
Abstract
Over the past 2 decades, rapid advances in molecular profiling and the development of targeted therapies have dramatically improved the clinical course of advanced non-small-cell lung cancer (NSCLC). Mutations in the epidermal growth factor receptor (EGFR) gene are found in about a third of patients with advanced NSCLC, and the approval of first-generation EGFR targeted kinase inhibitors significantly improved survival when compared with platinum-based doublet chemotherapy (PBC), the previous standard of care. Inevitably, selective pressure from first-generation EGFR inhibitors led to acquired resistance mechanisms, such as the T790M mutation. The advent of third-generation EGFR inhibitors (e.g., osimertinib) successfully overcame the T790M resistance mechanism, and osimertinib subsequently became the first-line therapy for EGFR mutant NSCLC. Currently, research in EGFR mutant NSCLC is primarily focused on targeting resistance mechanisms to osimertinib. Over the past several years, many important acquired and intrinsic mechanisms of resistance to osimertinib have been identified. Acquired resistance mechanisms include C797X, mesenchymal epithelial transition factor (MET) amplification, HER2/HER3 amplification, phosphoinositide 3-kinase (PI3K) pathway mutations, RAS/mitogen-activated protein kinase (MAPK) pathway mutations, cell–cycle gene alterations, oncogenic fusions, and histologic transformations. An important intrinsic resistance mechanism to osimertinib is the EGFR exon 20 insertion mutation, which is sensitive to the newly Food and Drug Administration (FDA)-approved tyrosine kinase inhibitor mobocertinib and the EGFR/MET bispecific antibody amivantamab. This review article aims to (1) summarize the advances in the treatment of EGFR mutant NSCLC, (2) delineate known resistance mechanisms to the current first-line therapy, osimertinib, and (3) describe the development of targeted drugs that aim to overcome these resistance mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Osimertinib, a third-generation epidermal growth factor receptor (EGFR) inhibitor, is the standard of care for advanced EGFR mutant non-small-cell lung cancer in the first-line setting. |
Identified mechanisms of resistance to osimertinib can be classified as EGFR-dependent versus EGFR-independent and acquired versus intrinsic. |
The EGFR exon 20 insertion mutation is more sensitive to novel tyrosine kinase inhibitor and antibody therapies than osimertinib or the earlier generation EGFR inhibitors. |
Several targeted therapies aiming to overcome resistance mechanisms to osimertinib are currently in development or being tested in clinical trials. |
1 Background
The clinical course of advanced non-small-cell lung cancer (NSCLC) has swiftly evolved over the past 20 years. Platinum-based doublet chemotherapy (PBC) was the standard of care for all patients with advanced NSCLC and a good performance status. However, PBC yielded disappointing results: an objective response rate (ORR) of about 30%, median progression-free survival (PFS) of 5–6 months, and median overall survival (OS) of 11–12 months [1, 2]. Fortunately, improvements in molecular profiling and the approval of various targeted therapies have drastically improved the prognosis for patients with targetable mutations [3,4,5,6].
The detection of oncogenic driver mutations in the epidermal growth factor receptor (EGFR) gene was a pivotal milestone in the diagnosis and treatment of NSCLC [7]. EGFR is a receptor tyrosine kinase that is commonly expressed in normal tissue and participates in cellular pathways leading to cell proliferation, migration, and survival. Activating mutations affecting the kinase domain of EGFR lead to ligand-independent downstream signaling of EGFR, thereby promoting cancer growth. Such mutations occur in up to half of patients with NSCLC, with the peak incidence in East-Asian, non-smoking, and female patients [8].
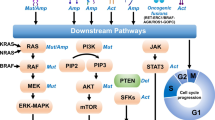
The classical EGFR L858R point mutation and exon 19 deletions comprise up to 90% of EGFR mutant NSCLC and lead to conformational changes that destabilize the dormant form of the EGFR protein, effectively shifting the equilibrium towards the active form [9, 10]. This conformational change in the adenosine triphosphate (ATP) pocket of EGFR is the target of first- and second-generation tyrosine kinase inhibitors (TKIs) [7]. EGFR TKIs bind the ATP pocket of EGFR, leading to inhibition of kinase phosphorylation and downstream pathways. The approval of first-generation reversible EGFR TKIs (e.g., gefitinib, erlotinib) for advanced and metastatic EGFR mutant NSCLC dramatically improved ORR as high as 80% and median PFS to greater than 10 months, exceeding that observed from PBC [1, 2]. The success of these agents was pivotal in transforming the management of NSCLC from a histology-based approach to a personalized, targeted approach. However, despite such advancements in molecular profiling and targeted therapeutics, selective pressure on the cancer cells inevitably led to drug resistance and disease progression [11]. While some of this was overcome in the landmark FLAURA trial with the T790M-active inhibitor osimertinib, up to 10% of EGFR mutant NSCLC had de novo or primary resistance either due to different EGFR mutations, such as EGFR exon 20 insertion (ex20ins), or poorly understood initial tumor biology [6]. This article will review the tumor biology of resistance (Fig. 1) and current research to overcome this common clinical challenge (Table 1).

Approximate distribution of resistance mechanisms to first-line osimertinib. EGFR epidermal growth factor receptor, ex20ins exon 20 insertion, MAPK mitogen-activated protein kinase, MET mesenchymal epithelial transition factor, PI3K phosphoinositide 3-kinase
2 Acquired Resistance: EGFR-Dependent Mechanisms
2.1 T790M
Soon after the advent of first-generation TKIs, a somatic mutation in EGFR, p.Thr790Met (T790M), was discovered. T790M alters a residue situated deep inside the ATP pocket of EGFR and thereby blocks the binding of first- and second-generation TKIs to the ATP-binding site [12]. Therefore, the presence of the T790M mutation prior to treatment with first- and second-generation TKIs led to intrinsic resistance and therefore poorer outcomes [13]. Additionally, 50–60% of patients who initially responded to first- or second-generation TKIs ultimately developed T790M mutations, leading to acquired resistance to therapy [14, 15].
The development of third-generation TKIs, particularly osimertinib, was essential in overcoming this resistance mechanism. Osimertinib covalently bonds with the C797 residue of the ATP-binding site of EGFR regardless of the T790M mutation. Osimertinib was initially approved for treatment of EGFR mutant, T790M-positive NSCLC after progression on first-line TKI. Subsequently, the FLAURA trial in 2018 showed that osimertinib led to increased PFS (19 vs. 10 months) when compared to gefitinib and erlotinib in the first-line setting for advanced EGFR mutant NSCLC [6]. As osimertinib improved the median OS for advanced EGFR mutant NSCLC to 38.6 months and demonstrated a favorable toxicity profile compared to earlier generation TKIs, it was approved as first-line therapy for all advanced EGFR mutant NSCLC regardless of T790M status. [6, 16]
Notably, osimertinib also demonstrated improved efficacy against central nervous system (CNS) disease, which is present in about 30% of EGFR mutant NSCLC at diagnosis [16]. First- and second-generation EGFR TKIs yielded variable activity against brain metastases [17,18,19], whereas in the FLAURA trial, front-line osimertinib demonstrated a PFS benefit in patients with CNS disease [6, 20]. In the phase I BLOOM study, 160 mg of osimertinib daily, which is double the normal dose, yielded an ORR of 62% and a median OS of 11 months in patients with leptomeningeal disease [21].
Recently, another third-generation TKI, lazertinib, also demonstrated a favorable safety profile and anticancer activity in a phase I/II trial of EGFR mutant, T790M-positive NSCLC [22], and there are other agents with similar activity in development worldwide. Because of these advances, T790M as a resistance mechanism has become less clinically relevant. When resistance develops to first-line osimertinib, plasma genotyping shows no evidence of emergence of T790M mutations [23]. Instead, due to selective pressure from osimertinib, acquired resistance is often associated with development of other EGFR-dependent and EGFR-independent bypass pathways.
2.2 C797S
Since osimertinib overcomes T790M resistance by binding to the C797 residue in the ATP pocket, it is not surprising that the most common EGFR-dependent mechanism of resistance to osimertinib are mutations at C797 [23]. C797 mutations also confer resistance to similar third-generation EGFR TKIs (e.g., rociletinib, olmutinib, and nazartinib) [24]. Mutations at C797 were detected in 15% of patients at disease progression to osimertinib in the second-line setting and 7% of patients at progression to first-line osimertinib [22]. Notably, similar resistance mutations at C797, including C797G, have also been reported. [24]
In the absence of a T790M mutation, tumors resistant to osimertinib due to C797S mutations retain sensitivity to first- and second-generation EGFR TKIs (e.g., gefitinib, erlotinib, afatinib) [25, 26]. In the presence of a T790M mutation, which is only observed in patients who had a prior earlier generation EGFR TKI, if the T790M and C797S mutations are on different alleles (trans), then the tumor will likely retain sensitivity to first- and second-generation EGFR TKIs [27]. Retrospective data show that two-thirds of progressed cases have cis presentations, which would remain resistant to both first- and second-generation EGFR TKIs [28].
Adding first- or second-generation EGFR TKIs to osimertinib in the first-line setting may prevent the clonal selection of C797S mutations [29]. Additionally, “fourth-generation” EGFR TKIs in development (e.g., EAI045, JBJ-04-125-02, BLU-945) may overcome both C797S and T790M mutations in vitro and in vivo, but have not been assessed in clinical trials yet and may be dependent on the underlying core driver mutation [30]. Additionally, a novel anaplastic lymphoma kinase (ALK)/EGFR inhibitor, brigatinib, in combination with a fourth-generation EGFR TKI, has also demonstrated in vivo efficacy against triple-mutant (EGFR mutant, T790M positive, C797S mutant) NSCLC [31]. BBT-176 is another novel EGFR TKI designed to allosterically inhibit EGFR with C797S mutations (NCT04820023).
2.3 Other EGFR-Dependent Acquired Resistance Mechanisms
While mutations at C797 are the most common on-target resistance mechanisms in EGFR mutant NSCLC, other tertiary EGFR mutations have also been detected. For example, G796D/R/S and L792H mutations in exon 20 of EGFR lead to conformational changes that sterically hinder osimertinib [31,32,33]. On exon 18 of EGFR, rare mutations at G719, L718, and G724 have been associated with osimertinib resistance, though in the absence of T790M mutation, they may also remain sensitive to first- and second-generation EGFR TKIs [24, 32]. Interestingly, G724S mutations generally only lead to osimertinib resistance in the presence of exon 19 deletion. but not in the presence of L858R [33]. EGFR amplification, which is correlated with EGFR immunohistochemistry (IHC), is also associated with osimertinib resistance, though this association may be confounded by concurrent off-target bypass pathways [34, 35]. A phase I trial employing the combination of osimertinib and necitumumab for patients demonstrated clinical activity against EGFR-dependent resistance (T790M+/C797S+) after progression on third-generation TKI (NCT02496663). Most recently, a phase I study utilizing the combination of amivantamab, a bispecific EGFR and c-mesenchymal epithelial transition factor (c-MET) antibody, with lazertinib, a third-generation EGFR TKI, demonstrated promising results with an ORR of 36% in patients who progressed on osimertinib and an ORR of 100% in TKI-naïve patients [36]. Overall, the lack of specific agents to target these EGFR-dependent acquired resistance mechanisms is an important area of future research and drug development.
3 Acquired Resistance: EGFR-Independent Mechanisms
3.1 MET Amplification
In classical EGFR mutant NSCLC, the most common EGFR-independent mechanism that confers resistance to osimertinib is MET amplification, which bypasses EGFR by leading to constitutive activation of downstream signaling pathways, such as those mediated by mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT), and phosphoinositide 3-kinase (PI3K)-Akt [37, 38]. Like EGFR amplification, MET amplification is strongly correlated with MET IHC [39]. MET amplifications can also be identified through routine circulating tumor DNA (ctDNA) analysis [38].
In the AURA3 study, MET amplification was found through plasma next-generation sequencing (NGS) in 19% of patient samples at disease progression [40]. Through NGS ctDNA analysis, after progression on first-line osimertinib, MET amplification was found in 15% of patient samples [41]. Because it is more challenging to detect amplifications than mutations diagnostically, the incidence of MET amplification may be underestimated by these data. Based on existing retrospective data, MET amplification occurs regardless of the presence or loss of the T790M mutation [42,43,44] and co-occurs with EGFR C797S in 5–10% of cases. [45]
To overcome resistance to osimertinib due to MET amplification, c-Met inhibitors may be utilized. Given the availability for other indications, crizotinib with osimertinib was initially tested and found to be efficacious against tumors that acquire resistance to osimertinib through MET amplification [46, 47]. In the phase Ib TATTON trial, the combination of the MET TKI savolitinib with osimertinib yielded an ORR of 30% and a PFS of 5.4 months in patients with acquired resistance to third-generation EGFR TKIs in the setting of MET amplification [48]. The phase II trial for this combination is currently underway (NCT03778229). The combination of another MET TKI (capmatinib) with gefitinib has also yielded favorable results in a phase II trial of patients with MET amplification who were previously treated with an EGFR TKI. In this trial, ORR for all patients was 27%, but it increased to 47% in the subset of patients who had six or more MET gene copies [49]. In another phase Ib/II trial of patients with MET overexpression or amplification who had progressed on a previous EGFR TKI, the combination of gefitinib and the MET TKI, tepotinib, led to higher ORR compared with standard PBC [50]. As previously mentioned, the combination of the bispecific EGFR and c-MET antibody amivantamab and the third-generation EGFR TKI lazertinib has recently demonstrated an ORR of 36% in patients who progressed on osimertinib and an ORR of 100% in TKI-naïve patients. [36]
3.2 HER2 and HER3 Amplification
The ErbB2 tyrosine kinase receptor is encoded by HER2 and is responsible for activating downstream PI3K-Akt and MAPK pathways. HER2 amplification is found in about 2% of patients with resistance to first-line osimertinib [41]. The anti-HER2 antibody-drug conjugated (ADC) trastuzumab-emtansine (T-DM1) has shown efficacy in preclinical models and in patients harboring concurrent HER2 amplification and EGFR mutation after progressing on an EGFR TKI [51]. Further clinical studies are needed to optimize the role of HER2 inhibitors in overcoming osimertinib resistance in EGFR mutant NSCLC.
HER3 (ERBB3) is another receptor that is often overexpressed in EGFR mutant NSCLC, and it leads to cell growth and proliferation through dimerization with either EGFR or HER2 [52]. Patritumab deruxtecan is a novel HER3 directed ADC that is demonstrating favorable results in patients previously treated with an EGFR TKI, yielding an ORR of 25% and a disease control rate of 70%. Interestingly, the performance of patritumab deruxtecan was not affected by the presence or absence of other oncogenic mutations, suggesting that HER3 antagonism may serve as a therapeutic approach that is relatively agnostic to the mechanism of resistance (NCT03260491).
3.3 PI3K Pathway Mutations
Activation of the PI3K pathway, either through PIK3CA mutation or PTEN deletion, is implicated in up to 5% of patients who develop resistance to first-generation EGFR TKIs and 5–12% of patients who develop resistance to osimertinib [30]. Though PIK3CA mutations commonly co-occur with other driver mutations in NSCLC and generally portend worse prognosis, evidence suggests that in EGFR mutant NSCLC, the presence of a concurrent PIK3CA mutation has no significant impact on the clinical benefit from EGFR TKI monotherapy [53]. Targeted therapies against PIK3CA mutations have not demonstrated clinical benefit thus far.
3.4 RAS-MAPK Pathway Mutations
Mutations along the RAS-MAPK pathway have also been implicated in TKI resistance in patients with EGFR mutant NSCLC. In the FLAURA trial, variable mutations in NRAS and KRAS were found in 1% of patients who progressed on first-generation TKIs and 3% of patients who progressed on first-line osimertinib [41]. NRAS mutations include the E63K mutation, while KRAS mutations include the G12S, G13D, Q61R, and G12D mutations [44]. BRAFV600E mutations were found in 3% of patients who progressed on first- or second-line osimertinib [54, 55]. There has also been a reported case of MAPK1 mRNA overexpression in one patient who progressed on second-line osimertinib [56]. BRAF inhibitors or the vascular endothelial growth factor receptor (VEGFR)/MET/AXL inhibitor cabozantinib may confer efficacy against osimertinib resistance due to such mutations, but robust clinical trial data are lacking [54, 57]. Similarly, MEK inhibitors such as selumetinib may help overcome this resistance mechanism to osimertinib. Indeed, the combination of selumetinib and osimertinib overcame TKI resistance attributed to NRAS mutations both in vitro and in vivo, but further clinical evidence supporting these combination strategies with EGFR and MAPK active TKIs are needed [58].
3.5 Cell-Cycle–Related Gene Mutations
In the AURA3 and FLAURA trials, alteration of cell-cycle–related genes was found in about 10% of patients who progressed on first-line osimertinib and 12% of patients who progressed on second-line osimertinib [40, 41]. The most common cell-cycle gene alterations are mutations or amplifications of genes encoding cyclin D1, D2, and E1, cyclin-dependent kinase (CDK) 4 and 6, and CDK inhibitor 2A. Such mutations have been reported in other studies and are associated with poorer prognosis after progression on osimertinib [59]. There is currently one phase Ib/II trial utilizing lerociclib, a CDK4/6 inhibitor, in conjunction with osimertinib in patients with EGFR mutant NSCLC (NCT03455829).
3.6 Oncogenic Fusions
Chromosomal rearrangements involving driver oncogenes, also known as oncogenic fusions, are rare events that have been identified in about 5% of patients who progress on first-line osimertinib [40]. These include FGFR3–TACC3, RET–ERC1, CCDC6–RET, NTRK1–TPM3, NCOA4–RET, GOPC-ROS1, AGK–BRAF, ESYT2–BRAF, and SPTBN1–ALK. [40, 44] In two patients with acquired resistance attributed to CCD6-RET fusion, the combination of osimertinib with the Ret inhibitor pralsetinib (BLU-667) was well-tolerated and led to rapid response in both patients [60]. The other fusions, while unusual, might be amenable to combination TKI therapy as well.
3.7 Histologic and Phenotypic Transformations
Histologic transformation from EGFR mutant NSCLC to small-cell lung cancer (SCLC) has been observed in up to 14% of patients who progressed on first-generation TKIs and between 4% and 15% of patients who progressed on first- or second-line osimertinib [61,62,63,64]. At time of transformation, the founder EGFR mutation is generally preserved [62, 65]. While the mechanism of transformation is unclear, concurrent loss of function mutations in TP53 and RB1 are associated with a significantly increased risk of transformation [66,67,68]. Therefore, patients with EGFR mutant NSCLC and concurrent pretreatment alterations in TP53 or RB1 may warrant monitoring for transformation into SCLC [66]. Unlike gene mutations, the occurrence of histologic or phenotypic transformation is not apparent through plasma analysis and therefore necessitates tissue biopsy. Unfortunately, there are no targeted therapies for such transformations, and treatment with standard PBC generally yields modest outcomes with chemotherapy and little to no observed efficacy with immunotherapy [65]. The combination of osimertinib with carboplatin and etoposide is being studied in a phase 1 study aiming to prevent transformation to SCLC in patients with EGFR mutant NSCLC and concurrent TP53 and RB1 alterations (NCT03567642). Transformation to squamous cell cancer has been similarly noted in about 15% of patients who progress on first- or second-line osimertinib, and the EGFR mutation is generally preserved in this scenario as well [69,70,71]. Lastly, resistance to osimertinib has also been attributed to epithelial-to-mesenchymal transition (EMT) and over-expression of TWIST-1 (an EMT transcription factor) by NSCLC cells, leading to active investigation of TWIST-1 inhibitors in animal models [72, 73].
3.8 Strategies to Prevent Resistance to Osimertinib
Simultaneously targeting EGFR as well as known bypass pathways may prevent EGFR-independent resistance. There are multiple clinical trials testing EGFR TKIs in combination with targeted inhibitors, and many more rational combinations, as described in the previous sections.
For commonly emerging mechanisms of resistance, moving the combination to the frontline may improve PFS. Since chemotherapy is a standard second-line approach now, there are trials combining chemotherapy in the first-line setting with EGFR TKIs. Concurrent use of chemotherapy with gefitinib versus gefitinib alone did not confer survival benefit in patients with untreated EGFR mutant NSCLC [74, 75]. However, for selected patients in the second-line setting, the combination of various chemotherapies with osimertinib appears to be tolerable and may better control CNS disease than chemotherapy alone [76]. Now, the same concept is being tested in the phase III FLAURA2 trial, comparing PBC plus osimertinib versus osimertinib alone in untreated EGFR mutant NSCLC (NCT04035486).
Increased vascular endothelial growth factor (VEGF) has been associated with EGFR TKI resistance in preclinical models [77], and some Japanese studies have shown that the combination of VEGF inhibitors with first-generation TKIs increase PFS [78, 79]. However, the combination of osimertinib with VEGF inhibitors has failed to prolong PFS or survival when compared to osimertinib alone [80]. The phase III EA5182 study is testing the combination of bevacizumab plus osimertinib with osimertinib alone in the frontline setting (NCT04181060).
Immune checkpoint inhibitors appear generally less effective in EGFR mutant NSCLC, without clear predictive biomarkers of response [81]. In pre-clinical studies, EGFR activation led to upregulated programmed death-ligand 1 (PD-L1), but the combination of EGFR inhibitors and programmed cell death protein 1 (PD-1) inhibitors did not lead to synergistic effects [82]. Early combination trials of osimertinib and durvalumab were halted due to high rates of immune-related adverse events, particularly pneumonitis, so only chemotherapy combinations are now being investigated [83]. Identifying effective combinations of targeted therapy and immunotherapy is an unmet need in treating EGFR mutant NSCLC, and the reduced efficacy of checkpoint inhibitors is likely from lower tumor immunogenicity, but may be augmented by future combination therapies [84]. In the IMpower130 trial, the addition of atezolizumab to PBC in the first-line setting conferred no benefit when compared to PBC alone [85]. Interestingly, in the IMpower150 trial, the addition of both atezolizumab and bevacizumab to PBC (ABCP regimen) demonstrated improved PFS and OS when compared to other arms, suggesting a synergistic efficacy of VEGF and immune checkpoint inhibitors [86].
Concurrent local radiotherapy with a third-generation EGFR inhibitor versus third-generation EGFR inhibitor alone has improved PFS and OS in patients with oligometastatic disease in the first-line setting (NCT02893332). This suggests that radiotherapy, when appropriate, may prevent or delay the development of resistance mechanisms.
4 Intrinsic Resistance to EGFR TKIs
Each of the acquired mechanisms of resistance to EGFR TKIs described in this paper can also present as intrinsic mechanisms of resistance prior to any treatment. For example, though T790M is considered an acquired mechanism of resistance to earlier generation TKIs, germline T790M mutations have been observed in 1% of NSCLC cases [87]. Though third-generation TKIs like osimertinib have made germline T790M mutations less clinically relevant, finding this mutation warrants a genetics evaluation and counseling for the patient and their family [88]. Another rare but well-described mechanism of intrinsic resistance to third-generation EGFR TKIs is MET amplification [89]. Beyond these, there are other types of EGFR mutations that have been relatively resistant to TKI therapy until recently.
4.1 EGFR Exon 20 Insertion
A rare but important subset of EGFR mutant NSCLC with intrinsic resistance to third-generation TKIs is EGFR ex20ins NSCLC. EGFR ex20ins comprises about 4% of all EGFR mutant NSCLC and is associated with intrinsic resistance to currently available EGFR TKIs and poorer outcomes for patients [90, 91]. EGFR proteins with ex20ins mutations have binding pockets that are inaccessible to existing EGFR TKIs [92]. Retrospective studies of first-generation EGFR TKIs in EGFR ex20ins NSCLC demonstrated ORR between 8% and 27% and median PFS of less than 3 months [93]. Third-generation EGFR inhibitors, such as osimertinib, show only slightly better activity against EGFR ex20ins NSCLC, and most patients have a short duration of response [37]. Interestingly, a few variants of EGFR ex20ins NSCLC, such as A763_Y764insFQEA insertion, are significantly more responsive to existing EGFR TKIs [94].
Therefore, most EGFR ex20ins NSCLC patients are treated with PBC with or without antiangiogenic therapy or immunotherapy as first-line therapy, though some patients may be prescribed first-line osimertinib, with variable results. In clinical trials of EGFR inhibitors versus PBC in classical EGFR mutant NSCLC, PBC yields an ORR of about 30% and median PFS of about 5–6 months [1, 10, 95]. Existing literature suggests that immunotherapy is relatively ineffective against EGFR mutant NSCLC, whereas data on the utility of antiangiogenic therapy in EGFR mutant NSCLC are mixed [96, 97]. Retrospective studies have described the clinical course of EGFR ex20ins NSCLC treated with first-line PBC, finding ORR of 20–30% and PFS of 6–7 months, similar to the course of classical EGFR mutant NSCLC treated with first-line platinum-based chemotherapy [98,99,100].
Amivantamab, a novel bispecific antibody targeting EGFR and MET receptor, was recently approved for patients with locally advanced or metastatic EGFR ex20ins NSCLC after progression on or after platinum-based chemotherapy [101]. This accelerated approval was based on results from the multicenter, multicohort, non-randomized, open-label clinical trial CHRYSALIS. In the subset of 81 patients with EGFR ex20ins NSCLC who had progressed on platinum-based chemotherapy, the ORR was 40% and the median duration of response was 11.1 months (NCT02609776).
TKIs targeting the ex20ins EGFR protein are also being tested against PBC in the first-line setting. Mobocertinib (TAK-788) is a novel TKI with higher affinity binding to the ex20ins mutant EGFR than other available TKIs. A phase II, open-label, cohort expansion demonstrated that mobocertinib leads to an ORR of 28% with a median duration of response of 17.5 months, leading to the recent Food and Drug Administration (FDA) approval of this agent (NCT02716116). Other TKIs are in clinical development as well, such as poziotinib, DZD9008, and CLN-081 (NCT03318939, NCT03974022, and NCT04036682)
While currently the EGFR ex20ins agents are approved in the second-line setting, both have ORR similar to what we observe with platinum-based chemotherapy in the first-line setting. In the EXCLAIM-2 study, mobocertinib is being tested against PBC in the frontline setting (NCT04129502). A phase III study of combination amivantamab and carboplatin-pemetrexed therapy compared with carboplatin-pemetrexed therapy in advanced EGFR ex20ins NSCLC is also currently underway (NCT04538664).
5 Future Directions
Upon the development of resistance to third-generation TKIs, most EGFR mutant NSCLC is treated with standard PBC. However, as targeted therapies against specific resistance mechanisms are developed, there will likely be a myriad of agents and their combinations that may be used to overcome resistance, as summarized in Fig. 2. Notably, the phase II ORCHARD trial follows a biomarker-driven approach to assigning targeted therapies to be given simultaneously with osimertinib when specific resistance mechanisms arise (e.g., add savolitinib for MET alteration, add gefitinib for C797X mutation) [102].

Overview of oncogenic pathways and examples of targeted inhibitors to overcome resistance to treatment in EGFR mutant NSCLC. CDK cyclin-dependent kinase, EGFR epidermal growth factor receptor, MAPK mitogen-activated protein kinase, MET mesenchymal epithelial transition factor, NSCLC non-small-cell lung cancer, PI3K phosphoinositide 3-kinase
To facilitate a biomarker-driven approach, we anticipate widespread utilization of liquid biopsy as a complement to repeat tissue biopsy or empiric PBC. Monitoring ctDNA, released from tumor cells into the bloodstream, is a non-invasive and feasible method of detecting tumor alterations pertinent to NSCLC [103]. Compared with tissue biopsy, monitoring ctDNA for EGFR mutations has a 67% sensitivity and 94% specificity [104]. Polymerase chain reaction (PCR)-based and NGS-based analysis of ctDNA has high specificity, but lower sensitivity due to lack of tumor shedding in up to 20% of patients with NSCLC [105]. The other limitations of NGS-based analysis are reduced sensitivity in detecting gene amplifications, which is ideally assessed through fluorescence in situ hybridization (FISH), and inability to detect histologic transformation, which requires tissue biopsy. Despite these limitations, NGS-based liquid biopsy is clinically useful and relatively feasible for not only detecting initial driver mutations, but also predicting recurrence and identifying genetic modifiers of resistance [106]. One study profiling ctDNA in patients with stage I–III lung cancer found that post-treatment ctDNA reliably identified minimal residual disease and preceded radiographic recurrence by a median of 5.2 months, suggesting that ctDNA profiling may allow for personalized adjuvant therapy while disease burden is at its lowest [107]. Importantly, NGS-based biopsy for patients with NSCLC appears to be more time efficient for personnel and more cost-effective for patients [108]. Standardization of NGS-based liquid biopsy in monitoring for resistance in NSCLC is likely to become more standard in the future as costs continue to drop.
Ultimately, further molecular profiling, active surveillance of resistance mechanisms, and development of targeted therapeutics will continue to transform the landscape of EGFR mutant NSCLC. Concurrent investigation of immune checkpoint inhibitor, antiangiogenic therapy, and radiation therapy will likely augment the efficacy of targeted treatment regimens and move toward the goal of personalized, gene-directed therapy in most patients with NSCLC.
References
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–46. https://doi.org/10.1016/S1470-2045(11)70393-X.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–42. https://doi.org/10.1016/S1470-2045(11)70184-X.
Shaw AT, Ou SHI, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–71. https://doi.org/10.1056/NEJMoa1406766.
Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16. https://doi.org/10.1016/S1470-2045(17)30679-4.
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–77. https://doi.org/10.1056/NEJMoa1408440.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–25. https://doi.org/10.1056/NEJMoa1713137.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. https://doi.org/10.1056/NEJMoa040938.
Saito S, Espinoza-Mercado F, Liu H, Sata N, Cui X, Soukiasian HJ. Current status of research and treatment for non-small cell lung cancer in never-smoking females. Cancer Biol Ther. 2017;18(6):359–68. https://doi.org/10.1080/15384047.2017.1323580.
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–7. https://doi.org/10.1126/science.1101637.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–57. https://doi.org/10.1056/NEJMoa0810699.
Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–9. https://doi.org/10.1093/annonc/mdx703.
Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev. 2018;65:1–10. https://doi.org/10.1016/j.ctrv.2018.02.006.
Attili I, Karachaliou N, Conte P, Bonanno L, Rosell R. Therapeutic approaches for T790M mutation positive non-small-cell lung cancer. Expert Rev Anticancer Ther. 2018;18(10):1021–30. https://doi.org/10.1080/14737140.2018.1508347.
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–92. https://doi.org/10.1056/NEJMoa044238.
Passaro A, Guerini-Rocco E, Pochesci A, Vacirca D, Spitaleri G, Catania CM, et al. Targeting EGFR T790M mutation in NSCLC: From biology to evaluation and treatment. Pharmacol Res. 2017;117:406–15. https://doi.org/10.1016/j.phrs.2017.01.003.
Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. https://doi.org/10.1056/NEJMoa1913662.
Hoffknecht P, Tufman A, Wehler T, Pelzer T, Wiewrodt R, Schütz M, et al. Efficacy of the irreversible ErbB family blocker afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)-pretreated non-small-cell lung cancer patients with brain metastases or leptomeningeal disease. J Thorac Oncol. 2015;10(1):156–63. https://doi.org/10.1097/JTO.0000000000000380.
Schuler M, Wu YL, Hirsh V, O’Byrne K, Yamamoto N, Mok T, et al. First-line afatinib versus chemotherapy in patients with non-small cell lung cancer and common epidermal growth factor receptor gene mutations and brain metastases. J Thorac Oncol. 2016;11(3):380–90. https://doi.org/10.1016/j.jtho.2015.11.014.
Wu YL, Zhou C, Cheng Y, Lu S, Chen GY, Huang C, et al. Erlotinib as second-line treatment in patients with advanced non-small-cell lung cancer and asymptomatic brain metastases: a phase II study (CTONG-0803). Ann Oncol. 2013;24(4):993–9. https://doi.org/10.1093/annonc/mds529.
Reungwetwattana T, Nakagawa K, Cho BC, Cobo M, Cho EK, Bertolini A, et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(33):3290–7. https://doi.org/10.1200/JCO.2018.78.3118.
Yang JCH, Kim SW, Kim DW, Lee JS, Cho BC, Ahn JS, et al. Osimertinib in patients with epidermal growth factor receptor mutation-positive non-small-cell lung cancer and leptomeningeal metastases: the BLOOM Study. J Clin Oncol. 2020;38(6):538–47. https://doi.org/10.1200/JCO.19.00457.
Ahn MJ, Han JY, Lee KH, Kim SW, Kim DW, Lee YG, et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet Oncol. 2019;20(12):1681–90. https://doi.org/10.1016/S1470-2045(19)30504-2.
Lazzari C, Gregorc V, Karachaliou N, Rosell R, Santarpia M. Mechanisms of resistance to osimertinib. J Thorac Dis. 2020;12(5):2851–8. https://doi.org/10.21037/jtd.2019.08.30.
Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating Novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–107. https://doi.org/10.1158/1078-0432.CCR-17-2310.
Rangachari D, To C, Shpilsky JE, VanderLaan PA, Kobayashi SS, Mushajiang M, et al. EGFR-mutated lung cancers resistant to osimertinib through EGFR C797S respond to first-generation reversible EGFR Inhibitors but eventually acquire EGFR T790M/C797S in preclinical models and clinical samples. J Thorac Oncol. 2019;14(11):1995–2002. https://doi.org/10.1016/j.jtho.2019.07.016.
Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21(17):3913–23. https://doi.org/10.1158/1078-0432.CCR-14-2789.
Arulananda S, Do H, Musafer A, Mitchell P, Dobrovic A, John T. Combination osimertinib and gefitinib in C797S and T790M EGFR-mutated non-small cell lung cancer. J Thorac Oncol. 2017;12(11):1728–32. https://doi.org/10.1016/j.jtho.2017.08.006.
Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–2. https://doi.org/10.1038/nm.3854.
Wang Z, Yang JJ, Huang J, Ye JY, Zhang XC, Tu HY, et al. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J Thorac Oncol. 2017;12(11):1723–7. https://doi.org/10.1016/j.jtho.2017.06.017.
Passaro A, Jänne PA, Mok T, Peters S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat Cancer. 2021;2(4):377–91. https://doi.org/10.1038/s43018-021-00195-8.
Maity S, Pai KSR, Nayak Y. Advances in targeting EGFR allosteric site as anti-NSCLC therapy to overcome the drug resistance. Pharmacol Rep PR. 2020;72(4):799–813. https://doi.org/10.1007/s43440-020-00131-0.
Fassunke J, Müller F, Keul M, Michels S, Dammert MA, Schmitt A, et al. Overcoming EGFRG724S-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat Commun. 2018;9(1):4655. https://doi.org/10.1038/s41467-018-07078-0.
Brown BP, Zhang YK, Westover D, Yan Y, Qiao H, Huang V, et al. On-target resistance to the mutant-selective egfr inhibitor osimertinib can develop in an allele-specific manner dependent on the original EGFR-activating mutation. Clin Cancer Res. 2019;25(11):3341–51. https://doi.org/10.1158/1078-0432.CCR-18-3829.
Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I, et al. Amplification of EGFR Wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 2017;77(8):2078–89. https://doi.org/10.1158/0008-5472.CAN-16-2359.
Li AR, Chitale D, Riely GJ, Pao W, Miller VA, Zakowski MF, et al. EGFR mutations in lung adenocarcinomas. J Mol Diagn. 2008;10(3):242–8. https://doi.org/10.2353/jmoldx.2008.070178.
Dhillon S. Lazertinib: First Approval. Drugs. 2021;81(9):1107–13. https://doi.org/10.1007/s40265-021-01533-x.
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–7. https://doi.org/10.1158/1078-0432.CCR-12-2246.
Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. https://doi.org/10.1038/ncomms11815.
Park S, Koh J, Kim DW, Kim M, Keam B, Kim TM, et al. MET amplification, protein expression, and mutations in pulmonary adenocarcinoma. Lung Cancer. 2015;90(3):381–7. https://doi.org/10.1016/j.lungcan.2015.10.022.
Papadimitrakopoulou VA, Wu YL, Han JY, Ahn MJ, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. 2018;29:viii741. https://doi.org/10.1093/annonc/mdy424.064
Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:viii740. https://doi.org/10.1093/annonc/mdy424.063
O’Kane GM, Barnes TA, Leighl NB. Resistance to epidermal growth factor receptor tyrosine kinase inhibitors, T790M, and clinical trials. Curr Oncol Tor Ont. 2018;25(Suppl 1):S28–37. https://doi.org/10.3747/co.25.3796.
Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22(19):4837–47. https://doi.org/10.1158/1078-0432.CCR-15-1915.
Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725–37. https://doi.org/10.1038/s41416-019-0573-8.
Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. 2018;24(24):6195–203. https://doi.org/10.1158/1078-0432.CCR-18-1542.
York ER, Varella-Garcia M, Bang TJ, Aisner DL, Camidge DR. Tolerable and effective combination of full-dose crizotinib and osimertinib targeting MET amplification sequentially emerging after T790M positivity in EGFR-mutant non-small cell lung cancer. J Thorac Oncol. 2017;12(7):e85–8. https://doi.org/10.1016/j.jtho.2017.02.020.
Deng L, Kiedrowski LA, Ravera E, Cheng H, Halmos B. Response to Dual crizotinib and osimertinib treatment in a lung cancer patient with MET amplification detected by liquid biopsy who acquired secondary resistance to EGFR tyrosine kinase inhibition. J Thorac Oncol. 2018;13(9):e169–72. https://doi.org/10.1016/j.jtho.2018.04.007.
Sequist LV, Han JY, Ahn MJ, Cho BC, Yu H, Kim SW, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020;21(3):373–86. https://doi.org/10.1016/S1470-2045(19)30785-5.
Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JCH, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J Clin Oncol. 2018;36(31):3101–9. https://doi.org/10.1200/JCO.2018.77.7326.
Wu YL, Cheng Y, Zhou J, Lu S, Zhang Y, Zhao J, et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): an open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir Med. 2020;8(11):1132–43. https://doi.org/10.1016/S2213-2600(20)30154-5.
Li BT, Michelini F, Misale S, Cocco E, Baldino L, Cai Y, et al. HER2-mediated internalization of cytotoxic agents in erbb2 amplified or mutant lung cancers. Cancer Discov. 2020;10(5):674–87. https://doi.org/10.1158/2159-8290.CD-20-0215.
Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res. 2014;20(6):1410–6. https://doi.org/10.1158/1078-0432.CCR-13-1549.
Eng J, Woo KM, Sima CS, Plodkowski A, Hellmann MD, Chaft JE, et al. Impact of concurrent PIK3CA mutations on response to EGFR tyrosine kinase inhibition in EGFR-mutant lung cancers and on prognosis in oncogene-driven lung adenocarcinomas. J Thorac Oncol. 2015;10(12):1713–9. https://doi.org/10.1097/JTO.0000000000000671.
Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Yang JCH. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12(3):567–72. https://doi.org/10.1016/j.jtho.2016.11.2231.
Lin CC, Shih JY, Yu CJ, Ho CC, Liao WY, Lee JH, et al. Outcomes in patients with non-small-cell lung cancer and acquired Thr790Met mutation treated with osimertinib: a genomic study. Lancet Respir Med. 2018;6(2):107–16. https://doi.org/10.1016/S2213-2600(17)30480-0.
Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, et al. Mechanisms of acquired resistance to AZD9291: A Mutation-Selective Irreversible EGFR Inhibitor. J Thorac Oncol. 2015;10(12):1736–44. https://doi.org/10.1097/JTO.0000000000000688.
Tian Y, Zhang Z, Miao L, Yang Z, Yang J, Wang Y, et al. Anexelekto (AXL) increases resistance to EGFR-TKI and activation of AKT and ERK1/2 in Non-small cell lung cancer cells. Oncol Res. 2016;24(5):295–303. https://doi.org/10.3727/096504016X14648701447814.
Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75(12):2489–500. https://doi.org/10.1158/0008-5472.CAN-14-3167.
Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet. 2017;49(12):1693–704. https://doi.org/10.1038/ng.3990.
Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8(12):1529–39. https://doi.org/10.1158/2159-8290.CD-18-1022.
Minari R, Bordi P, Del Re M, Facchinetti F, Mazzoni F, Barbieri F, et al. Primary resistance to osimertinib due to SCLC transformation: Issue of T790M determination on liquid re-biopsy. Lung Cancer. 2018;115:21–7. https://doi.org/10.1016/j.lungcan.2017.11.011.
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. https://doi.org/10.1126/scitranslmed.3002003.
Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4(11):1527–34. https://doi.org/10.1001/jamaoncol.2018.2969.
Lee J, Kim HS, Lee B, Kim HK, Sun JM, Ahn JS, et al. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer. 2020;126(11):2704–12. https://doi.org/10.1002/cncr.32809.
Marcoux N, Gettinger SN, O’Kane G, Arbour KC, Neal JW, Husain H, et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J Clin Oncol. 2019;37(4):278–85. https://doi.org/10.1200/JCO.18.01585.
Lee JK, Lee J, Kim S, Kim S, Youk J, Park S, et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol. 2017;35(26):3065–74. https://doi.org/10.1200/JCO.2016.71.9096.
Offin M, Chan JM, Tenet M, Rizvi HA, Shen R, Riely GJ, et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J Thorac Oncol. 2019;14(10):1784–93. https://doi.org/10.1016/j.jtho.2019.06.002.
Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. https://doi.org/10.1038/ncomms7377.
Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. 2020;26(11):2654–63. https://doi.org/10.1158/1078-0432.CCR-19-3563.
Hakozaki T, Kitazono M, Takamori M, Kiriu T. Combined Small and Squamous Transformation in EGFR-mutated Lung Adenocarcinoma. Intern Med. 2020;59(10):1291–4. https://doi.org/10.2169/internalmedicine.3542-19.
Izumi H, Yamasaki A, Ueda Y, Sumikawa T, Maeta H, Nakamoto S, et al. Squamous cell carcinoma transformation from EGFR-mutated lung adenocarcinoma: a case report and literature review. Clin Lung Cancer. 2018;19(1):e63–6. https://doi.org/10.1016/j.cllc.2017.10.005.
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC, Tseng RY, et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene. 2019;38(4):455–68. https://doi.org/10.1038/s41388-018-0454-2.
Yochum ZA, Cades J, Wang H, Chatterjee S, Simons BW, O’Brien JP, et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene. 2019;38(5):656–70. https://doi.org/10.1038/s41388-018-0482-y.
Sugawara S, Oizumi S, Minato K, Harada T, Inoue A, Fujita Y, et al. Randomized phase II study of concurrent versus sequential alternating gefitinib and chemotherapy in previously untreated non-small cell lung cancer with sensitive EGFR mutations: NEJ005/TCOG0902. Ann Oncol. 2015;26(5):888–94. https://doi.org/10.1093/annonc/mdv063.
Oizumi S, Sugawara S, Minato K, Harada T, Inoue A, Fujita Y, et al. Updated survival outcomes of NEJ005/TCOG0902: a randomised phase II study of concurrent versus sequential alternating gefitinib and chemotherapy in previously untreated non-small cell lung cancer with sensitive EGFR mutations. ESMO Open. 2018;3(2): e000313. https://doi.org/10.1136/esmoopen-2017-000313.
White MN, Piotrowska Z, Stirling K, Liu SV, Banwait MK, Cunanan K, et al. Combining osimertinib with chemotherapy in EGFR-mutant NSCLC at progression. Clin Lung Cancer. 2021;22(3):201–9. https://doi.org/10.1016/j.cllc.2021.01.010.
Naumov GN, Nilsson MB, Cascone T, Briggs A, Straume O, Akslen LA, et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res. 2009;15(10):3484–94. https://doi.org/10.1158/1078-0432.CCR-08-2904.
Saito H, Fukuhara T, Furuya N, Watanabe K, Sugawara S, Iwasawa S, et al. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): interim analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2019;20(5):625–35. https://doi.org/10.1016/S1470-2045(19)30035-X.
Nakagawa K, Garon EB, Seto T, Nishio M, Ponce Aix S, Paz-Ares L, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(12):1655–69. https://doi.org/10.1016/S1470-2045(19)30634-5.
Yu HA, Schoenfeld AJ, Makhnin A, Kim R, Rizvi H, Tsui D, et al. Effect of osimertinib and bevacizumab on progression-free survival for patients with metastatic EGFR-mutant lung cancers: a phase 1/2 single-group open-label trial. JAMA Oncol. 2020;6(7):1048–54. https://doi.org/10.1001/jamaoncol.2020.1260.
Krieger T, Pearson I, Bell J, Doherty J, Robbins P. Targeted literature review on use of tumor mutational burden status and programmed cell death ligand 1 expression to predict outcomes of checkpoint inhibitor treatment. Diagn Pathol. 2020;15(1):6. https://doi.org/10.1186/s13000-020-0927-9.
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10(6):910–23. https://doi.org/10.1097/JTO.0000000000000500.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–35. https://doi.org/10.1056/NEJMoa1504627.
Listì A, Barraco N, Bono M, Insalaco L, Castellana L, Cutaia S, et al. Immuno-targeted combinations in oncogene-addicted non-small cell lung cancer. Transl Cancer Res. 2018;8(S1):S55–63. https://doi.org/10.21037/tcr.2018.10.04.
West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):924–37. https://doi.org/10.1016/S1470-2045(19)30167-6.
Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med. 2019;7(5):387–401. https://doi.org/10.1016/S2213-2600(19)30084-0.
Gazdar A, Robinson L, Oliver D, Xing C, Travis WD, Soh J, et al. Hereditary lung cancer syndrome targets never smokers with germline EGFR gene T790M mutations. J Thorac Oncol. 2014;9(4):456–63. https://doi.org/10.1097/JTO.0000000000000130.
Oxnard GR, Miller VA, Robson ME, Azzoli CG, Pao W, Ladanyi M, et al. Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol. 2012;7(6):1049–52. https://doi.org/10.1097/JTO.0b013e318250ed9d.
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–7. https://doi.org/10.1073/pnas.0710370104.
Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13(1):e23-31. https://doi.org/10.1016/S1470-2045(11)70129-2.
Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12(2):220–9. https://doi.org/10.1158/1535-7163.MCT-12-0620.
Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med. 2018;24(5):638–46. https://doi.org/10.1038/s41591-018-0007-9.
Naidoo J, Sima CS, Rodriguez K, Busby N, Nafa K, Ladanyi M, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: clinical outcomes and response to erlotinib. Cancer. 2015;121(18):3212–20. https://doi.org/10.1002/cncr.29493.
Voon PJ, Tsui DWY, Rosenfeld N, Chin TM. EGFR exon 20 insertion A763–Y764insFQEA and response to erlotinib–Letter. Mol Cancer Ther. 2013;12(11):2614–5. https://doi.org/10.1158/1535-7163.MCT-13-0192.
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–8. https://doi.org/10.1056/NEJMoa0909530.
Cavanna L, Citterio C, Orlandi E. Immune checkpoint inhibitors in EGFR-mutation positive TKI-treated patients with advanced non-small-cell lung cancer network meta-analysis. Oncotarget. 2019;10(2):209–15. https://doi.org/10.18632/oncotarget.26541.
Alexander M, Halmos B. VEGF inhibitors in EGFR-mutated lung cancer: a never-ending story? Ann Transl Med. 2018;6(23):446. https://doi.org/10.21037/atm.2018.11.20.
Shah MP, Aredo JV, Padda SK, Ramchandran KJ, Wakelee HA, Das MS, et al. EGFR exon 20 insertion NSCLC and response to platinum-based chemotherapy. Clin Lung Cancer. 2021. https://doi.org/10.1016/j.cllc.2021.07.001.
Yang G, Li J, Xu H, Yang Y, Yang L, Xu F, et al. EGFR exon 20 insertion mutations in Chinese advanced non-small cell lung cancer patients: Molecular heterogeneity and treatment outcome from nationwide real-world study. Lung Cancer. 2020;145:186–94. https://doi.org/10.1016/j.lungcan.2020.03.014.
Cardona AF, Rojas L, Zatarain-Barrón ZL, Freitas HC, Granados ST, Castillo O, et al. EGFR exon 20 insertion in lung adenocarcinomas among Hispanics (geno1.2-CLICaP). Lung Cancer. 2018;125:265–72. https://doi.org/10.1016/j.lungcan.2018.10.007.
Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR exon 20 insertion-mutated non–small-cell lung cancer progressing on platinum chemotherapy: initial results from the CHRYSALIS Phase I Study. J Clin Oncol. 2021;39(30):3391–402. https://doi.org/10.1200/JCO.21.00662.
Yu H, Goldberg S, Le X, Piotrowska Z, Smith P, Mensi I, et al. P.201–22 ORCHARD: a phase II Platform Study in Patients with Advanced NSCLC Who Have Progressed on First-Line Osimertinib Therapy. J Thorac Oncol. 2019;14(10):S647. https://doi.org/10.1016/j.jtho.2019.08.1366.
Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical implications of plasma-based genotyping with the delivery of personalized therapy in metastatic non-small cell lung cancer. JAMA Oncol. 2019;5(2):173–80. https://doi.org/10.1001/jamaoncol.2018.4305.
Luo J, Shen L, Zheng D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: a systematic review and meta-analysis. Sci Rep. 2014;4:6269. https://doi.org/10.1038/srep06269.
Sacher AG, Komatsubara KM, Oxnard GR. Application of plasma genotyping technologies in non-small cell lung cancer: a practical review. J Thorac Oncol. 2017;12(9):1344–56. https://doi.org/10.1016/j.jtho.2017.05.022.
Russo A, Incorvaia L, Del Re M, Malapelle U, Capoluongo E, Gristina V, et al. The molecular profiling of solid tumors by liquid biopsy: a position paper of the AIOM-SIAPEC-IAP-SIBioC-SIC-SIF Italian Scientific Societies. ESMO Open. 2021;6(3): 100164. https://doi.org/10.1016/j.esmoop.2021.100164.
Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7(12):1394–403. https://doi.org/10.1158/2159-8290.CD-17-0716.
Pisapia P, Pepe F, Baggi A, Barberis M, Galvano A, Gristina V, et al. Next generation diagnostic algorithm in non-small cell lung cancer predictive molecular pathology: the KWAY Italian multicenter cost evaluation study. Crit Rev Oncol Hematol. 2022;169: 103525. https://doi.org/10.1016/j.critrevonc.2021.103525.
Acknowledgments
The authors would like to thank Isabelle Blanchard for assistance with figure illustrations and manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this article.
Conflict of interest
M.P. Shah declares that he has no conflicts of interest that might be relevant to the contents of this article. J.W. Neal declares the following: consulting or advisory role for AstraZeneca, Genentech/Roche, Exelixis, Jounce Therapeutics, Takeda Pharmaceuticals, Eli Lilly and Company, Calithera Biosciences, Amgen, Iovance Biotherapeutics, Blueprint Pharmaceuticals, Regeneron Pharmaceuticals, Natera, Sanofi/Regeneron, D2G Oncology, Surface Oncology, Turning Point Therapeutics; research funding from Genentech/Roche, Merck, Novartis, Boehringer Ingelheim, Exelixis, Nektar Therapeutics, Takeda Pharmaceuticals, Adaptimmune, GSK, Janssen, and AbbVie.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors' contributions
MPS and JWN outlined the manuscript. MPS conducted literature review and composed the manuscript. MPS and JWN revised the manuscript.
Rights and permissions
About this article

Cite this article
Shah, M.P., Neal, J.W. Targeting Acquired and Intrinsic Resistance Mechanisms in Epidermal Growth Factor Receptor Mutant Non-Small-Cell Lung Cancer. Drugs 82, 649–662 (2022). https://doi.org/10.1007/s40265-022-01698-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01698-z