clinical response (decrease from baseline in MMS by ≥ 30% and ≥ 2 points, with either a ≥ 1 point decrease from baseline in the RB subscore or a RB subscore = 0 or 1) at week 12. A significantly higher proportion of patients treated with guselkumab achieved clinical response compared with patients treated with placebo at week 12 (Table 1). Statistically higher proportions of patients treated with guselkumab (any dose) also achieved key secondary outcome measures including clinical remission, symptomatic remission, endoscopic improvement, and histo-endoscopic mucosal improvement compared with patients treated with placebo. No consistent dose-response relationship was observed for any outcome. A total of 47.3% (148/313) of patients had prior inadequate response or intolerance to biologics/tofacitinib, and approximately half of these patients had prior inadequate response or intolerance to two or more classes of advanced therapies. Clinical response at week 12 in these patients was achieved by a higher proportion of patients treated with guselkumab compared with placebo (50.5% versus 25.5%) [68]. Greater reductions in CRP and fecal calprotectin concentrations at week 12 were also observed in guselkumab treated patients compared with patients treated with placebo, and these reductions were observed as early as week 4.
In a phase 2a, proof of concept trial (VEGA), 214 patients with moderate-to-severe UC who failed conventional therapy and were naïve to anti-TNF, anti-IL-12/23p40 antagonist, and IL-23p19 antagonist treatment (prior exposure to vedolizumab and tofacitinib were permitted) were randomized in 1:1:1 ratio to treatment with a combination of guselkumab and golimumab (guselkumab 200 mg IV and SC golimumab 200 mg at week 0, SC golimumab 100 mg at weeks 2, 6, and 10, and IV guselkumab 200 mg at weeks 4 and 8, followed by SC guselkumab 100 mg q8w until week 32) or monotherapy with either guselkumab (200 mg at weeks 0, 4, and 8, followed by SC 100 mg q8w weeks until week 32) or golimumab (200 mg SC at week 0, followed by 100 mg SC at week 2 and q4w thereafter until week 34) [28]. The primary endpoint was clinical response (≥ 30% decrease from baseline in the full Mayo Clinic score and a decrease of ≥ 3 points with either a decrease in RB subscore ≥ 1 point or a RB subscore 0 or 1) at week 12. A higher number of patients in the combination therapy group (61%, 43/71) and guselkumab monotherapy group (66%, 47/71) had endoscopically severe disease compared with the golimumab monotherapy group (51, 37/72). At week 12, a greater proportion of patients treated with combination therapy achieved clinical response (83%) at week 12 compared with patients treated with guselkumab (75%, p = 0.21) or golimumab (61%, p = 0.003). Similarly, a greater proportion of patients treated with combination therapy achieved clinical remission (full Mayo Clinic score ≤ 2 with no individual subscore > 1) at week 12 (37%) compared with patients treated with either guselkumab (21%, p = 0.04) or golimumab (22%, p = 0.05) monotherapy. At week 38, 69% of patients in the combination therapy group had clinical response compared with 58% of patients in the golimumab monotherapy group and 72% of patients in the guselkumab monotherapy group. Clinical remission at week 38 was achieved in a greater proportion of patients treated with combination induction therapy followed by guselkumab maintenance therapy [44% (31/71)] compared with patients who were treated with guselkumab [31% (22/71), p = 0.109] or golimumab [22% (16/72), p = 0.006]. The proportion of patients who achieved key secondary endpoints including endoscopic improvement, histological remission, composite histological remission, and endoscopic improvement were higher in the combination therapy group than either monotherapy group at both week 12 and 38.
5 Safety of Biologics Targeting IL-23
The roles of IL-12 and IL-23 in mucosal defense have been well characterized in animal models, and IL-12 deficiency has been shown to be associated with increased risk of infections in animals as well as human beings [69, 70, 71]. Despite these observations, treatment with the IL-12/23p40 antagonist ustekinumab has not been associated with increased risk of infections. Moreover, long-term follow-up data from clinical trials of IBD have not identified any negative safety signals with ustekinumab therapy [72, 73]. This is supported by animal studies where IL-23 knockout mice have been shown to be immune competent compared with IL-12 knockout mice [74].
5.1 Safety of Anti-IL23p19 Agents from Psoriasis Clinical Trials
Unlike IL-17 antagonists, which have been associated with worsening of CD disease activity, IL-23 antagonists have been shown to be safe and effective for the treatment of CD. This may be explained by the role of IL-17 in maintaining intestinal mucosal barrier integrity by promoting expression of occludin, a cellular tight junction protein. Although IL-23 is a key cytokine in the production of IL-17 from Th17 cells, blocking IL-23 does not affect IL-17 due to IL-23-independent production of IL-17 from γδ-T cells [75]. Long-term data are lacking; however, evidence from phase 2 and 3 clinical trials of IL-23p19 antagonists for the treatment of IBD have not demonstrated a significant increase in the rates of serious adverse events compared with placebo with these agents, as described in earlier paragraphs. Safety data from psoriasis and psoriatic arthritis trials are also reassuring. For example, short- and long-term treatment with risankizumab in patients with moderate-to-severe psoriasis was associated with an overall favorable safety profile [76,77,78]. In a recent meta-analysis of risankizumab safety data from clinical trials in moderate-to-severe plaque psoriasis with 7927 patient-years (PY) of follow-up, the rates of serious adverse events, serious infections, nonmelanoma skin cancers (NMSC), malignant tumors excluding NMSC, and adjudicated major adverse cardiovascular events (MACE) were 7.8, 1.2, 0.7, 0.5, and 0.3 per 100 PY, respectively, with no important identified risks [79]. Additionally, a pooled analysis of safety data based on 5 years of continuous treatment with guselkumab in the VOYAGE 1 and 2 moderate-to-severe psoriasis trials reported adverse and serious adverse events rates of 149/100 PY and 5.01/100 PY, respectively [80]. The rates of serious infections (0.85/100 PY), NMSC (0.34/100 PY), malignancies other than NMSC (0.45/100 PY), and major adverse cardiovascular events (0.29/100 PY) were also low.
5.2 Safety of Anti-IL23p19 Agents from IBD Clinical Trials
Although large phase 3 trials with long-term follow-up are needed to confirm favorable safety profiles, phase 2 trials including patients with CD and UC, have not identified increased rates of adverse events with mirikizumab, guselkumab, and brazikumab treatment compared with placebo. In a long-term, open-label phase 2 extension study of risankizumab with a median of 33 months of therapy (167.0 PY), the rate of serious adverse events was 24.6 events/100 PY, the majority of which were gastrointestinal in nature and the rates of serious infections, opportunistic infections, and fungal infections were 4.2, 1.8, and 6.6 events per 100 PY, respectively [81]. No deaths, malignancies, adjudicated major adverse cardiovascular events, latent/active tuberculosis, or herpes zoster were reported in this study.
Similar rates of adverse events were observed with risankizumab and placebo treatment in the phase 3 ADVANCE and MOTIVATE induction studies [22]. Three deaths occurred in these studies: one patient treated with 1200 mg risankizumab in MOTIVATE, and two patients treated with placebo in ADVANCE. The cause of death for the patient treated with risankizumab in the MOTIVATE study was not considered drug related. The serious adverse event rate in the phase 3 risankizumab induction trials was higher in patients treated with placebo, likely due to worsening of CD. Three serious infections were reported in risankizumab-treated patients in the MOTIVATE study (Escherichia coli gastroenteritis, viral pharyngitis, and sepsis), and five serious infections were reported in the ADVANCE study (appendicitis, leptospirosis, lower respiratory tract infection, pneumonia, and urinary tract infection). None of the serious infections were considered by the investigator as related to risankizumab, and no serious infection led to treatment discontinuation. One patient treated with risankizumab in the ADVANCE study with a history of tuberculosis developed active tuberculosis after 8 weeks of therapy. No adjudicated MACE, adjudicated extended MACE, or adjudicated anaphylactic reactions were reported in any treatment group in either study. In the FORTIFY risankizumab maintenance study, similar rates of adverse events were reported at week 52 in both the placebo and SC risankizumab treatment groups [23]. The most commonly reported adverse events included worsening CD, nasopharyngitis, arthralgias, headache, nausea, diarrhea, abdominal pain, and anemia. The incidence of infectious adverse events was lower in both risankizumab treatment groups compared with the placebo group (180 mg risankizumab, 51.4/100 PY; 360 mg risankizumab, 57.7/100 PY; placebo, 76.0/100 PY). The incidence of serious infections was similar across treatment groups. Two herpes zoster, two opportunistic, and one oral fungal infection (oral candidiasis) were reported in the 180 mg risankizumab group. One intestinal Aeromonas infection was reported in the 360 mg risankizumab group. All events were mild to moderate in severity and resolved without discontinuation of the study drug. A single case of malignancy (breast cancer) was reported in a patient treated with 360 mg risankizumab, although this event was considered unrelated to the study drug by the investigator.
6 Positioning of Newer IL-23p19 Antagonists in IBD Management Algorithms
Therapeutic options for IBD have greatly expanded in the last two decades with the discovery of new molecules targeting different immune pathways. Nevertheless, decision-making is increasingly difficult given the availability of multiple pharmacological agents and the paucity of direct comparisons. Risankizumab is currently the only IL-23p19 inhibitor approved for the treatment of moderate-to-severe CD, although other IL-23p19 antagonists are in phase 3 clinical development trials and are also likely to gain regulatory approval in the future. Appropriate positioning of these agents in the IBD therapeutic algorithm is crucial to achieve optimal outcomes, and the choice of agent will depend on many factors, such as disease characteristics, affordability, and patient preference. In this section we are going to provide a comprehensive overview of existing direct and indirect data on comparative efficacy of drugs targeting IL-23 and other biologics, which might help in positioning of these agents.
6.1 Comparative Efficacy of Drugs Targeting IL-23 Compared with Drugs Targeting TNF
Data from phase 2 and 3 studies suggest that targeting IL-23 is safe and effective for the treatment of both CD and UC. In the absence of well-designed head-to-head trials, efficacy comparisons among agents are difficult. Comparative studies in psoriasis showed that agents targeting IL-23p19 were superior to the TNF antagonist adalimumab. In the phase 3 VOYAGE 1 and 2 RCTs comparing guselkumab and adalimumab, guselkumab was associated with higher PASI90 scores (90% or greater reduction in the baseline Psoriasis Area and Severity Index; VOYAGE 1: 76.3% versus 47.9% at week 48, p < 0.001; and VOYAGE 2: 75.2% versus 54.8% at week 24, p < 0.001) [82, 83]. In a head-to-head trial of risankizumab and adalimumab for the treatment of plaque psoriasis (IMMvent), 72% of patients treated with risankizumab achieved a PASI90 score compared with 47% of patients treated with adalimumab at week 16 [84]. There is a paucity of data comparing the efficacy of IL-23p19 antagonists and TNF antagonists in IBD. Similar endoscopic and clinical outcomes were observed in patients with moderate-to-severe UC who were treated with guselkumab (anti-IL-23p19) or golimumab (anti-TNF) monotherapy arms in the VEGA trial [28]. The SEAVUE trial compared ustekinumab (anti-IL-12/23p40) and adalimumab (anti-TNF) for treatment of patients with moderate-to-severe CD naïve to biologic therapy [85]. Although a statistically significant treatment difference was not observed for efficacy outcomes, a numerically higher proportion of infections were reported with adalimumab treatment (41% versus 34%). However, the rates of serious infections were similar between the adalimumab and ustekinumab treatment groups (2% versus 3%) [85].
Therapies targeting IL-23p19 have several potential advantages over TNF antagonists. IL-23p19 blockers are associated with negligible rates of neutralizing antibody formation [86]. Moreover, there does not appear to be a clear dose-response relationship for the agents studied to date, and therapeutic drug monitoring is therefore not likely to be required. TNF antagonists have been associated with increased risk of opportunistic infections and lymphoma in patients with IBD, especially when combined with immunosuppressives [87, 88]. The limited available data have not supported an increased risk of serious infections and malignancy with IL-23p19 antagonists. Long-term follow-up data from registries and real-world evidence are needed to support these preliminary observations. TNF antagonists are the preferred agents for treatment of special patient populations, including those with perianal fistula and extraintestinal manifestations, as well as for pregnant patients. Additional data on the safety and efficacy of IL-23p19 antagonists in these populations are required.
6.2 IL-12/23p40 versus IL-23p19 Agents
Earlier animal models suggested that IL-23, as opposed to IL-12, is an essential driver of inflammation and it was observed that IL-12p35 deficient IL-10 knockout mice but not IL-23p19 deficient IL-10 knockout mice could develop spontaneous colitis [51]. It is yet to be determined in head-to-head IBD trials whether selectively blocking IL-23 is more beneficial than blocking both IL-12 and IL-23. In psoriasis, a phase 3 head-to-head comparison of risankizumab and ustekinumab for the treatment of moderate-to-severe plaque psoriasis demonstrated the superiority of risankizumab [16]. Although there is insufficient evidence to support the superiority of risankizumab over ustekinumab for the treatment of CD, the phase 2 guselkumab GALAXI-1 study included a relatively small ustekinumab treatment arm. Although there was no statistical comparison, the proportion of patients achieving various outcomes at week 12 was similar between the pooled guselkumab treatment groups and patients treated with ustekinumab (clinical remission: 53.0% versus 46.0%; PRO2 remission: 42.7% versus 39.7%; endoscopic response: 35.7% versus 28.6%; and clinical-biomarker response: 47.0% versus 46.0%) [20]. Similar rates of clinical and PRO2 remission at week 48 of the GALAXI-1 study were also recently reported for guselkumab and ustekinumab [62]. Risankizumab has been directly compared with ustekinumab for the treatment of moderate-to-severe plaque psoriasis. The ongoing phase 3, randomized, head-to-head SEQUENCE trial (NCT04524611) comparing risankizumab and ustekinumab in patients with moderate-to-severe CD who have failed prior treatment with a TNF antagonist (primary endpoints: clinical remission (CDAI < 150) at week 24, endoscopic remission (SES-CD ≤ 4 and at least a 2-point reduction from baseline and no subscore > 1) at week 48, adverse events up to 220 weeks) will provide more definitive data regarding the comparative efficacy of targeting IL-23p19 and IL-12/23p40.
6.3 Drugs Targeting IL-23 as First- and Second-Line Agents
6.3.1 Crohn’s Disease
As described earlier, data from phase 3 trials of risankizumab and phase 2 trials of mirikizumab and guselkumab demonstrate that these agents are effective and safe for inducing and maintaining clinical and endoscopic remission in bio-naïve and bio-experienced patients with moderate-to-severe CD. A subgroup analysis of the ADVANCE, MOTIVATE, and FORTIFY trials found that risankizumab was effective regardless of the number of prior failed biologics [89]. In a recently updated network meta-analysis by Barberio et al. that pooled data from 25 CD induction trials including more than 8700 patients, infliximab 5 mg/kg was ranked highest for induction of clinical remission compared with other agents. However, analyses based upon biologic exposure ranked risankizumab (600 mg) highest for induction of clinical remission in patients both naïve and with prior exposure to biologics [90]. It should be noted that comparing outcomes across IBD trials is challenging as the definitions of endoscopic and clinical outcomes vary among studies. Furthermore, eligibility criteria for clinical trials involving patients with IBD have significantly evolved over the years, with more recent trials enrolling more refractory patients. For example, approximately 40% of patients enrolled in the risankizumab phase 3 trials had previously failed two or more biologics [22, 23]. Therefore, anti-IL23p19 agents may be considered as first-line therapy or second-line therapy when previous biologic treatment has failed.
6.3.2 Ulcerative Colitis
Phase 3 trial data on the efficacy and safety of anti-IL-23p19 agents in UC are limited. Available evidence suggests that anti-IL-23p19 agents are safe and effective in both biologic-naïve and biologic-experienced patients with UC. In the VEGA trial, there was no significant difference between guselkumab and golimumab in anti-TNF-naïve individuals with UC. Therefore, anti-IL-23p19 agents may be considered as first-line or second-line therapy. However, it is premature to make a firm recommendation given the lack of data.
7 Conclusions
Recent scientific discoveries have revealed additional biologic pathways and molecules important to the immunopathogenesis of IBD with potential utility for drug development, including IL-23. Targeting IL-23 was associated with significant therapeutic benefits in clinical trials involving patients with IBD and has led to the approval of ustekinumab (an IL-12/23 antagonist) for the treatment of both CD and UC, and risankizumab (a selective IL-23p19 antagonist), for the treatment of moderate-to-severe CD. Additional agents selectively targeting IL-23p19 are also in clinical development for the treatment of IBD. The availability of multiple classes of pharmacotherapies with different mechanisms of actions may confound therapeutic decision-making, and appropriate positioning of these agents in treatment algorithms for IBD will only be possible based on data from well-designed head-to-head trials. These algorithms may be expected to further evolve with the development of newer drugs including oral IL-23 agents and oral small molecules targeting TYKs.
References
Knowles SR, Graff LA, Wilding H, Hewitt C, Keefer L, Mikocka-Walus A. Quality of life in inflammatory bowel disease: a systematic review and meta-analyses-part I. Inflamm Bowel Dis. 2018;24:742–51.
Knowles SR, Keefer L, Wilding H, Hewitt C, Graff LA, Mikocka-Walus A. Quality of life in inflammatory bowel disease: a systematic review and meta-analyses-part II. Inflamm Bowel Dis. 2018;24:966–76.
Burisch J, Jess T, Martinato M, Lakatos PL. The burden of inflammatory bowel disease in Europe. J Crohns Colitis. 2013;7:322–37.
Kappelman MD, Rifas-Shiman SL, Porter CQ, Ollendorf DA, Sandler RS, Galanko JA, et al. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135:1907–13.
Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769–78.
The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5:17–30.
Alsoud D, Verstockt B, Fiocchi C, Vermeire S. Breaking the therapeutic ceiling in drug development in ulcerative colitis. Lancet Gastroenterol Hepatol. 2021;6:589–95.
Kayal M, Ungaro RC, Bader G, Colombel JF, Sandborn WJ, Stalgis C. Net remission rates with biologic treatment in Crohn's disease: a reappraisal of the clinical trial data. Clin Gastroenterol Hepatol. 2023;21:1348-1350.
Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–9.
Gisbert JP, Marín AC, McNicholl AG, Chaparro M. Systematic review with meta-analysis: the efficacy of a second anti-TNF in patients with inflammatory bowel disease whose previous anti-TNF treatment has failed. Aliment Pharmacol Ther. 2015;41:613–23.
Singh S, Murad MH, Fumery M, Sedano R, Jairath V, Panaccione R, et al. Comparative efficacy and safety of biologic therapies for moderate-to-severe Crohn’s disease: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2021;6:1002–14.
Schmitt H, Billmeier U, Dieterich W, Rath T, Sonnewald S, Reid S, et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut. 2019;68:814–28.
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3.
Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2016;375:1946–60.
Sands BE, Sandborn WJ, Panaccione R, O’Brien CD, Zhang H, Johanns J, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381:1201–14.
Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551–60.
Feagan BG, Sandborn WJ, D’Haens G, Panés J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389:1699–709.
Sands BE, Chen J, Feagan BG, Penney M, Rees WA, Danese S, et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe Crohn’s disease: a phase 2a study. Gastroenterology. 2017;153:77–86.
Sands BE, Peyrin-Biroulet L, Kierkus J, Higgins PDR, Fischer M, Jairath V, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with Crohn’s disease. Gastroenterology. 2022;162:495–508.
Sandborn WJ, D’Haens GR, Reinisch W, Panés J, Chan D, Gonzalez S, et al. Guselkumab for the treatment of Crohn’s disease: induction results from the phase 2 GALAXI-1 study. Gastroenterology. 2022;162:1650–64.
D'Haens G, Rubin DT, Panes J, Gonzalez S, Chan D, Johanns J, et al. The effect of guselkumab induction therapy on endoscopic outcome measures in patients with moderately to severely active Crohn's disease: week 12 results from the phase 2 GALAXI 1 study. Gastroenteorlogy. 2021;160:S-91.
D’Haens G, Panaccione R, Baert F, Bossuyt P, Colombel JF, Danese S, et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. 2022;399:2015–30.
Ferrante M, Panaccione R, Baert F, Bossuyt P, Colombel JF, Danese S, et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet. 2022;399:2031–46.
Sandborn WJ, Ferrante M, Bhandari BR, Berliba E, Feagan BG, Hibi T, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with ulcerative colitis. Gastroenterology. 2020;158:537–49.
D’Haens G, Kobayashi T, Morris N, Lissoos T, Hoover A, Li X, et al. Efficacy and safety of mirikizumab as induction therapy in patients with moderately to severely active ulcerative colitis: results from the phase 3 LUCENT-1 study. United Eur Gastroenterol J. 2022;10:710–1.
NCT03524092. A maintenance study of mirikizumab in participants with moderately to severely active ulcerative colitis (LUCENT 2). https://clinicaltrials.gov/ct2/show/NCT03524092. Accessed 1 Dec 2022.
Dignass A, Rubin D, Bressler B, Huang KH, Shipitofsky N, Germinaro M, et al. The efficacy and safety of guselkumab induction therapy in patients with moderately to severely active ulcerative colitis: phase 2b QUASAR study results through week 12. J Crohns Colitis. 2022;16:i025-i26.
Sands BE, Feagan BG, Sandborn WJ, Shipitofsky N, Marko M, Sheng S, et al. OP36 efficacy and safety of combination induction therapy with guselkumab and golimumab in participants with moderately-to-severely active ulcerative colitis: results through week 12 of a phase 2a randomized, double-blind, active-controlled, parallel-group, multicenter, proof-of-concept study. J Crohns Colitis. 2022;16:i042-i43.
Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med. 1989;170:827–45.
Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13:722–8.
Pirhonen J, Matikainen S, Julkunen I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J Immunol. 2002;169:5673–8.
Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–52.
Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–9.
Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25.
Lupardus PJ, Garcia KC. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J Mol Biol. 2008;382:931–41.
Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–8.
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69.
Jouanguy E, Döffinger R, Dupuis S, Pallier A, Altare F, Casanova JL. IL-12 and IFN-gamma in host defense against mycobacteria and salmonella in mice and men. Curr Opin Immunol. 1999;11:346–51.
Sun R, Abraham C. IL23 promotes antimicrobial pathways in human macrophages, which are reduced with the IBD-protective IL23R R381Q variant. Cell Mol Gastroenterol Hepatol. 2020;10:673–97.
Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–708.
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33.
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40.
Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21:719–29.
Becker C, Wirtz S, Blessing M, Pirhonen J, Strand D, Bechthold O, et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J Clin Investig. 2003;112:693–706.
Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, et al. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J Immunol. 2002;168:1804–12.
Zundler S, Neurath MF. Interleukin-12: functional activities and implications for disease. Cytokine Growth Factor Rev. 2015;26:559–68.
Segal BM, Dwyer BK, Shevach EM. An interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J Exp Med. 1998;187:537–46.
McIntyre KW, Shuster DJ, Gillooly KM, Warrier RR, Connaughton SE, Hall LB, et al. Reduced incidence and severity of collagen-induced arthritis in interleukin-12-deficient mice. Eur J Immunol. 1996;26:2933–8.
Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–90.
Fuss IJ, Marth T, Neurath MF, Pearlstein GR, Jain A, Strober W. Anti-interleukin 12 treatment regulates apoptosis of Th1 T cells in experimental colitis in mice. Gastroenterology. 1999;117:1078–88.
Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Investig. 2006;116:1310–6.
Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18.
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8.
Karaboga İ, Demirtas S, Karaca T. Investigation of the relationship between the Th17/IL-23 pathway and innate-adaptive immune system in TNBS-induced colitis in rats. Iran J Basic Med Sci. 2017;20:870–9.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62.
Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. 2015;43:739–50.
Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–700.
Targan SR, Feagan B, Vermeire S, Panaccione R, Melmed GY, Landers C, et al. A randomized, double-blind, placebo-controlled phase 2 study of brodalumab in patients with moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2016;111:1599–607.
Aden K, Rehman A, Falk-Paulsen M, Secher T, Kuiper J, Tran F, et al. Epithelial IL-23R signaling licenses protective IL-22 responses in intestinal inflammation. Cell Rep. 2016;16:2208–18.
Cox JH, Kljavin NM, Ota N, Leonard J, Roose-Girma M, Diehl L, et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 2012;5:99–109.
Pai R, De Hertogh G, Reinisch W, Harpaz N, Feagan B, Agada N, et al. Impact of mirikizumab therapy on histologic measures of intestinal inflammation in a phase 2 study of patients with moderately to severely active Crohn’s disease. J Crohns Colitis. 2021;15:S404–6.
Danese S, Panaccione R, Rubin DT, Sands BE, Reinisch W, D’Haens G, et al. Clinical efficacy and safety of guselkumab maintenance therapy in patients with moderately to severely active Crohn’s disease: week 48 analyses from the phase 2 GALAXI 1 study. J Crohns Colitis. 2022;16:i026-i27.
D’Haens GR, Panaccione R, Panés J, Hisamatsu T, Bossuyt P, Danese S, et al. 52-weeks risankizumab subcutaneous maintenance dosing Is efficacious and well tolerated in patients with moderate to severe Crohn’s disease who had delayed response to 12-weeks IV risankizumab induction. United Eur Gastroenterol J. 2022;10:98–9.
Sandborn WJ, Ferrante M, Bhandari BR, Berliba E, Hibi T, D’Haens GR, et al. Efficacy and safety of continued treatment with mirikizumab in a phase 2 trial of patients with ulcerative colitis. Clin Gastroenterol Hepatol. 2022;20:105–15.
Efficacy and safety of mirikizumab as induction therapy in patients with moderately to severely active ulcerative colitis: results from the phase 3 LUCENT-1 study. Gastroenterol Hepatol (N Y). 2022;18:7–8.
Magro F, Pai RK, Kobayashi T, Jairath V, Rieder F, Redondo I, et al. Efficacy of mirikizumab in resolving active histologic inflammation in ulcerative colitis in LUCENT-1 induction and LUCENT-2 maintenance trials. United Eur Gastroenterol J. 2022;10:335–6.
Dignass A, Danese S, Matsuoka K, Ferrante M, Long M, Redondo I, et al. Sustained symptom control with mirikizumab in patients with moderately to severely active ulcerative colitis in the LUCENT-2 maintenance trial. United Eur Gastroenterol J. 2022;10:331.
Panés J, Allegretti J, Sands BE, Huang K-HG, Kavalam M, Germinaro M, et al. The effect of guselkumab induction therapy in patients with moderately to severely active ulcerative colitis: QUASAR phase 2b induction results at week 12 by prior inadequate response or intolerance to advanced therapy. United Eur Gastroenterol J. 2022;10:87–8
Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–61.
MacLennan C, Fieschi C, Lammas DA, Picard C, Dorman SE, Sanal O, et al. Interleukin (IL)-12 and IL-23 are key cytokines for immunity against Salmonella in humans. J Infect Dis. 2004;190:1755–7.
Ouederni M, Sanal O, Ikinciogullari A, Tezcan I, Dogu F, Sologuren I, et al. Clinical features of Candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis. 2014;58:204–13.
Sandborn WJ, Feagan BG, Danese S, O’Brien CD, Ott E, Marano C, et al. Safety of ustekinumab in inflammatory bowel disease: pooled safety analysis of results from phase 2/3 studies. Inflamm Bowel Dis. 2021;27:994–1007.
Papp KA, Griffiths CE, Gordon K, Lebwohl M, Szapary PO, Wasfi Y, et al. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: final results from 5 years of follow-up. Br J Dermatol. 2013;168:844–54.
Chackerian AA, Chen SJ, Brodie SJ, Mattson JD, McClanahan TK, Kastelein RA, et al. Neutralization or absence of the interleukin-23 pathway does not compromise immunity to mycobacterial infection. Infect Immun. 2006;74:6092–9.
Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43:727–38.
Singh S, Singh S, Thangaswamy A, Thangaraju P, Varthya SB. Efficacy and safety of Risankizumab in moderate to severe psoriasis: a systematic review and meta-analysis. Dermatol Ther. 2021;34: e14487.
Bai F, Li GG, Liu Q, Niu X, Li R, Ma H. Short-term efficacy and safety of IL-17, IL-12/23, and IL-23 inhibitors brodalumab, secukinumab, ixekizumab, ustekinumab, guselkumab, tildrakizumab, and risankizumab for the treatment of moderate to severe plaque psoriasis: a systematic review and network meta-analysis of randomized controlled trials. J Immunol Res. 2019;2019:2546161.
Östör A, Van den Bosch F, Papp K, Asnal C, Blanco R, Aelion J, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 52-week results from the KEEPsAKE 2 study. Rheumatology (Oxford). 2022 (e-pub ahead of print).
Gordon KB, Lebwohl M, Papp KA, Bachelez H, Wu JJ, Langley RG, et al. Long-term safety of risankizumab from 17 clinical trials in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2022;186:466–75.
Blauvelt A, Tsai TF, Langley RG, Miller M, Shen YK, You Y, et al. Consistent safety profile with up to 5 years of continuous treatment with guselkumab: pooled analyses from the phase 3 VOYAGE 1 and VOYAGE 2 trials of patients with moderate-to-severe psoriasis. J Am Acad Dermatol. 2022;86:827–34.
Ferrante M, Feagan BG, Panés J, Baert F, Louis E, Dewit O, et al. Long-term safety and efficacy of risankizumab treatment in patients with Crohn’s disease: results from the phase 2 open-label extension study. J Crohns Colitis. 2021;15:2001–10.
Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405–17.
Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76:418–31.
Reich K, Gooderham M, Thaçi D, Crowley JJ, Ryan C, Krueger JG, et al. Risankizumab compared with adalimumab in patients with moderate-to-severe plaque psoriasis (IMMvent): a randomised, double-blind, active-comparator-controlled phase 3 trial. Lancet. 2019;394:576–86.
Sands BE, Irving PM, Hoops T, Izanec JL, Gao LL, Gasink C, et al. Ustekinumab versus adalimumab for induction and maintenance therapy in biologic-naive patients with moderately to severely active Crohn’s disease: a multicentre, randomised, double-blind, parallel-group, phase 3b trial. Lancet. 2022;399:2200–11.
Norden A, Moon JY, Javadi SS, Munawar L, Maul JT, Wu JJ. Anti-drug antibodies of IL-23 inhibitors for psoriasis: a systematic review. J Eur Acad Dermatol Venereol. 2022;36:1171–7.
Bonovas S, Fiorino G, Allocca M, Lytras T, Nikolopoulos GK, Peyrin-Biroulet L, et al. Biologic therapies and risk of infection and malignancy in patients with inflammatory bowel disease: a systematic review and network meta-analysis. Clin Gastroenterol Hepatol. 2016;14:1385–97.
Chupin A, Perduca V, Meyer A, Bellanger C, Carbonnel F, Dong C. Systematic review with meta-analysis: comparative risk of lymphoma with anti-tumour necrosis factor agents and/or thiopurines in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2020;52:1289–97.
Ferrante M, Peyrin-Biroulet L, Dignass A, Rubin DT, Danese S, D’Haens GR, et al. Clinical and endoscopic improvements with risankizumab induction and maintenance dosing versus placebo are observed irrespective of number of prior failed biologics. United Eur Gastroenterol J. 2022;10:101–2.
Barberio B, Gracie DJ, Black CJ, Ford AC. Efficacy of biological therapies and small molecules in induction and maintenance of remission in luminal Crohn’s disease: systematic review and network meta-analysis. Gut. 2023;72:264–74.
Acknowledgements
We would like to thank Alexa Zayadi for helping to prepare the figure presented in this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
None.
Conflict of interest
SKV is an employee of Alimentiv Inc. LMS has received consulting fees from Alimentiv Inc. JH has received speaker’s fees from Abbvie, Janssen, and Takeda, and consulting fees from Alimentiv Inc. CM has received consulting fees from AbbVie, Alimentiv, Amgen, AVIR Pharma Inc, BioJAMP, Bristol Myers Squibb, Celltrion, Ferring, Fresenius Kabi, Janssen, McKesson, Mylan, Pendopharm, Pfizer, Prometheus Biosciences Inc., Roche, Sanofi, Takeda, Tillotts Pharma; speaker's fees from AbbVie, Amgen, AVIR Pharma Inc, Alimentiv, Bristol Myers Squibb, Ferring, Fresenius Kabi, Janssen, Organon, Pendopharm, Pfizer, Takeda; royalties from Springer Publishing; research support from Ferring, Pfizer. VJ has received consulting/advisory board fees from AbbVie, Alimentiv Inc (formerly Robarts Clinical Trials), Arena pharmaceuticals, Asieris, Bristol Myers Squibb, Celltrion, Eli Lilly, Ferring, Fresenius Kabi, Galapagos, GlaxoSmithKline, Genetech, Gilead, Janssen, Merck, Mylan, Pandion, Pendopharm, Pfizer, Reistone Biopharma, Roche, Sandoz, Takeda, Topivert; and speaker’s fees from Abbvie, Ferring, Janssen Pfizer Shire, and Takeda. BGF has received grant/research support from Millennium Pharmaceuticals, Merck, Tillotts Pharma AG, AbbVie, Novartis Pharmaceuticals, Centocor Inc., Elan/Biogen, UCB Pharma, Bristol-Myers Squibb, Genentech, ActoGenix, and Wyeth Pharmaceuticals Inc.; consulting fees from Millennium Pharmaceuticals, Merck, Centocor Inc., Elan/Biogen, Janssen-Ortho, Teva Pharmaceuticals, Bristol-Myers Squibb, Celgene, UCB Pharma, AbbVie, Astra Zeneca, Serono, Genentech, Tillotts Pharma AG, Unity Pharmaceuticals, Albireo Pharma, Given Imaging Inc., Salix Pharmaceuticals, Novonordisk, GSK, Actogenix, Prometheus Therapeutics and Diagnostics, Athersys, Axcan, Gilead, Pfizer, Shire, Wyeth, Zealand Pharma, Zyngenia, GiCare Pharma Inc., and Sigmoid Pharma; and speakers bureaux fees from UCB, AbbVie, and J&J/Janssen. Alimentiv Inc. is an academic gastrointestinal contract research organization (CRO), operating under the Alimentiv Health Trust. Alimentiv Inc. provides comprehensive clinical trial services, precision medicine offerings, and centralized imaging solutions for endoscopy, histopathology, and other imaging modalities. The beneficiaries of the Alimentiv Health Trust are the employees of the enterprises it holds. LMS, JH, CM, VJ, and BGF are consultants to Alimentiv Inc.; they do not hold equity positions or shares in Alimentiv Inc. JH, CM, VJ, and BGF have a primary academic appointment.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors contributions
VJ and BGF supervised the work. All authors drafted and critically advised the manuscript for important intellectual content, and approved the final submitted version of the work.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vuyyuru, S.K., Shackelton, L.M., Hanzel, J. et al. Targeting IL-23 for IBD: Rationale and Progress to Date. Drugs 83, 873–891 (2023). https://doi.org/10.1007/s40265-023-01882-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-023-01882-9