
Abstract
Up to 25% of patients with metastatic prostate cancer present with germline or somatic DNA damage repair alterations, some of which are associated with aggressive disease and poor outcomes. New data have brought poly(ADP-ribose) polymerase (PARP) inhibitors into sharp focus in the treatment of metastatic castrate-resistant prostate cancer (mCRPC). Olaparib improved survival after at least one new hormonal therapy (NHT) in a cohort of patients harboring BRCA1, BRCA2 or ATM mutations in the PROfound trial, while rucaparib, talazoparib and niraparib demonstrated compelling activity in phase II trials. While patients with prostate cancer and BRCA1 or BRCA2 mutations may derive greatest benefit of PARP inhibition, the magnitude of benefit seems much lower in the context of most other homologous recombination gene mutations. Several PARP inhibitors are currently developed in combination with conventional therapy, including chemotherapy, NHT, and alpha-particle emitters, at different disease stages. Herein, we review the rationale for PARP inhibition in patients with prostate cancer, discuss the impact of PARP inhibitors on outcomes, and explore underlying challenges for future developments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Poly(ADP-ribose) polymerase (PARP) inhibitors demonstrated improvement in survival in metastatic castration-resistant prostate cancer with homologous recombination repair alterations. |
Multiple PARP inhibitors are now in development as single-agent or combination therapies at multiple prostate cancer stages. |
Assessing the optimal screening procedures, patient selection, treatment sequences, and overcoming resistances to PARP inhibitors are new challenges lying ahead. |
1 Introduction
Prostate adenocarcinoma is one of the most frequent types of cancer worldwide, accounting for nearly 400,000 deaths each year [1]. While constant progress is achieved with regard to overall survival (OS), this disease remains incurable in the metastatic setting, and the onset of castration resistance marks a turning point in cancer evolution [2]. Prostate cancer also features a rising incidence in younger males, associated with adverse outcomes and resistance to therapy [3].
Therapeutic strategies in the metastatic castrate-resistant prostate cancer (mCRPC) setting rely mostly on cytotoxic agents, in the form of docetaxel and cabazitaxel [4, 5], as well as inhibitors of the androgen axis, including androgen receptor inhibitors such as enzalutamide [6] or androgen biosynthesis inhibitors such as abiraterone acetate [7]. Despite advances in molecular characterization of prostate cancer and identification of adverse molecular alterations [8], molecular selection of patients and targeted molecular therapies had not been routinely available until recently.
This paradigm is currently shifting with the advent of poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi), which target prostate adenocarcinoma harboring alterations in DNA damage repair (DDR) pathways and demonstrate potent antitumor activity in advanced settings, now also entering the arena in earlier, hormone-sensitive stages. Herein, we discuss the biological rationale behind PARP inhibition in prostate adenocarcinoma, the projected impact of PARPi on outcomes of selected patients, and challenges associated with targeted strategies in a shifting treatment landscape.
2 Rationale for Poly(ADP-Ribose) Polymerase (PARP) Inhibition in Advanced Prostate Cancer
2.1 DNA Damage Repair (DDR), PARP Inhibition and Synthetic Lethality
Genomic instability is an essential hallmark of cancer, as cancer cells acquire genomic abnormalities leading to selective advantages for cell growth and survival. Maintaining cellular genomic integrity involves sophisticated DDR mechanisms that detect and correct DNA damage and ultimately prevent cell death [9]. These pathways of sensors of genetic damage and replication stress, transducers of signal and effectors of DNA repair are intricate and interacting and involve a large number of genes [10]. Their classification is functional, based on genetic and mechanistic criteria [11], as genes involved in each pathway encode for proteins that function collaboratively to repair specific types of DNA damage [12].
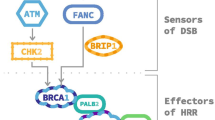
Various genotoxic factors induce DNA damage by lethal double-strand breaks (DSBs) or single-strand breaks (SSBs), which may alter cells through a cumulative effect. The DDR system for DSBs involves two main mechanisms—the homologous recombination repair (HRR) and the non-homologous end-joining (NHEJ) system. The HRR is a high-fidelity pathway that uses DNA repair damage proteins, such as BRCA1, BRCA2, PALB2 and RAD51, to excise the damaged DNA sequence and synthesize a novel homologous DNA sequence using the normal sister chromatid as a template. The NHEJ pathway differs in that it directly binds the ends of a DSB together as a quick-fix mechanism throughout the cell cycle, without using a guidance template, which is more prone to errors and could be mutagenic by itself. Repair mechanisms for SSBs involve three main mechanisms: the base excision repair (BER), which corrects damaged single bases or nucleotides, including oxidative lesions and alkylation products; the nucleotide excision repair (NER), which corrects bulky, helix-distorting damage of nucleotide sequences; and the mismatch repair (MMR) pathway. Similar to homologous recombination, both BER and NER involve excision of the damaged base or sequence and replacement with newly synthetized DNA. The MMR pathway corrects base mismatches, erroneous insertions/deletions or small loops often found in repetitive sequences of DNA. Should lesions persist despite the above corrective mechanisms, the cell utilizes DNA polymerases in a DNA damage tolerance process called translesion synthesis (TLS), which allows replication past such lesions, to ensure genome replication and cell survival, allowing potential increase in mutagenesis. Alterations of these DDR pathways by mutations, chromosomal deletions or epigenetic silencing may impact the ability of cells to repair DNA damage, and ultimately lead to acquisition of adverse cancer features and resistance to therapy [13].
The PARP1 and 2 enzymes play an essential role in the BER pathway, while PARP1 is also involved in NHEJ [14]. PARP enzymes detect and bind to SSBs and act through poly-ADP-ribosylation (PARsylation) of a series of proteins to promote DNA repair. PARP inhibitors allow persistence and accumulation of SSBs, either through inhibition of PARP activity or by trapping of PARP at the DNA binding site [15, 16]. Data stemming from pharmacology studies suggest that median inhibitory concentration (IC50) for PARP1, PARP2, and trapping activity are independent and that both participate in PARPi activity [17, 18].
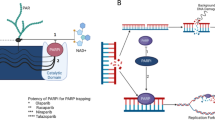
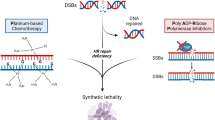
Both mechanisms of action of PARPi may lead to the development of lethal DSBs by hampering the replication fork. In cells with normal HRR, the resultant DSB may be successfully repaired, as opposed to cells with defective HRR, which will eventually undergo apoptosis (Fig. 1) and may thus be used as a therapeutic strategy in cancers harboring HRR alterations. This concept has been labeled as synthetic lethality, and its proof of concept has been first demonstrated in ovarian cancers. In these tumor types known for recurrent HRR alterations, including germline alterations of BRCA1 or BRCA2 in up to 15% of cases, the PARPi olaparib demonstrated improved outcomes using maintenance strategies after chemotherapy [19]. Several PARPi have been developed, harboring distinct IC50 for PARP1 and PARP2, as well as distinct trapping activity, with a clinical relevance that is still to be investigated [18]. Translation of these concepts in other tumor types harboring HRR alterations, such as prostate cancer, have been explored since with successful developments.

Adapted from Sonnenblick et al. [27]
Mechanism of action of PARP inhibitors. Functional PARP repair DNA single-strand DNA break occurring following genomic instability. PARPi may trap or inhibit PARP activity, inducing replication fork collapse and double-strand breaks. In patients with homologous recombination defects, double-strand breaks cannot be repaired, leading to cancer cell death. PARP poly(ADP-ribose) polymerase, PARPi PARP inhibitors, HRR homologous recombination repair.
2.2 DDR Alterations in Prostate Cancer
The prevalence of germline and somatic DDR mutations ranges from 19% up to 30% in molecular studies of advanced prostate cancer [20,21,22,23,24]. The most frequently altered gene, consistently across reports, is BRCA2. Additional data from whole genome sequencing on metastatic tissue biopsies from 197 patients with mCRPC revealed that BRCA2 inactivation was biallelic in 25/32 tumors (78%), while of the remaining 7 without biallelic activation, 4 had at least one deleterious aberration in other HRR-related genes [25]. Biallelic BRCA2 inactivation even reached 90% of BRCA2-mutated patients in an independent cohort of 150 mCRPCs [20]. Additional cohorts showed varying results with liquid biopsy assessments that identified 7.8% of men with biallelic alterations in ATM, BRCA1, BRCA2, BRIP1, CHEK2, FANCA, HDAC2, and PALB2 genes in the GALAHAD trial [21].
Initial reports were conflicting in regard to the discrepancy in the frequency of DDR alterations between primary and metastatic disease. While a higher frequency of DDR alterations has consistently been reported in metastatic samples compared with primary tumors, this can be mitigated by the fact that primary tumors were not matched to metastatic samples and that the presence of DDR alteration may mostly reflect a more aggressive natural history [20, 23]. The largest DDR screening in mCRPC focused on 15 HRR genes and was performed within the PROfound study through sequencing of tissue biopsy, and identified 778 (28%) mutated patients from a total of 2792. The most frequent alterations were loss-of-function alterations of BRCA2 (9.7%), followed by CDK12 (7.1%) and ATM (6.3%), while other alterations, including BRCA1, CHEK2, PPP2R2A, PALB2, BRIP1, RAD54L, BARD1, RAD51B, RAD51D, CHEK1, FANCL and RAD51C had a lower prevalence [21]. The PROfound study, as it focused exclusively on mCRPC patients, provided clarification on the previously reported discrepancies between primary tumors and metastatic disease. It reported a similar frequency in primary tumors (27%) and in biopsy samples from metastatic sites (32%), suggesting that HRR alterations are early features of this subset of more aggressive tumors that will eventually give rise to metastatic disease. This is indeed consistent with previous reports, indicating worse outcomes for patients with HRR alterations and, notably, BRCA2 alterations [20, 26].
Germline mutations alone account for nearly half of all DDR alterations [22, 27], in 7.5–13% of metastatic patients, irrespective of family history of cancer or age at diagnosis, with the most frequently altered genes being BRCA2, ATM, CHEK2 and BRCA1 [20,21,22,23, 28,29,30]. Although the frequency is higher in cases of family history of cancer or early-onset disease, germline DDR alterations may occur in prostate cancer independently of these [28, 31]. In 60–70% of cases, the second allele is defective by the acquisition of either a second loss-of-function mutation or a gene-copy loss [27, 28, 30]. Some DDR genes were shown to be preferentially affected by germline alterations (e.g., BRCA1/2, CHEK2, FANCM, PALB2), whereas others (e.g., ATM, BAP1, CDK12) were preferentially acquired as somatic events [21, 22, 32]. Several of these genes are reportedly involved in the acquisition of castration resistance. Notably, ATM as well as FANCA alterations are enriched in mCRPC compared with metastatic castration-sensitive prostate cancer (mCSPC) [22, 31]. Alterations in CDK12, an indirect regulator of HRR [33], have been repeatedly reported in aggressive tumors with complex androgen receptor rearrangements [34, 35].
Germline DDR alteration carriers have a higher risk to develop prostate cancer compared with non-carriers, with the highest risk for gBRCA2 (2.64-fold) [36]. Overall, DDR-positive (DDR+) patients seem to have a more aggressive disease, with higher Gleason scores associated with worse clinical outcomes [28, 30]. Patients with BRCA1/2 and ATM mutations have a shorter time to death after diagnosis of prostate cancer, and death occurs at an earlier age than non-mutated patients. Furthermore, BRCA1/2, ATM and CHEK2 c.1100delC germline mutations are more frequently encountered in lethal prostate cancer than localized cancer patients [37, 38]. Patients with DDR-altered tumors might have a higher risk of developing visceral metastases, while BRCA mutations are associated with a higher risk of nodal involvement and metastases at diagnosis [39]. Indeed, time to progression to mCRPC from the start of androgen deprivation therapy in DDR-mutated patients was only 11.8 months in a population with both synchronous and asynchronous metastatic presentation, compared with 19 months in non-germline mutated patients, consistent with a short interval of castration sensitivity [29]. However, OS from the castration-resistance phase has been reported to be similar, regardless of the DDR status: 3 years for DDR-, 3 years for BRCA2- and 3.2 years for non-DDR-mutated patients, demonstrating a maintained activity of conventional therapies, including taxanes and new hormonal therapies (NHT), regardless of Gleason score or age at diagnosis [40].
3 Clinical Activity of PARP Inhibitors in Prostate Cancer
3.1 Proof of Concept of PARP Inhibition in Patients with Metastatic Castration-Resistant Prostate Cancer
The TOPARP A phase II trial evaluated olaparib in mCRPC men in both DDR+ and DDR-negative (DDR−) patients, and paved the way toward precision medicine in prostate cancer by proving for the first time that DDR alterations were predictive for response to olaparib. Patients were considered to be biomarker-positive in tissue if homozygous deletions, deleterious mutations, or both were detected in DNA repair genes (but not single-copy deletions without events detected in the second allele) [41]. TheTOPARP-B phase II trial, evaluating olaparib in DDR+ only mCRPC patients, showed a median radiographic progression-free survival (rPFS) of 8.3 months and an objective response rate (ORR) of 52% [42].
Other PARPi, including niraparib, talazoparib, and rucaparib, underwent investigation in phase II trials in patients who had not responded to prior taxane therapy and NHT, which demonstrated promising preliminary results (Table 1) [43,44,45]. Together, these studies show that PARP inhibitors may achieve up to 50% of ORR as well as a median rPFS up to 11 months in patients with BRCA1 or BRCA2 mutations.
3.2 Olaparib as a New Standard of Care in Metastatic, Castration-Resistant Prostate Cancer
The most clinically relevant data to date were provided from the phase III PROfound study of olaparib versus NHT in patients who already received one previous NHT and whose tumors harbored DDR alterations. Olaparib showed a reduction in the risk of progression or death in comparison with abiraterone or enzalutamide in mCRPC patients previously treated with NHT and harboring BRCA2, BRCA1 or ATM alterations (cohort A), achieving a median rPFS of 7.4 versus 3.6 months (hazard ratio [HR] 0.34) [46]. The ORR was also improved with olaparib in cohort A patients, at 33% versus 2%. Longer follow-up confirmed the activity of olaparib in this setting with a clear improvement in OS, from 14.7 months with NHT to 19.1 months with olaparib. The reduction of the risk of death was 31% and rose up to 58% when accounting for the crossover (81% of patients within the control arm) [47], with a magnitude of benefit that seemed to favor patients who did receive prior taxanes. Olaparib overall had a manageable safety profile: anemia was the most frequent adverse event (46%), including grade 3 or 4 in 26%, followed by asthenia and nausea in 41% (all grades). Dose reduction and discontinuation occurred respectively in 22% and 18% of patients [46].
However, these practice-changing data seemed not to apply to patients who harbored alterations in 12 other HRR prespecified genes (cohort B). While benefit in rPFS was still reported when accounting for both cohorts A and B (5.8 months vs. 3.5 months; HR 0.49) [46], the magnitude of benefit appeared lesser in cohort B, with no significant benefit in OS even when accounting for crossover [47]. Of note, post hoc subgroup analyses within cohort A also demonstrated that patients who present with ATM mutations may not derive similar benefit compared with patients harboring BRCA1 or BRCA2 mutations, although instances of individual benefit have been reported [42, 44,45,46, 48,49,50].
Following the results of the PROfound trial, olaparib received European Medicines Agency (EMA) approval, which was restricted for tumors showing BRCA1/2 alterations based on the cohort A results. US FDA approval was obtained for men with mCRPC who progressed after at least one NHT and harboring any HRR alterations, except for PPP2R2A mutations.
4 Optimizing the Potential of PARP Inhibition in Prostate Cancer
4.1 A Differential Benefit of PARP Inhibitors According to Genomic Alterations
Parsing the data from trials of PARPi in monotherapy, it becomes clear that all studies demonstrate compelling activity of PARPi in germline or somatic BRCA-mutated patients (Table 1). However, not all DDR mutations seem to be equal in terms of response to PARPi across trials. A recent exploratory analysis of the PROfound trial reported on the most frequently found alterations (BRCA1/2, ATM and CDK12) and confirmed superior olaparib efficacy for the BRCA1/2 tumors, with a 5.7-month absolute OS benefit, a 6.8-month absolute rPFS benefit, and a 43.9% ORR [48]. However, analyzing patients with ATM alterations within cohort A, PARPi did not provide such compelling activity compared with the standard treatment arm, with a reported median rPFS of 5.4 versus 4.7 months, with similar ORR (10%) for both the experimental and control arms [48]. The notion of limited activity of PARPi in patients with ATM alterations has been corroborated by other PARPi studies showing modest ORR and prostate-specific antigen (PSA) responses of < 15% (Table 1), supporting further exploration of combinations exploiting synthetic lethality in this setting [51]. Further data are also needed to clarify the activity of PARPi in BRCA1+ patients as only eight BRCA1+ patients were treated with olaparib in the PROfound trial, although subgroup analyses of OS tend to show a trend for benefit similar to BRCA2+ patients [46, 52].
The predictive value of other DDR genes is less clear. Patients with CDK12 alterations, which appear to be the most frequent DDR alteration after BRCA2, only rarely achieved objective responses with PARPi, with median rPFS consistently < 6 months and no clear trend for benefit in OS based on the PROfound trial [42, 52]. Still, while OS data for PROfound showed limited efficacy of olaparib in patients with non-BRCA mutations, prolonged survival has been reported in individual patients with ATM, CDK12, and other HRR gene alterations such as RAD54L and CHEK2 [48]. Similarly, an ad hoc analysis of patients with deleterious alterations in non-BRCA DDR genes included in the TRITON-2 trial showed a limited number of radiographic and PSA responses with niraparib and ATM, CDK12 or CHEK2 alterations, as well as with DDR genes less frequently altered in mCRPC, such as PALB2, BRIP1, FANCA, and RAD51B [49]. In the GALAHAD study of niraparib, of 35 patients having non-BRCA1/2 biallelic alterations, only two patients had objective responses and both harbored a FANCA alteration [50]. Overall, these results stem from small populations, and the relevance of PARPi in patients with non-BRCA DDR mutations remains to be defined, thanks to larger cohorts and a better understanding of determinants of response to PARPi (Table 1).
It is also unknown whether additional non-DDR gene alterations may impact response to PARPi in patients with known DDR alterations. Exploratory analyses performed in the TALAPRO-1 trial showed that additional alterations in TP53, PTEN, AR or MYC, associated with adverse outcomes in historical series [53], did not impact the prognosis of patients treated with talazoparib for mCRPC [54]. Additional studies will be needed to assess gene-specific genotype/phenotype correlations in this population.
4.2 PARP Inhibitors as Potential Candidates for Combinations?
4.2.1 Combinations of PARP Inhibitors and New Hormonal Therapy
Combinations of PARPi and NHT may harbor synergistic effects, with hints at synthetic lethality, as inhibition of androgen receptor signaling is associated with lower HRR gene expression [55]. The phase I trial of enzalutamide plus rucaparib (RAMP) yielded interesting results, as four of eight patients exhibited confirmed PSA response >50% despite being heavily pretreated and without HRR alterations [56]. In a randomized phase II trial, the combination of abiraterone plus olaparib in patients pretreated with docetaxel for mCRPC demonstrated a benefit of nearly 6 months for rPFS over abiraterone (13.8 vs. 8.2 months; HR 0.65) in the intent-to-treat population (with or without HRR alterations), albeit similar response rates were observed in both groups (27% vs. 32%) [57]. However, this combination was also associated with numerically higher rates of adverse events, including nausea, fatigue, decreased appetite, pyrexia, and cytopenia; up to 54% of grade 3–5 adverse events were reported with abiraterone plus olaparib, compared with 28% with abiraterone plus placebo [57]. Discordant results emerged from another phase II randomized trial assessing veliparib plus abiraterone versus abiraterone, which failed to demonstrate any improvement in antitumor response and PFS in the same setting [58].
More robust data have now emerged from the randomized phase III MAGNITUDE and PROpel trials, evaluating the combination of abiraterone plus niraparib and abiraterone plus olaparib, respectively, versus abiraterone plus placebo (Table 2). Both trials had radiographic PFS as their primary endpoint but differed in their patient population: the MAGNITUDE trial included two distinct cohorts of patients, with or without HRR alterations (HRR+/−), including specific assessment of patients with BRCA mutations. The PROpel trial included all-comers with HRR status assessed retrospectively. The MAGNITUDE trial demonstrated improved rPFS in HRR+ patients, with an HR of 0.73 in all HRR+ patients and 0.53 in BRCA+ patients [59]; however, outcomes were not improved in HRR− patients. Nonethless, the PROpel trial demonstrated rPFS benefit in the entire patient population, with an HR of 0.66 [60]. Interestingly, benefit in the PROpel trial was sustained across subgroups in HRR+ (HR 0.50) and HRR− (HR 0.76) patients. These combinations yielded increased treatment-related adverse events, at 50% and 70% of grade 3–4 in the PROpel and MAGNITUDE trials, respectively. Most common adverse events regardless of severity included fatigue, gastrointestinal disorders, and cytopenia. Anemia was the most common grade 3/4 event in both trials (15% and 30% in PROpel and MAGNITUDE, respectively), while the PROpel trial reported numerically increased rates of thromboembolic events [59, 60].
The first reports of these trials tend to support the use of abiraterone in combination with PARPi in mCRPC in selected populations, but raise several questions as benefit in HRR− patients differ between trials. Subgroup analyses assessing individual gene mutations will likely be useful to better identify patients who will benefit from these combinations, which seem to be especially relevant in HRR+ and, notably, BRCA+ patients. It is also unknown whether respective pharmacologic properties of niraparib and olaparib come into play regarding activity across subgroups. Overall, longer follow-up as well as mature OS data will be needed to confirm the role of these combinations in the early mCRPC setting.
4.2.2 Other Developments of PARP Inhibitor Combinations
PARPi are investigated in combination with agents that directly impact DSB formation or DDR pathways. Both PARPi and ionizing radiation promote DSBs, suggesting that PARPi may act as radiosensitizers [61]. Additional data support the fact that DDR defects may improve response to radiation-based therapy. Notably, the assessment of radium-223 activity in patients with or without DDR mutations demonstrated that DDR alterations were associated with improved OS (36.3 vs. 17.0 months) [62]. While these results are encouraging, they remain retrospective and based on a small number of patients, prompting the need for larger, prospective assessments. Current developments involve radium-223 plus niraparib, a combination regimen that showed preliminary activity in a phase I trial recruiting chemo-naïve mCRPC patients [63], while evaluation of PARPi and LU177-PSMA is ongoing in patients with unselected mCRPC (NCT03874884).
Other potential combinations include PARPi and immune checkpoint inhibitors, as PARPi may increase genomic instability and, as a consequence, promote immunogenicity of tumor cells [64]. To date, the combination of pembrolizumab plus olaparib only led to a confirmed ORR of 7% in an unselected population [65], with phase III trial data pending (NCT03834519). Similarly, the combination of nivolumab plus rucaparib demonstrated an ORR of only 16% in unselected patients, but subgroup analysis demonstrated that patients with homologous recombination deficiency (HRD) (ORR 25%) and, most importantly, BRCA1/2 alterations (ORR 33%) derived greater benefit to that regimen [66]. Study of PSA response in this small phase II trial (n = 66) corroborates these data, with a proportion of patients with PSA decline ≥50% of 27% in unselected patients, 42% in HRD+ patients, and 85% in BRCA1/2 patients [66]. The combination of durvalumab plus olaparib is also investigated in selected patients with biochemically recurrent prostate cancer (NCT04336943, NCT03810105). However, it remains unclear whether men with BRCA1 and BRCA2 alterations really derive greater benefit from immune checkpoint inhibitors.
Tumor angiogenesis may also impact the activity of PARPi, considering that hypoxic conditions are reported to impair HRR gene expression [67, 68]. In a phase II trial including men with mCRPC, olaparib associated with the angiogenesis inhibitor cediranib increased rPFS over olaparib alone [69]. This benefit has been seemingly driven by patients with HRR alterations, albeit the small number of patients and short follow-up invite for further confirmation [70].
5 Refining Therapeutic Strategies in Patients with DDR Alterations
5.1 Activity of Other Antitumor Agents in Patients Harboring DDR Defects
5.1.1 New Hormonal Therapies and Taxanes in Patients with DDR Defects
Understanding the activity of conventional therapies in patients harboring DDR mutations may inform therapeutic strategies in the era of PARPi. To date, the magnitude of benefit of conventional therapies and optimal treatment sequence have not been completely established, with conflicting results for first-line NHT or docetaxel in heterogeneous populations (Table 3). A large-scale retrospective effort has shown that patients have similar outcomes in terms of PSA response, ORR, PFS, and cancer-specific survival (CSS) when treated with NHT or docetaxel at castration resistance onset [40], including for BRCA2 carriers, which contrasts with other reports identifying BRCA2 mutations as an adverse prognostic factor compared with patients harboring other DDR mutations [31]. Conflicting results were obtained by Annala et al., who showed a dismal biochemical PFS of only 3.3 months for all patients harboring DDR mutations, in a population enriched for patients with poor prognosis and harboring a high tumor burden [29]. Some data suggest that patients eligible for upfront NHT in the mCRPC setting and harboring BRCA2 mutations may derive compelling benefit to therapy compared with non BRCA2-mutated patients, but the small number of patients studied in these studies precludes any robust analysis of optimal treatment sequence in this setting [29, 30].
Exploring further lines of therapy, cabazitaxel seems to offer sustained activity with similar activity irrespective of DDR alterations. In a large, international, retrospective study including DDR+ patients and matched DDR− controls, response rates (32% and 36%, respectively) and rPFS (5.3 and 5.7 months, respectively) were similar [71]. However, in 10 patients with BRCA1/2 alterations previously treated with PARPi, no biochemical response was observed with cabazitaxel. While this may challenge the role of cabazitaxel after PARPi in selected patients, these findings must be confirmed in larger prospective cohorts.
Most of these studies were retrospective in nature and included a limited number of patients with DDR alterations. In addition, most focused on germline DDR mutations, while somatic mutations may be as frequent and may also impact outcomes. These data highlight the need for molecular-based trials and widespread integration of molecular testing, which could improve therapeutic sequences in line with individual molecular pictures.
5.1.2 Platinum-Based Therapy in Patients with DDR Defects
Platinum-based compounds commonly induce DSBs, and as such have been reported to be active in several BRCA-mutated tumor subtypes, including ovarian and breast cancer [72, 73]. While activity of carboplatin is limited in prostate cancer and did not demonstrate improvement in survival, more recent data show increased activity in patients with mCRPC and DDR defects [74, 75]. Two retrospective studies demonstrated biochemical responses (PSA decline of 50% or more) in up to 50% of patients with mCRPC and DDR defects, and up to 75% in patients with BRCA mutations, with a twofold increase in survival compared with non-BRCA carriers [74, 75]. These data have been reported before the advent of PARPi in prostate cancer and activity of platinum compounds after PARPi is still unkown. Assessment of carboplatin versus PARPi in mCRPC is ongoing in a phase II randomized trial (NCT04038502).
5.2 PARP Inhibition in Early Prostate Cancer Settings
Multiple trials are now evaluating PARPi in earlier prostate cancer stages (Table 4). Olaparib without androgen deprivation therapy led to PSA response >50% in 15% of patients with biochemically recurrent prostate cancer, with a PSA doubling time <6 months, and unselected for molecular alterations [76]. Among these, two had complete PSA response and harbored BRCA2 mutations. This proof-of-concept trial suggests that PARPi may be active even in the absence of androgen deprivation therapy in selected patients. Other trials are now investigating PARPi alone in patients with DDR alterations, either in the metastatic castration-sensitive (NCT03413995) or biochemically recurrent setting (NCT03533946).
5.3 Sequencing PARP Inhibition in a Moving Treatment Landscape
Development of PARPi is now thriving in multiple disease stages (Tables 2, 3, 4), in a landscape that has shown numerous advancement over the past few years. Triplets including docetaxel and next-generation hormonal therapies on top of castration are challenging the standard of care in the castration-sensitive setting [77]. In the castration-resistance setting, new targeted compounds include 177LU-PSMA-617-targeted radiation therapy [78], while other innovative compounds such as bispecific T-cell engagers are in development [79]. To date, olaparib is approved after at least one NHT, but it is yet unclear what will be the optimal sequence for patients with HRR alterations among these novel therapeutic options. This question will be all the more important for patients with non-BRCA mutations, for which olaparib showed limited clinical benefit in the PROfound phase III trial compared with BRCA alterations carriers. As trials evaluating treatment sequences may be difficult to undertake in a dynamic landscape, more real-life data could help better define the future role of PARPi in patients with prostate cancer.
6 Improving Biomarker-Based Patient Selection
6.1 Assessment of DDR Alterations
Technical aspects of molecular testing are important to understand as some limitations may impact DDR assessment and adequate therapeutic orientation. Assessment of HRR alterations may differ between circulating tumor DNA and tissue assessments: only 81% of positive percentage agreement for BRCA or ATM mutations in the PROfound trial [80, 81], while this percentage rose to 93% in patients who underwent prescreening for TRITON-2/3 trials [24]. Molecular assays also do not always indicate whether DDR gene alterations are mono- or bi-allelic, which may impact the functional consequences of the alteration and thus response to therapy. Data from the TRITON-2 trial indicate that more PSA responses were observed with rucaparib in patients with biallelic DDR gene alterations [45, 82]. Other hurdles may lie in the detection of DDR gene alterations that do not stem from prostate cancer but from clonal hematopoiesis, which may account for up to 10% of patients tested for circulating tumor DNA (ctDNA) [83]. Tissue assessments (although not always easy to perform in men with bone metastases) could overcome some of these hurdles by avoiding clonal hematopoiesis detection or by providing the possibility of assessing the functional impact of these mutations on the HRR pathway, a strategy that has been in use for ovarian cancer [84]. Refinements in ctDNA analyses are still urgently needed though, as tissue availability may be an issue in patients with bone-only disease and as repeated assessments may be needed to detect acquired DDR gene alterations. It is thus likely that tissue and liquid biopsy assessments are bound to co-exist in the near future, prompting more streamlined strategies to assess DDR alterations in patients with prostate cancer.
As PARPi are now standard of care in patients with mCRPC, molecular testing should now be part of routine clinical practice in this population. Approvals may however differ regarding both the PARPi indication as well as the molecular companion test. For instance, olaparib is approved in HRR-mutated patients in the US, but is only approved in the context of BRCA1 or BRCA2 mutations in the European Union. Regarding molecular assessments, the FDA only approved somatic HRR alterations testing on tissue, while the EMA allows BRCA testing including somatic alteration on tissue or blood. These differences may affect current integration of molecular testing and therapeutic strategies on a per region basis. However, it is likely that these aspects will evolve as more data emerge on testing performance and novel indications for PARPi in prostate cancer arise.
6.2 Understanding and Overcoming Mechanisms of Resistance to PARP Inhibitors
All patients with mCRPC and DDR alterations will ultimately experience disease progression on PARPi, with mechanisms of resistance that are gradually uncovered. Acquired mutation in PARP genes have been described and may alter the trapping ability of PARPi [85]. Other resistance mechanisms may rely on the alteration of negative regulators of HRR [86] or genes involved in the degradation of the replication fork in the event of a DSB [87]. Several acquired alterations in HRR may also restore its functionality, such as demethylation of a silenced BRCA gene, intragenic mutations in an altered BRCA gene that may produce new functional BRCA isoforms, or restoration of the open reading frame [88,89,90]. Such mutations, called reversion mutations, have been reported in prostate cancer patients previously treated with PARPi or platinum-based compounds [91, 92]. These may be acquired in up to 40% of treated patients and could represent a key resistance mechanism for both PARPi and platinum-based compounds [91].
Strategies to overcome resistance to PARPi in prostate cancer may rely on concurrent inhibition of other DDR pathways, pushing forward the concept of synthetic lethality. ATR is a DDR protein that can act as a sensor of both SSBs and DSBs, and, as such, one promising target for such strategies [93]. ATR inhibition has been reported to be synergistic with PARPi in platinum-resistant ovarian cancer models [94], and PARPi and ATR inhibitors are now evaluated in both HRR-proficient and HRR-deficient mCRPC (NCT03787680, NCT03682289) [95].
7 Conclusion
The era of precision medicine in prostate cancer opens with the advent of PARPi in patients with mCRPC harboring DDR alterations. While single-agent data have been compelling enough to warrant approval of PARPi after previous NHT, their evaluation in combination for earlier settings has shown compelling activity. Optimal timing of PARPi use remains unknown to date, and several ongoing phase III trials are likely to further refine therapeutic strategies using PARPi. Routine clinical practice ought to now include genetic testing for all prostate cancer patients entering castration resistance. Determining optimal techniques to assess DDR status, bringing biomarker-driven strategies upfront, and assessing sensitivity profiles to PARPi based on molecular profiling are now the new challenges for clinical and translational studies in patients with advanced prostate cancer.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Global cancer statistics, et al. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2020;71(3):209–49.
Rebello RJ, Oing C, Knudsen KE, Loeb S, Johnson DC, Reiter RE, et al. Prostate cancer. Nat Rev Dis Primers. 2021;7:1–27.
Salinas CA, Tsodikov A, Ishak-Howard M, Cooney KA. Prostate cancer in young men: an important clinical entity. Nat Rev Urol. 2014;11:317–23.
Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12.
de Wit R, de Bono J, Sternberg CN, Fizazi K, Tombal B, Wülfing C, et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med. 2019;381:2506–18.
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–33.
Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48.
Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45.
Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16:35–42.
Friedberg EC. DNA damage and repair. Nature. 2003;421:436–40.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23:239-254.e6.
Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108:3406–11.
Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94.
Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20:698–714.
D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018;71:172–6.
Carney B, Kossatz S, Lok BH, Schneeberger V, Gangangari KK, Pillarsetty NVK, et al. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat Commun. 2018;9:176.
Moore K, Colombo N, Scambia G, Kim B-G, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379:2495–505.
Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28.
de Bono JS, Fizazi K, Saad F, Shore N, Sandhu SK, Mehra N, et al. Central, prospective detection of homologous recombination repair gene mutations (HRRm) in tumour tissue from >4000 men with metastatic castration-resistant prostate cancer (mCRPC) screened for the PROfound study. Ann Oncol. 2019;30:v328–9.
Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. 2017;2017.
Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018;50:645–51.
Tukachinsky H, Madison RW, Chung JH, Gjoerup OV, Severson EA, Dennis L, et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin Cancer Res. 2021;27:3094–105.
van Dessel LF, van Riet J, Smits M, Zhu Y, Hamberg P, van der Heijden MS, et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat Commun. 2019;10:5251.
Castro E, Goh C, Leongamornlert D, Saunders E, Tymrakiewicz M, Dadaev T, et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localised prostate cancer. Eur Urol. 2015;68:186–93.
Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571:576–9.
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53.
Annala M, Struss WJ, Warner EW, Beja K, Vandekerkhove G, Wong A, et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair–deficient prostate cancer. Eur Urol. 2017;72:34–42.
Antonarakis ES, Lu C, Luber B, Liang C, Wang H, Chen Y, et al. Germline DNA-repair gene mutations and outcomes in men with metastatic castration-resistant prostate cancer receiving first-line abiraterone and enzalutamide. Eur Urol. 2018;74:218–25.
Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, et al. PROREPAIR-B: a prospective cohort study of the impact of Germline DNA repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2019;37:490–503.
Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, et al. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8:857.
Dubbury SJ, Boutz PL, Sharp PA. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature. 2018;564:141–5.
Antonarakis ES, Isaacsson Velho P, Fu W, Wang H, Agarwal N, Santos VS, et al. CDK12-altered prostate cancer: clinical features and therapeutic outcomes to standard systemic therapies, poly (ADP-Ribose) polymerase inhibitors, and PD-1 inhibitors. JCO Precis Oncol. 2020;4:370–81.
Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. 2018;174:433-447.e19.
Oh M, Alkhushaym N, Fallatah S, Althagafi A, Aljadeed R, Alsowaida Y, et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: a meta-analysis. Prostate. 2019;79:880–95.
Wu Y, Yu H, Zheng SL, Na R, Mamawala M, Landis T, et al. A comprehensive evaluation of CHEK2 germline mutations in men with prostate cancer. Prostate. 2018;78:607–15.
Na R, Zheng SL, Han M, Yu H, Jiang D, Shah S, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71:740–7.
Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–57.
Mateo J, Cheng HH, Beltran H, Dolling D, Xu W, Pritchard CC, et al. Clinical outcome of prostate cancer patients with germline DNA repair mutations: retrospective analysis from an international study. Eur Urol. 2018;73:687–93.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–74.
Smith MR, Fizazi K, Sandhu SK, Kelly WK, Efstathiou E, Lara P, et al. Niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD): correlative measures of tumor response in phase II GALAHAD study. J Clin Oncol. 2020;38:118–118.
de Bono JS, Mehra N, Scagliotti GV, Castro E, Dorff T, Stirling A, et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol. 2021;22:1250–64.
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38:3763–72.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–102.
Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with olaparib in metastatic castration-resistant prostate cancer. N Engl J Med. 2020;383:2345–57.
De Bono JS, Matsubara N, Penel N, Mehra N, Kolinsky MP, Bompas E, et al. Exploratory gene-by-gene analysis of olaparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): PROfound. J Clin Oncol. 2021;39:126–126.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene Alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26:2487–96.
Smith MR, Sandhu SK, Kelly WK, Scher HI, Efstathiou E, Lara PN, et al. Pre-specified interim analysis of GALAHAD: A phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD). Ann Oncol. 2019;30:v884–5.
Reichert ZR, Daignault S, Teply BA, Devitt ME, Heath EI. Targeting resistant prostate cancer with ATR and PARP inhibition (TRAP trial): a phase II study. J Clin Oncol. 2020;38:TPS254–TPS254.
de Bono JS, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. 610O Final overall survival (OS) analysis of PROfound: Olaparib vs physician’s choice of enzalutamide or abiraterone in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and homologous recombination repair (HRR) gene alterations. Ann Oncol. 2020;31:S508.
Hamid AA, Gray KP, Shaw G, MacConaill LE, Evan C, Bernard B, et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur Urol. 2019;76:89–97.
Mehra N, de Bono J, Laird AD, Barthélémy P, Delva R, Dorff T, et al. 580P TALAPRO-1: Talazoparib (TALA) monotherapy in metastatic castration-resistant prostate cancer (mCRPC) with DNA damage response alterations (DDRm)—exploration of non-DDR mutational landscape and potential associations with antitumor activity. Ann Oncol. 2021;32:S630–1.
Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8:374.
Rao A, Ryan CJ, Morris D, et al. Genomic characteristics and response to rucaparib and enzalutamide in the phase 1b RAMP study of metastatic castration-resistant prostate cancer (mCRPC) patients [abstract 445]. Cancer Res. 2021;81(13 Suppl):445.
Clarke N, Wiechno P, Alekseev B, Sala N, Jones R, Kocak I, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19:975–86.
Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results From NCI 9012. J Clin Oncol. 2018;36:991–9.
Chi KN, Rathkopf DE, Smith MR, Efstathiou E, Attard G, Olmos D, et al. Phase 3 MAGNITUDE study: first results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol. 2022;40:12–12.
Saad F, Armstrong AJ, Thiery-Vuillemin A, Oya M, Loredo E, Procopio G, et al. PROpel: Phase III trial of olaparib (ola) and abiraterone (abi) versus placebo (pbo) and abi as first-line (1L) therapy for patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2022;40:11.
Laird JH, Lok BH, Ma J, Bell A, de Stanchina E, Poirier JT, et al. Talazoparib is a potent radiosensitizer in small cell lung cancer cell lines and xenografts. Clin Cancer Res. 2018;24:5143–52.
van der Doelen MJ, Velho PI, Slootbeek PHJ, Naga SP, Bormann M, van Helvert S, et al. Impact of DNA damage repair defects on response to radium-223 and overall survival in metastatic castration-resistant prostate cancer. Eur J Cancer. 2020;136:16–24.
Kelly WK, Leiby B, Einstein DJ, Szmulewitz RZ, Sartor AO, Yang ES-H, et al. Radium-223 (Rad) and niraparib (Nira) treatment (tx) in castrate-resistant prostate cancer (CRPC) patients (pts) with and without prior chemotherapy (chemo). J Clin Oncol. 2020;38:5540–5540.
Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64.
Yu E, Piulats JM, Gravis G, Fong PCC, Todenhöfer T, Laguerre B, et al. 612P Pembrolizumab (pembro) plus olaparib in patients with docetaxel-pretreated metastatic castration-resistant prostate cancer (mCRPC): Update of KEYNOTE-365 cohort A with a minimum of 11 months of follow-up for all patients. Ann Oncol. 2021;32:S652–3.
Petrylak DP, Perez-Gracia JL, Lacombe L, Bastos DA, Mahammedi H, Kwan EM, et al. 579MO CheckMate 9KD cohort A2 final analysis: Nivolumab (NIVO) + rucaparib for chemotherapy (CT)-naïve metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol. 2021;32:S629–30.
Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Science Translational Medicine. 2019;11(492):eaav4508.
Liu JF, Barry WT, Birrer M, Lee J-M, Buckanovich RJ, Fleming GF, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann Oncol. 2019;30:551–7.
Kim JW, McKay RR, Taplin M-E, Davis NB, Monk P, Appleman LJ, et al. Randomized phase II study of olaparib with or without cediranib in men with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2020;38:111–111.
McKay RR, Radke MR, Shyr Y, Zhao S, Taplin M-E, Davis NB, et al. Biomarker analysis from a randomized phase II study of olaparib with or without cediranib in men with metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2021;39:7–7.
Aldea M, Lam L, Orillard E, Llacer Perez C, Saint-Ghislain M, Gravis G, et al. Cabazitaxel activity in men with metastatic castration-resistant prostate cancer with and without DNA damage repair defects. Eur J Cancer. 2021;159:87–97.
Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med. 2018;24:628–37.
Mirza MR, Coleman RL, González-Martín A, Moore KN, Colombo N, Ray-Coquard I, et al. The forefront of ovarian cancer therapy: update on PARP inhibitors. Ann Oncol. 2020;31:1148–59.
Pomerantz MM, Spisák S, Jia L, Cronin AM, Csabai I, Ledet E, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer. 2017;123:3532–9.
Schmid S, Omlin A, Higano C, Sweeney C, Martinez Chanza N, Mehra N, et al. Activity of platinum-based chemotherapy in patients with advanced prostate cancer with and without DNA Repair gene aberrations. JAMA Network Open. 2020;3:e2021692.
Antonarakis ES, Wang H, Teply BA, Kelly WK, Willms J, Sullivan R, et al. Interim results from a phase 2 study of olaparib (without ADT) in men with biochemically-recurrent prostate cancer after prostatectomy, with integrated biomarker analysis. J Clin Oncol. 2019;37:5045–5045.
Fizazi K, Carles Galceran J, Foulon S, Roubaud G, McDermott R, Fléchon A, et al. LBA5 A phase III trial with a 2x2 factorial design in men with de novo metastatic castration-sensitive prostate cancer: Overall survival with abiraterone acetate plus prednisone in PEACE-1. Ann Oncol. 2021;32:S1299.
Sartor O, de Bono J, Chi KN, Fizazi K, Herrmann K, Rahbar K, et al. Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med. 2021;385:1091–103.
Tran B, Horvath L, Dorff T, Rettig M, Lolkema MP, Machiels J-P, et al. 609O Results from a phase I study of AMG 160, a half-life extended (HLE), PSMA-targeted, bispecific T-cell engager (BiTE®) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol. 2020;31:S507.
Matsubara N, De Bono JS, Olmos D, Procopio G, Kawakami S, Urun Y, et al. Olaparib efficacy in patients with metastatic castration-resistant prostate cancer (mCRPC) carrying circulating tumor (ct) DNA alterations in BRCA1, BRCA2 or ATM: results from the PROfound study. J Clin Oncol. 2021;39:27–27.
Chi KN, Barnicle A, Sibilla C, Lai Z, Corcoran C, Williams JA, et al. Concordance of BRCA1, BRCA2 (BRCA), and ATM mutations identified in matched tumor tissue and circulating tumor DNA (ctDNA) in men with metastatic castration-resistant prostate cancer (mCRPC) screened in the PROfound study. J Clin Oncol. 2021;39:26–26.
Loehr A, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Response to rucaparib in BRCA-mutant metastatic castration-resistant prostate cancer identified by genomic testing in the TRITON2 study. Clin Cancer Res. 2021;27(24):6677–86.
Jensen K, Konnick EQ, Schweizer MT, Sokolova AO, Grivas P, Cheng HH, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol. 2021;7:107–10.
Ngoi NYL, Tan DSP. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: do we need it? ESMO Open. 2021;6:100144.
Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9:1849.
Lee EK, Matulonis UA. PARP inhibitor resistance mechanisms and implications for post-progression combination therapies. Cancers (Basel). 2020;12(8):2054.
Kharat SS, Ding X, Swaminathan D, Suresh A, Singh M, Sengodan SK, et al. Degradation of 5hmC-marked stalled replication forks by APE1 causes genomic instability. Sci Signal. 2020;13:eaba8091.
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5.
Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 2017;7:999–1005.
Sorrells S, McKinnon KE, McBratney A, Sumey C. Longitudinal and multi-tissue molecular diagnostics track somatic BRCA2 reversion mutations that correct the open reading frame of germline alteration upon clinical relapse. NPJ Genom Med. 2021;6(1):17.
Carneiro BA, Collier KA, Nagy RJ, Pamarthy S, Sagar V, Fairclough S, et al. Acquired resistance to poly (ADP-ribose) polymerase inhibitor olaparib in BRCA2-associated prostate cancer resulting from biallelic BRCA2 reversion mutations restores both germline and somatic loss-of-function mutations. JCO Precis Oncol. 2018;2.
Cheng HH, Salipante SJ, Nelson PS, Montgomery B, Pritchard CC. Polyclonal BRCA2 reversion mutations detected in circulating tumor DNA after platinum chemotherapy in a patient with metastatic prostate cancer. JCO Precis Oncol. 2018;2.
Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8.
Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. 2020;11:3726.
Abida W, Sanay E, Lukashchuk N, Pierce A, de Graaf W, Lau A, et al. PLANETTE: A modular phase IIa multicenter open-label study evaluating the ATR inhibitor ceralasertib (AZD6738) in ATM mutant advanced solid tumors. J Clin Oncol. 2021;39:TPS189.
Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. An update on PAARP inhibitors: moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12(1):27–41.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflicts of Interest
Ronan Flippot has received honoraria from Janssen, Ipsen, Bristol-Myers Squibb, MSD, and Pfizer, and has received travel expenses from Merck. Anna Patrikidou has received consulting fees from Basilea. Mihaela Aldea has received travel expenses from Sandoz. Emeline Colomba is a member of the Advisory Boards of Bristol-Myers Squibb, Ipsen, Sanofi, Glaxo-Smith Kline, MSD, Pfizer, Clovis, Novartis, and Eisai. Pernelle Lavaud has received travel expenses from Astellas, Ipsen, Janssen, and Mundipharma. Laurence Albigès has received consulting fees from Amgen, Astellas, AstraZeneca, Bristol-Myers Squibb, Corvus Pharmaceuticals, Exelixis, Ipsen, Janssen, Merck, MSD, Novartis, Peloton Therapeutics, Pfizer, and Roche; research funding from Bristol-Myers Squibb; and travel expenses from Bristol-Myers Squibb and MSD. Bernard Escudier has received honoraria from Bristol-Myers Squibb, EUSA Pharma, Ipsen, Novartis, Oncorena, Pfizer, and Roche/Genentech; consulting or advisory role fees from AVEO, Bristol-Myers Squibb, EUSA Pharma, Ipsen, Novartis, Pfizer, and Roche/Genentech; research funding from Bristol-Myers Squibb; and travel, accommodations and expenses from Bristol-Myers Squibb, Ipsen, MSD, Pfizer, and Roche/Genentech. Yohann Loriot has received honoraria from Pfizer and Sanofi; consulting or advisory role fees from Astellas Pharma, AstraZeneca, Bristol-Myers Squibb, Clovis Oncology, Janssen, MSD Oncology, Roche, Seattle Genetics; research funding from AstraZeneca, Boehringer Ingelheim, Clovis Oncology, CureVac, Exelixis, Incyte, Janssen Oncology, Medivation, MSD Oncology, Nektar, Oncogenex, Pfizer, and Sanofi; and travel, accommodations and expenses from Astellas Pharma, AstraZeneca, Janssen Oncology, MSD Oncology, Roche, and Seattle Genetics. Giulia Baciarello has received honoraria from Astellas Pharma, Janssen Oncology, Roche, and Sanofi; consulting or advisory role fees from Astellas Pharma, Europharma, Janssen Oncology, Modra Pharmaceuticals, Roche, Sanofi, Simon-Kucher and Partners; and travel, accommodations and expenses from Amgen, Astellas Pharma, AstraZeneca, Ipsen, Janssen Oncology, and Sanofi. Karim Fizazi has received advisory fees from Amgen, Astellas, Astrazeneca, Bayer, Clovis, Janssen, MSD, Novartis/AAA, Sanofi, CureVac, and Orion. Natacha Naoun, Pierre Blanchard, Mario Terlizzi, Camilo Garcia, Alice Bernard-Tessier, Alina Fuerea, and Mario Di Palma have no conflicts of interest to declare.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors' contributions
Data collection: Mihaela Aldea, Anna Patrikidou, Giulia Baciarello, Ronan Flippot. Data analysis, redaction, proofing: all authors.
Rights and permissions
About this article

Cite this article
Flippot, R., Patrikidou, A., Aldea, M. et al. PARP Inhibition, a New Therapeutic Avenue in Patients with Prostate Cancer. Drugs 82, 719–733 (2022). https://doi.org/10.1007/s40265-022-01703-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01703-5