
Abstract
Erdafitinib (Balversa™, Janssen Pharmaceutical Companies) is a pan-fibroblast growth factor receptor (FGFR) inhibitor that was recently approved in the USA for the treatment of locally advanced or metastatic FGFR3 or FGFR2 urothelial carcinoma. The drug is also being investigated as a treatment for other cancers including cholangiocarcinoma, liver cancer, non-small cell lung cancer, prostate cancer, lymphoma and oesophageal cancer. This article summarizes the milestones in the development of erdafitinib leading to this first approval for the treatment of urothelial carcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Erdafitinib (Balversa™) is an orally administered, small-molecule, pan-fibroblast growth factor receptor (FGFR) inhibitor being developed by Janssen Pharmaceutical Companies for the treatment of cancers expressing activating mutations, amplifications and over-expression of fibroblast growth factors (FGFs) [1].The FGF family consists of 22 ligands that regulate processes including cell migration, proliferation, differentiation and survival; FGF activity is mediated by four transmembrane receptor kinases (FGFR1–4) [2]. FGFR-mediated signalling has a role in tumour cell growth, survival, migration and angiogenesis; FGFs and FGFRs have both been recognised as oncogenic drivers in a range of cancers [2, 3]. Erdafitinib binds to and inhibits FGFR, resulting in inhibition of FGFR phosphorylation and suppression of FGFR-related signal transduction pathways, leading to the inhibition of tumour cell proliferation and cell death in FGFR-overexpressing tumour cells [1]. Erdafitinib was recently approved in the US for the treatment of urothelial carcinoma [1, 4] and is being developed as a treatment for cholangiocarcinoma, liver cancer, non-small cell lung cancer (NSCLC), prostate cancer, lymphoma and oesophageal cancer.

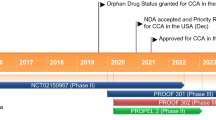
Key milestones in the development of erdafitinib in the treatment of urothelial carcinoma. NDA New Drug Application
The recommended starting dose of erdafitinib is 8 mg once daily, increased to 9 mg once daily according to serum phosphate levels and tolerability at 14–21 days [1]. The manufacturer’s prescribing information carries a warning regarding the risk of ocular disorders, hyperphosphataemia and embryo-fetal toxicity with erdafitinib [1].
2 Company Agreements
Erdafitinib was discovered in collaboration with Astex Pharmaceuticals from a partnership with Janssen that commenced in June 2008. Astex’s FGFR inhibitor program originated from a collaboration initiated in 2005 with the Cancer Research UK Drug Discovery Group at the Newcastle Cancer Centre (Newcastle University UK), and Cancer Research Technology Limited [5, 6].
Under the June 2008 contract, Astex granted Janssen a worldwide license to develop and commercialize FGFR inhibitors emerging from the research programme. In return, Astex received an upfront payment and equity investment, research funding with further payments due on achievement of development and regulatory milestones. The agreement stated that Janssen would be responsible for preclinical and clinical development and global commercialization of products arising from the collaboration [5].
In April 2011 Astex Therapeutics was purchased by US-based SuperGen, Inc. with the combined entity named Astex Pharmaceuticals Inc. [7]. In October 2013 Astex Pharmaceuticals was purchased by Otsuka Pharmaceuticals, becoming a wholly owned subsidiary of the latter [8].
3 Scientific Summary
3.1 Pharmacodynamics
Erdafitinib demonstrated potent anti-tyrosine kinase inhibitory activity against FGFR1, 2, 3 and 4 (IC50 values of 1.2, 2.5, 3.0 and 5.7 nmol/L, respectively) in time-resolved fluorescence assays in vitro, but was less potent against the closely related vascular endothelial growth factor receptor (VEGFR) 2 kinase (IC50 36.8 nmol/L). A similar pattern was observed in binding affinity (measured using the KINOMEscan platform), with erdafitinib Kd values of 0.24, 1.1, 1.4 and 2.2 nmol/L for FGFR1, 3, 4 and 2, respectively, and 6.6 nmol/L for VEGFR2. In BaF3 cell lines engineered to express FGFR family members, erdafitinib had IC50 values of 22.1, 13.2 and 25 nmol/L against FGFR1, 3 and 4, respectively. This activity was absent in the presence of interleukin 3, confirming the specificity of erdafitinib. The drug had considerably less activity against BaF3 cells expressing VEGFR2 (IC50 1160 nmol/L). In NCI-H1581 cells (a lung cancer cell line with a focal FGFR1 gene amplification) stimulated with FGF2 ligand to activate downstream signalling, pretreatment with erdafitinib was associated with reductions in phosphorylated (p)FGFR, phosphorylated fibroblast growth factor receptor substrate (pFRS2), phosphorylated phospholipase C γ 1 (pPLCγ1) and phosphorylated extracellular signal-regulated kinase (pErk)1/2 levels [9].
In vivo, administration of erdafitinib to mice bearing xenograft tumours derived from SNU-16 human gastric cancer cells harbouring FGFR2 amplifications inhibited tumour growth in a dose dependent manner (tumour growth inhibition of 37.8% and 59.4% after treatment with erdafitinib 10 and 30 mg/kg, respectively). Similar results were observed in other tumour models with various FGFR alterations (MDA-MB-453, SNU-16, NCI-H1581, A-204, HuH-7, NCI-H716, and RT112). The drug also demonstrated pronounced antitumour activity in mice bearing xenograft tumours derived from LUX001 cells (a human NSCLC cell that contains a translocation resulting in expression of a FGFR3-TACC3 fusion protein), indicating activity against cells with FGFRs constitutively activated by chromosomal rearrangement [9].
FGFR activity in the renal tubules controls the homeostasis of phosphate; consequently, FGFR inhibition increases serum phosphate levels [1, 2]. In patients with advanced solid tumours given erdafitinib 0.5 to 12 mg daily on a continuous schedule, or 10 or 12 mg on a 7 day on/7 day off cycle in a phase 1 study (NCT01703481), hyperphosphatemia emerged at the 4 mg daily dosage and peaked at ≈ 9 mg daily. No significant changes in calcium levels, and no dose-dependent changes in parathyroid hormone, vitamin D or FGF23 levels were observed. Parathyroid hormone levels were decreased at all doses > 4 mg, and vitamin D levels increased with doses ≥ 2 mg, with a potential plateau effect evident at higher doses [3].

Chemical structure of erdafitinib
In a further phase I study (NCT01962532) in Japanese patients with advanced or refractory solid tumours, administration of single 2, 4 and 6 mg oral doses of erdafitinib was associated with increased plasma phosphate concentrations but no clear dose-response relationship was observed. Calcitriol concentrations were increased at ≈ 4 h post dose, with maximum levels observed 24 to 48 h post dose. No clinically meaningful changes were observed in pS6, pErk, or pFGFR levels [10].
3.2 Pharmacokinetics
Erdafitinib 0.5 to 12 mg once daily continuously or 10 or 12 mg once daily on a 7 day on/7 day off cycle had a linear, dose proportional, and predictable pharmacokinetic profile in a phase I trial in patients with advanced solid tumours (n = 65) [NCT01703481] [3]. In a subset analysis of patients with urothelial cancer (n = 5) treated with erdafitinib 9 mg once daily continuously, or 10 or 12 mg once daily on a 7 day on/7 day off cycle in this study, peak plasma concentrations (Cmax) and area under the concentration-time curve from time 0 to 24 h (AUC24) for total erdafitinib were 1130–2690 ng/mL and 19,900–52,600 ng·h/mL, respectively [11].
In Japanese patients with advanced or refractory solid tumours, erdafitinib 2, 4 or 6 mg/day (n = 3 per dose), or 10 (n = 3) or 12 mg/day (n = 7) administered according to a 7 day on/7 day off cycle, was associated with a dose dependent increase in plasma concentration, with Cmax occurring 2–3 (median) h after the first dose and 2–6 h after multiple doses. The AUC for unbound drug also increased in a dose dependent manner after both single and multiple doses (NCT01962532) [10].
Erdafitinib had a mean apparent volume of distribution of 29 L in patients and was 99.8% bound to plasma protein. The mean total apparent clearance and effective half-life were 0.362 L/h and 59 h, respectively, in patients. The drug is primarily metabolised by CYP2C9 (estimated 39% of total clearance) and CYP3A4 (20%). Sixty nine percent of a single oral dose of radiolabelled erdafitinib was recovered in faeces (19% as unchanged drug) and 19% in urine (13% as unchanged drug) [1].
3.3 Therapeutic Trials
3.3.1 Urothelial Cancer
3.3.1.1 Phase II
Treatment with erdafitinib yielded a robust response rate in patients with metastatic or unresectable urothelial carcinoma and FGFR alterations in a global open-label phase II study (NCT02365597). In the initial part of the trial, the optimal schedule of erdafitinib was determined to be 8 mg/day continuously in 28 day cycles with up-titration to 9 mg/day if necessary, according to serum phosphate levels and tolerability. 96 patients received 5 (median) cycles of optimized erdafitinib therapy; of these 10% were chemotherapy naïve, 47% had received ≥ 2 prior lines of therapy and 80% had visceral metastases. The confirmed overall response rate (RECIST 1.1) was 42% (3 and 39% complete and partial responses, respectively) and 38% of patients had stable disease. The confirmed overall response rate in patients who had previously received immune checkpoint inhibitors (n = 21) was 70% [12].
3.3.2 Cholangiocarcinoma
3.3.2.1 Phase II
Erdafitinib had encouraging clinical activity in patients with advanced cholangiocarcinoma and FGFR alterations who had failed ≥ 1 prior systemic treatment in a preliminary analysis of an open-label phase IIa study (NCT02699606). At data cutoff (20 March 2018) 150 patients had been molecularly screened, with FGFR alterations identified in 25 patients. Of these, 11 patients received erdafitinib 8 mg once daily continuously for 4 (median) treatment cycles (treatment duration 3.5 [median] months). The overall response rate was 45.5% comprising three patients with confirmed partial responses and two patients with unconfirmed partial responses. Four patients had stable disease and two (both with FGFR3 mutations) had progressive disease. Six patients remained on treatment at the time of analysis [13].
3.4 Adverse Events
The most common adverse reactions in the phase II trial in patients with urothelial cancer treated with erdafitinib described above [12] were increased phosphate levels, stomatitis, fatigue, increased creatinine, diarrhoea, dry mouth, onycholysis, increased alanine aminotransferase, increased alkaline phosphatase, decreased sodium, decreased appetite, decreased albumin, dysgeusia, decreased haemoglobin, dry skin, increased aspartate aminotransferase, decreased magnesium, dry eye, alopecia, palmar-plantar erythrodysaesthesia syndrome, constipation, decreased phosphate, abdominal pain, increased calcium, nausea, and musculoskeletal pain [1].
Grade 3 or higher adverse events occurring in >1% of patients included stomatitis, nail dystrophy, palmar-plantar erythrodysaesthesia syndrome, paronychia, nail disorder, keratitis, onycholysis, and hyperphosphataemia. Forty one percent of patients experienced serious adverse reactions including eye disorders (10%). Fatal acute myocardial infarction occurred in 1% of patients [1].
Thirteen percent of patients permanently discontinued erdafitinib treatment because of adverse reactions, most commonly eye disorders (6%). A total of 68% of patients required treatment interruptions because of adverse events, most commonly hyperphosphataemia (24%), stomatitis (17%), eye disorders (17%), and palmar-plantar erythrodysaesthesia syndrome (8%). Dose reductions because of adverse events were required in 53% of patients and were most commonly due to eye disorders (23%), stomatitis (15%), hyperphosphataemia (7%), palmar-plantar erythrodysaesthesia syndrome (7%), paronychia (7%), and nail dystrophy (6%) [1].
3.5 Companion Diagnostic
The FDA-approved test for selection of patients with locally advanced or metastatic urothelial carcinoma for treatment with erdafitinib is the QIAGEN therascreen® FGFR RGQ RT-PCR Kit [1].
3.6 Ongoing Clinical Trials
A phase III trial comparing erdafitinib with vinflunine or docetaxel or pembrolizumab in patients with advanced urothelial cancer and FGFR gene aberrations (NCT03390504) is currently recruiting patients [14]. The efficacy of erdafitinib as a treatment for FGFR-mutated and translocated squamous NSCLC is being investigated in the phase II FIND trial [15].
The US National Cancer Institute (NCI) is conducting 3 phase II trials in patients with solid tumours, lymphomas, myelomas or histiocytic disorders that include erdafitinib-containing treatment arms; the paediatric MATCH screening (NCT03155620) and treatment (NCT03210714) studies, and the MATCH (NCT02465060) screening study. The institute is also conducting a phase Ib trial evaluating a combination of erdafitinib, fulvestrant, and palbociclib in patients with ER+/HER2-/FGFR-amplified metastatic breast cancer (NCT03238196).
The Multiple Myeloma Research Consortium is conducting the phase I/II MyDRUG (Myeloma-Developing Regimens Using Genomics) trial (NCT03732703) in patients with relapsed refractory multiple myeloma, which includes an erdafitinib-containing treatment arm, and a trial evaluating erdafitinib in combination with dexamethasone in patients with multiple myeloma that returned after a period of improvement (NCT02952573).
4 Current Status
Erdafitinib received its first global approval on April 12 2019 in the USA as a treatment for adult patients with locally advanced or metastatic urothelial carcinoma that has susceptible FGFR3 or FGFR2 genetic alterations and has progressed during or following at least one line of prior platinum-containing chemotherapy, including within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy. Patients are selected for therapy based on the FDA-approved companion diagnostic for erdafitinib [1].
References
Janssen Pharmaceuticals. BALVERSATM (erdafitinib): US prescribing information. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212018s000lbl.pdf. Accessed 7 May 2019.
Karkera JD, Cardona GM, Bell K, et al. Oncogenic characterization and pharmacologic sensitivity of activating fibroblast growth factor receptor (FGFR) genetic alterations to the selective FGFR inhibitor erdafitinib. Mol Cancer Ther. 2017;16(8):1717–26.
Tabernero J, Bahleda R, Dienstmann R, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33(30):3401–8.
FDA. FDA approves first targeted therapy for metastatic bladder cancer [media release]. 12 Apr 2019. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm635906.htm.
Astex Therapeutics. Astex announces new drug discovery alliance with Janssen Pharmaceutica N.V [media release]. 9 Jun 2008. http://www.astex-therapeutics.com.
Astex Therapeutics. Astex announces new drug discovery collaboration with University of Newcastle upon Tyne and Cancer Research Technology Limited [media release]. 13 Mar 2006. http://www.astex-therapeutics.com.
Astex Pharmaceuticals Inc. SuperGen and Astex Therapeutics enter definitive merger agreement [media release]. 6 Apr 2011. https://astx.com/.
Otsuka Holdings Co Ltd. Otsuka Pharmaceutical completes acquisition of Astex Pharmaceuticals [media release]. 11 Oct 2013. http://www.otsuka.co.jp.
Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ-42756493 (erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol Cancer Ther. 2017;16(6):1010–20.
Nishina T, Takahashi S, Iwasawa R, et al. Safety, pharmacokinetic, and pharmacodynamics of erdafitinib, a pan-fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in patients with advanced or refractory solid tumors. Invest New Drugs. 2018;36(3):424–34.
Tabernero J, Infante JR, Mita A, et al. Pharmacokinetics (PK) of the pan-FGFR inhibitor erdafitinib in urothelial carcinoma [abstract no. 789P]. Ann Oncol. 2016;27(Supplement 6).
Siefker-Radtke AO, Necchi A, Park SH, et al. First results from the primary analysis population of the phase 2 study of erdafitinib (ERDA; JNJ-42756493) in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRalt) [abstract no. 4503]. J Clin Oncol. 2018;36(15 Supplement):4503.
Chen YY, Park JO, Su WC, et al. Preliminary results of a ph2a study to evaluate the clinical efficacy and safety of erdafitinib in Asian patients with biomarker-selected advanced cholangiocarcinoma (CCA) [abstract no. 624PD]. Ann Oncol. 2018;29(Supplement 8):viii209.
Loriot Y, Necchi A, Park SH, et al. Erdafitinib compared with vinflunine or docetaxel or pembrolizumab in patients (pts) with metastatic or surgically unresectable (M/UR) urothelial carcinoma (UC) and selected FGFR gene alterations (FGFRalt): the phase III THOR study [abstract no. 920TiP]. Ann Oncol. 2018;29(Supplement 8):viii327–viii8.
Nogova L, Malchers F, Zadoyan G, et al. FIND trial: a phase II study to evaluate the efficacy of the FGFR-inhibitor erdafitinib in FGFR-mutated and -translocated squamous NSCLC [abstract no. P1.01-72]. J Thorac Oncol. 2018;13 (10 Suppl.):S490.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflicts of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. A. Markham, a contracted employee of Adis International Ltd/Springer Nature, is responsible for the article content and declares no relevant conflicts of interest.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Markham, A. Erdafitinib: First Global Approval. Drugs 79, 1017–1021 (2019). https://doi.org/10.1007/s40265-019-01142-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01142-9