
Abstract
Futibatinib (Lytgobi®) is an oral, covalently binding, irreversible inhibitor of fibroblast growth factor receptor (FGFR)1–4 that is being developed by Taiho Oncology and Taiho Pharmaceutical for the treatment of cancers, including cholangiocarcinoma, breast cancer, gastric cancer, urothelial cancer, oesophageal cancer and non-small cell lung cancer. Futibatinib was approved in the USA on 30 September 2022 for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harbouring FGFR2 gene fusions or other rearrangements. This article summarizes the milestones in the development of futibatinib leading to this first approval.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.21508350. |
An oral, covalently binding irreversible FGFR1–4 inhibitor is being developed by Taiho Oncology and Taiho Pharmaceutical for the treatment of cancers |
Received its first approval on 30 September 2022 in the USA |
Approved for use in adult patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harbouring FGFR2 gene fusions or other rearrangements |
1 Introduction
Fibroblast growth factor receptor (FGFR) signalling plays an important role in cell proliferation, differentiation, migration and survival. Deregulated FGFR signalling (resulting from upregulation, mutation, amplification and chromosomal rearrangements of FGFR) often leads to oncogenesis [1]. Thus, FGFR inhibition is a viable therapeutic approach for cancers that involve these abnormalities [2]. A number of small-molecule tyrosine kinase inhibitors (TKIs) that competitively bind to the adenosine triphosphate (ATP) pocket of FGFR1–4 are under clinical development [3]. Toxicities associated with non-selective TKIs led to the development of FGFR-specific reversible TKIs [3]. However, most selective TKIs do not competitively bind to FGFR4 and drug resistance remains a challenge even with selective TKIs. Thus, irreversible FGFR inhibitors are being developed [3, 4].
Futibatinib (Lytgobi®) is an oral, small-molecule, covalently binding, irreversible TKI of FGFR1-4 that is being developed by Taiho Oncology and Taiho Pharmaceutical for the treatment of cancers, including cholangiocarcinoma (bile duct cancer). In September 2022, futibatinib received its first approval in the USA for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harbouring FGFR2 gene fusions or other rearrangements [5]. The accelerated approval was based on results from a phase II trial (FOENIX-CCA2). Futibatinib is available as 4 mg oral tablets and the recommended dosage is 20 mg once daily taken with or without food at approximately the same time each day, until disease progression or unacceptable toxicity occurs [5].

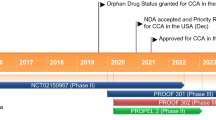
Key milestones in the development of futibatinib for cholangiocarcinoma. Est. estimated, NDA new drug application
Futibatinib as second- or later-line therapy for cholangiocarcinoma is filed for approval in the EU [6] and Japan [7]. Phase I or II trials of futibatinib are underway for breast cancer with FGFR1 and FGFR2 amplification, urothelial and oesophageal cancers (in combination with pembrolizumab) and non-small cell lung cancer (NSCLC) with KRAS mutations (in combination with binimetinib).
1.1 Company Agreements
In January 2020, the National Comprehensive Cancer Network entered into a collaboration with Taiho Oncology to conduct preclinical, translational and clinical trials of futibatinib as monotherapy and in combination with other drugs for malignancies with FGFR1–4 aberrations, supported by a $US2 million grant from Taiho Oncology [8]. In July 2022, the following projects were selected for the funding: a phase Ib trial of olaparib plus futibatinib in all solid tumours with BRCA1/2 alterations; a phase II trial of futibatinib plus pembrolizumab in metastatic microsatellite stable endometrial carcinoma; a phase II trial of futibatinib plus pembrolizumab in advanced or metastatic hepatocellular carcinoma with FGF19 expression after first line therapy; and, developing futibatinib for use on novel FGFR genomic alterations and in therapy combinations [9].
2 Scientific Summary
2.1 Pharmacodynamics
Futibatinib inhibits all four FGFR subtypes with IC50 values < 4 nM [10]. Unlike reversible ATP-competitive inhibitors, futibatinib rapidly forms a covalent adduct with a cysteine side chain in the P-loop of the FGFR tyrosine kinase domain, with an inherent ability to capture multiple FGFR P-loop conformations [11]. Through FGFR inhibition, futibatinib inhibits FGFR phosphorylation and downstream signalling, resulting in decreased cell viability in cancer cell lines harbouring FGFR alterations (fusions, rearrangements, amplifications and mutations) [10]. Futibatinib exhibited potent antitumour activity in FGFR-deregulated cancer cell lines and xenograft models [10]. In animal models, futibatinib showed synergistic antitumour effects with cytotoxic agents [12], PI3K pathway inhibitors [13] and a selective AKT inhibitor [14].
Futibatinib provided clinical benefits in patients with cholangiocarcinoma harbouring FGFR2 fusion and other rearrangements (Sect. 2.3.1). It demonstrated antitumour activity in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma who developed resistance to ATP-competitive FGFR inhibitors due to multiple FGFR2 mutations in the kinase domain [15]. Futibatinib also showed clinical activity in patients with intrahepatic cholangiocarcinoma harbouring FGFR2 extracellular domain in-frame deletions [16].
Since FGFRs play a crucial role in phosphorus homeostasis, FGFR inhibition by futibatinib dose-dependently increases serum phosphate levels, with increased risk of hyperphosphatemia at higher futibatinib exposure [4, 5].

Chemical structure of futibatinib
2.2 Pharmacokinetics
The pharmacokinetic properties of oral futibatinib 20 mg once daily have been evaluated in patients with advanced solid tumours [4, 5]. Futibatinib exposure increased in a dose-proportional manner over a dose range of 4–24 mg [5]. Following a 20 mg dose, the median time to maximum futibatinib plasma concentration was 2 h. The geometric mean apparent volume of distribution of futibatinib was 66 L. Futibatinib is 95% bound to human plasma protein, primarily to albumin and α1-acid glycoprotein. The drug is metabolized mainly by CYP3A, and to a lesser extent by CYP2C9 and CYP2D6, with unchanged drug being the main active moiety. Following a single 20 mg oral dose of radiolabelled futibatinib, ≈ 91% of the total radioactivity was recovered in faeces and 9% in urine, with an insignificant proportion of unchanged drug in urine or faeces. The mean elimination half-life of futibatinib is 2.9 h and the geometric mean apparent clearance is 20 L/h [5].
There were no clinically meaningful differences in futibatinib exposure based on age (18–82 years), sex, race (White, Asian, and African American), body weight (36–152 kg), mild to moderate kidney function impairment or mild liver function impairment [5]. The effects of severe kidney function impairment, kidney failure and moderate or severe liver function impairment on futibatinib pharmacokinetics have not been studied [5].
Concomitant use of futibatinib with dual P-glycoprotein (P-gp) and strong CYP3A inhibitors (which may increase futibatinib exposure) or with dual P-gp and strong CYP3A inhibitors (which may decrease futibatinib exposure) should be avoided [5]. As futibatinib is an inhibitor of P-gp and BCRP, it may increase the exposure of drugs that are substrates of these proteins [5].
Features and properties of futibatinib
Alternative names | LYTGOBI; TAS-120 |
Class | Amines; antineoplastics; ketones; phenyl ethers; pyrazoles; pyrimidines; pyrrolidines; small molecules |
Mechanism of action | Fibroblast growth factor receptor (FGFR) antagonist |
Route of administration | Oral |
Pharmacodynamics | Inhibits FGFR phosphorylation and downstream signalling; exhibits antitumour activity in FGFR-deregulated cancer cell lines and xenograft models; increases serum phosphate levels, with increased risk of hyperphosphatemia at higher drug exposure |
Pharmacokinetics | Tmax 2 h, Vd/F 66 L, plasma protein binding 95%, primarily metabolised by CYP3A, t1/2 2.9 h, CL/F 20 L/h, faecal excretion is the major route of elimination |
Adverse events | |
Most frequent grade ≥ 3 treatment-related | Hyperphosphatemia, increased ALT, increased AST, fatigue, stomatitis and palmar-plantar erythrodysesthesia syndrome |
ATC codes | |
WHO ATC code | L01-EN04 (Futibatinib) |
EphMRA ATC code | L1 (Antineoplastics) |
Chemical name | 1-[(3S)-3-[4-amino-3-[2-(3,5-dimethoxyphenyl)ethynyl]pyrazolo[3,4-d]pyrimidin-1-yl]pyrrolidin-1-yl]prop-2-en-1-one |
2.3 Therapeutic Trials
Phase I pharmacological and safety data supported futibatinib 20 mg once daily as the recommended phase II dose [4, 17, 18].
2.3.1 Intrahepatic Cholangiocarcinoma
Futibatinib demonstrated efficacy in patients with advanced/metastatic, unresectable, intrahepatic cholangiocarcinoma harbouring FGFR2 fusion/rearrangements in the pivotal, single-arm, phase II part (FOENIX-CCA2) of an open-label, multicentre phase I/II trial (NCT02052778) [5, 19]. Eligible patients had disease progression after one or more lines of systemic therapy (including gemcitabine plus platinum-based chemotherapy), no prior FGFR inhibitor treatment and an Eastern Cooperative Oncology Group performance status of 0 or 1. Patients received futibatinib 20 mg once daily until disease progression or unacceptable toxicity. The primary endpoint of objective response rate (ORR; per RECIST 1.1 criteria, assessed by an independent central review, with a target ORR of 20% for futibatinib) was 42% (95% CI 32–52) in 103 evaluable patients. ORRs were consistent in patients with FGFR2 fusions, FGFR2 rearrangements, BICC1 and non-BICC1 fusion partners, and co-occurring mutations. In the overall population, the median duration of response (DoR) was 9.7 months (95% CI 7.6–17.1); DoR was ≥ 6 and ≥ 12 months in 72% and 14% of responders, respectively. Disease control rate was 82.5%, median progression-free survival (PFS) was 9.0 months and median overall survival (OS) was 21.7 months (12-month OS rate 72%) [5, 19]. The primary analysis findings were confirmed by a final analysis, which included an additional 8 months’ follow-up [20].
In FOENIX-CCA2, physical, cognitive and emotional functioning, and overall health status were maintained during 9 months of futibatinib treatment, based on patient-reported outcomes assessed using the EORTC QLQ-C30, EQ-5D-3L and EQ visual analogue scale [21].
A simulated indirect treatment comparison using patient-level data from FOENIX-CCA2 and other published aggregated data suggests that futibatinib may be similar to pemigatinib and is better than chemotherapy in terms of PFS and OS benefits [22].
2.3.2 Advanced Solid Tumours
Futibatinib showed preliminary activity in patients with advanced solid tumours in the first-in-human, phase I dose-escalation part of the NCT02052778 trial [4]. In the dose-expansion part, 170 patients with advanced solid tumours (cholangiocarcinoma and gastric, urothelial, CNS, head and neck, and breast cancer) received futibatinib 20 mg [23]. The ORR was 13.7% in the overall cohort and 25.4% in patients with FGFR2 fusion/rearrangement-positive intrahepatic cholangiocarcinoma [23].
In a phase I trial (JapicCTI-142552), futibatinib showed antitumour activity in Japanese patients with advanced solid tumours, including gastric cancer [18, 24]. In a phase Ib trial (JapicCTI-195063), futibatinib in combination with pembrolizumab [an immune checkpoint inhibitor (ICI)] showed antitumour activity in patients advanced or metastatic solid tumours, including oesophageal cancer; the activity was seen in ICI-naïve as well as in ICI-refractory patients [25].
Key clinical trials of futibatinib sponsored by Taiho Oncology
Drug(s) | Indication | Phase | Status | Location(s) | Identifier |
|---|---|---|---|---|---|
Futibatinib | Cholangiocarcinoma (2nd line) | I/II | Active, not recruiting | Global | NCT02052778, EudraCT2013-004810-16, FOENIX-101, FOENIX-CCA2 |
Futibatinib | Cholangiocarcinoma (1st line) | III | Active, not recruiting | Global | NCT04093362, EudraCT2019-004630-42, FOENIX-CCA3 |
Futibatinib, fulvestrant | Breast cancer with FGFR-1, -2 amplification | II | Recruiting | Global | NCT04024436, EudraCT2019-001164-30, FOENIX-MBC2 |
Futibatinib | Tumours with FGFR aberrations | II | Active, not recruiting | Global | NCT04189445, EudraCT2019-004084-49, JapicCTI205312 |
Futibatinib, pembrolizumab | Urothelial cancer | II | Recruiting | France, Spain, USA | NCT04601857 |
Futibatinib, binimetinib | Non-small cell lung cancer with KRAS mutations | I/II | Recruiting | USA | NCT04965818 |
Futibatinib, pembrolizumab | Solid tumours (including oesophageal cancer) | Ib | Recruiting | Japan | JapicCTI-195063 |
2.4 Adverse Events
2.4.1 Intrahepatic Cholangiocarcinoma
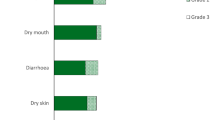
In FOENIX-CCA2, two patients (1.9%) discontinued treatment because of treatment-related adverse events (TRAEs) [19]. Adverse reactions led to dose interruption in 66% of patients and dose reductions in 58% of patients [5]. The most common (incidence ≥ 20%) adverse reactions with futibatinib were nail toxicity, musculoskeletal pain, constipation, diarrhoea, fatigue, dry mouth, alopecia, stomatitis, abdominal pain, dry skin, arthralgia, dysgeusia, dry eye, nausea, decreased appetite, urinary tract infection, palmar-plantar erythrodysesthesia syndrome (PPES) and vomiting. The most common (incidence ≥ 2%) grade 3 adverse reactions were fatigue (8%), stomatitis (6%), PPES (4.9%), musculoskeletal pain (3.9%), weight loss (3.9%), abdominal pain (2.9%), decreased appetite (2.9%) and urinary tract infection (2.9%). The most common (incidence ≥ 5%) grade 3 or 4 laboratory abnormalities were decreased haemoglobin, decreased lymphocytes, decreased phosphate, increased phosphate, increased aspartate aminotransferase (AST), increased creatine kinase and increased activated partial thromboplastin time. Serious adverse reactions occurred in 39% of futibatinib recipients; the most common (incidence ≥ 2%) of these were pyrexia (3.9%), gastrointestinal haemorrhage (3.9%), ascites (2.9%), musculoskeletal pain (2.9%) and bile duct obstruction (2.9%) [5]. Results of the final analysis of FOENIX-CCA2 were consistent with that of the primary analysis, with no new safety signals [20].
2.4.2 Breast Cancer
In an ongoing phase II trial (NCT04024436), eight patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative, metastatic breast cancer harbouring high-level FGFR1 amplification were treated with futibatinib 20 mg once daily in combination with intramuscular fulvestrant 500 mg on days 1 and 15 of cycle 1, and on day 1 of every subsequent 28-day cycle (median treatment duration 8 weeks) [26]. All patients experienced TRAEs, with 25% experiencing grade ≥ 3 TRAEs. The most common TRAEs were hyperphosphatemia (88%; grade ≥ 3: 12%), constipation (62%), transaminase elevation (50%), dry mouth (38%) and alopecia (38%). TRAEs led to treatment discontinuation in one patient, dose reductions in four patients and dosing interruptions in three patients. There were no dose-limiting toxicities (in five evaluable patients), serious adverse events (AEs) or fatal AEs [26].
2.4.3 Urothelial Carcinoma
In an ongoing phase II trial (NCT04601857), six patients with advanced or metastatic urothelial carcinoma were treated with futibatinib 20 mg once daily in combination with intravenous pembrolizumab 200 mg every 21 days (median treatment duration 48 days for futibatinib and 35 days pembrolizumab) [27]. AEs occurred in all patients and they led to any study drug discontinuation, dose interruption and dose modification in three patients each. AEs occurring in more than two patients included diarrhoea, hyperphosphatemia, increased AST and pruritis. Grade 3 AEs (increased AST, maculopapular rash, myositis) occurred in two patients. There were no dose-limiting toxicities or grade 4–5 AEs [27].
2.4.4 Pooled Analysis
An integrated safety analysis showed that futibatinib 20 mg once daily (median treatment duration 111 days) had a manageable tolerability profile and an acceptable safety profile in 318 patients with advanced solid tumours harbouring FGFR aberrations [28]. There was no grade 5 TRAE; grade 3 and 4 TRAEs (incidence 45% and 1%) were manageable with dosage reduction (36%) and/or interruptions (42%). TRAEs led to treatment discontinuation in 3% of patients. The most frequent grade ≥ 3 TRAEs were hyperphosphatemia (23%), increased alanine transaminase (ALT; 6%), increased AST (5%), fatigue (3%), stomatitis (3%) and PPES (≈ 3%). AEs of special interest with futibatinib were hyperphosphatemia, hepatotoxicity, nail toxicities, PPES, retinal disorders and rash. There were no grade 4 hyperphosphatemia and no patient discontinued treatment because of hyperphosphatemia. Grade 3–4 nail toxicity occurred in 1% of patients and there were no grade 3–4 retinal disorders or rash. Most grade ≥ 3 AEs of special interest resolved to grade < 3 within 7–8 days [28].
2.5 Ongoing Clinical Trials
The FOENIX-CCA2 trial in patients with cholangiocarcinoma is still ongoing. A phase III trial (NCT04093362; FOENIX-CCA3) is evaluating futibatinib versus gemcitabine-cisplatin as first-line therapy in patients with advanced cholangiocarcinoma. A phase II trial (NCT04189445) is evaluating futibatinib in tumours with specific FGFR aberrations. The phase II trials in patients with breast cancer (NCT04024436; FOENIX-MBC2) and urothelial cancer (NCT04601857) are ongoing.
A phase Ib/II trial (NCT04965818) is evaluating futibatinib plus binimetinib (a MEK inhibitor) in patients with KRAS mutation-positive advanced NSCLC. The phase Ib trial (JapicCTI-195063) of futibatinib plus pembrolizumab in patients advanced solid tumours, including oesophageal cancer, is ongoing.
3 Current Status
Futibatinib received its first approval on 30 September 2022 in the USA for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harbouring FGFR2 gene fusions or other rearrangements [29].
References
Haugsten EM, Wiedlocha A, Olsnes S, et al. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8(11):1439–52.
Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. 2016;22(1):259–67.
Krook MA, Reeser JW, Ernst G, et al. Fibroblast growth factor receptors in cancer: genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br J Cancer. 2021;124(5):880–92.
Bahleda R, Meric-Bernstam F, Goyal L, et al. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann Oncol. 2020;31(10):1405–12.
Taiho Oncology. LYTGOBI® (futibatinib) tablets, for oral use: US prescribing information. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214801s000lbl.pdf. Accessed 21 Oct 2022.
European Medicines Agency. Medicines for human use under evaluation. 2022. https://www.ema.europa.eu/en/medicines/medicines-human-use-under-evaluation. Accessed 21 Oct 2022.
Taiho Pharmaceutical Co. Taiho Pharmaceutical submits new drug application of FGFR inhibitor futibatinib (TAS-120) for biliary tract cancer [media release]. 28 Jul 2022. http://www.taiho.co.jp.
National Comprehensive Cancer Network. NCCN Oncology Research Program to oversee trials of the FGFR inhibitor futibatinib in tumors with aberrant FGFR expression, in collaboration with Taiho Oncology [media release]. 27 Jan 2020. http://www.nccn.org.
National Comprehensive Cancer Network. NCCN Oncology Research Program announces projects selected for funding to study futibatinib in tumors with aberrant FGFR expression, in collaboration with Taiho Oncology [media release]. 23 Jul 2020. http://www.nccn.org.
Sootome H, Fujita H, Ito K, et al. Futibatinib Is a novel irreversible FGFR 1–4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020;80(22):4986–97.
Kalyukina M, Yosaatmadja Y, Middleditch MJ, et al. TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. ChemMedChem. 2019;14(4):494–500.
Sootome H, Miura A, Komori T, et al. Futibatinib (TAS-120) plus chemotherapy demonstrates a synergistic effect across various FGFR-deregulated cancer cell lines and xenograft models [abstract no. 564]. Cancer Res. 2020;80(Suppl 16).
Miura A, Sootome H, Komori T, et al. Synergistic antitumor activity of futibatinib (TAS-120), a FGFR1-4 inhibitor, and PI3K pathway inhibitors [abstract no. 659]. Cancer Res. 2020;80(Suppl 16).
Iwasaki J, Kuramoto T, Komori T. Synergistic antitumor activity of futibatinib, an FGFR1-4 inhibitor, and TAS-117, a selective AKT inhibitor, in FGFR-deregulated cancer models [abstract no. 661]. Cancer Res. 2020;80(Suppl 16).
Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion–positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9(8):1064–79.
Cleary JM, Raghavan S, Wu Q, et al. FGFR2 extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov. 2021;11(10):2488–505.
Hollebecque A, Bridgewater JA, Meric-Bernstam F, et al. Assessment of futibatinib exposure-response (E-R) relationships in patients with advanced solid tumors, including cholangiocarcinoma (CCA) [abstract no. 52P]. Ann Oncol. 2021;32(Suppl 5):S378.
Morizane C, Kojima T, Kuboki Y, et al. Phase I study of the irreversible FGFR inhibitor (i) futibatinib (FBN; TAS-120) in Japanese patients (pts) with advanced (adv) solid tumours [abstract no. 544P]. Ann Oncol. 2020;31(Suppl 4):S475.
Goyal L, Meric-Bernstam F, Hollebecque A, et al. Primary results of phase 2 FOENIX-CCA2: the irreversible FGFR1-4 inhibitor futibatinib in intrahepatic cholangiocarcinoma (iCCA) with FGFR2 fusions/rearrangements [abstract no. CT010]. Cancer Res. 2021;81(Suppl 13).
Goyal L, Meric-Bernstam F, Hollebecque A, et al. Updated results of the FOENIX-CCA2 trial: efficacy and safety of futibatinib in intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 fusions/ rearrangements [abstract no. 4009]. J Clin Oncol. 2022;40(Suppl 16).
Valle JW, Hollebecque A, Furuse J, et al. FOENIX-CCA2 quality of life data for futibatinib-treated intrahepatic cholangiocarcinoma (iCCA) patients with FGFR2 fusions/rearrangements [abstract no. 4097]. J Clin Oncol. 2021;39(Suppl 15).
Borad MJ, Paine A, Wacheck V, et al. Indirect treatment comparison of futibatinib with chemotherapy and pemigatinib in cholangiocarcinoma with FGFR2 fusions/rearrangements [abstract no. 440]. J Clin Oncol. 2022;40(Suppl 4).
Meric-Bernstam F, Bahleda R, Hierro C, et al. Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: a phase I dose-expansion study. Cancer Discov. 2022;12(2):402–15.
Kuboki Y, Shitara K, Morizane C, et al. Phase I study of the irreversible FGFR inhibitor futibatinib in Japanese patients with advanced solid tumors: updated dose expansion results and activity in gastric cancer [abstract no. 1383P]. Ann Oncol. 2021;32(Suppl 5):S1047–8.
Muro K, Kato K, Chin K, et al. Phase Ib study of futibatinib plus pembrolizumab in patients with advanced or metastatic solid tumors: tolerability results and antitumor activity in esophageal carcinoma [abstract no. 1241P]. Ann Oncol. 2022;33(Suppl 7):S1116.
Damodaran S, Unni N, Giridhar KV, et al. Futibatinib in combination with fulvestrant in patients with metastatic breast cancer (MBC) harboring high-level FGFR1 amplification: preliminary data from a phase 2 study [abstract no. P1-18-35]. Cancer Res. 2022;82(Suppl 4).
Koshkin VS, Sonpavde GP, Hwang C, et al. Futibatinib plus pembrolizumab in patients (pts) with advanced or metastatic urothelial carcinoma (mUC): preliminary safety results from a phase 2 study [abstract no. 501]. J Clin Oncol. 2022;40(Suppl 6).
Meric-Bernstam F, Furuse J, Oh D, et al. Pooled analysis safety profile of futibatinib in patients with advanced solid tumors, including intrahepatic cholangiocarcinoma (iCCA) [abstract no. 51P]. Ann Oncol. 2021;32(Suppl 5):S378.
Taiho Oncology. FDA approves Taiho's LYTGOBI® (futibatinib) tablets for previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma [media release]. 30 Sep 2022 2022. http://www.taihooncology.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Yahiya Y. Syed is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Syed, Y.Y. Futibatinib: First Approval. Drugs 82, 1737–1743 (2022). https://doi.org/10.1007/s40265-022-01806-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01806-z