
Abstract
Relebactam (formerly known as MK-7655) is a non-β-lactam, bicyclic diazabicyclooctane, β-lactamase inhibitor that is structurally related to avibactam, differing by the addition of a piperidine ring to the 2-position carbonyl group. Vaborbactam (formerly known as RPX7009) is a non-β-lactam, cyclic, boronic acid-based, β-lactamase inhibitor. The structure of vaborbactam is unlike any other currently marketed β-lactamase inhibitor. Both inhibitors display activity against Ambler class A [including extended-spectrum β-lactamases (ESBLs), Klebsiella pneumoniae carbapenemases (KPCs)] and class C β-lactamases (AmpC). Little is known about the potential for relebactam or vaborbactam to select for resistance; however, inactivation of the porin protein OmpK36 in K. pneumoniae has been reported to confer resistance to both imipenem–relebactam and meropenem–vaborbactam. The addition of relebactam significantly improves the activity of imipenem against most species of Enterobacteriaceae [by lowering the minimum inhibitory concentration (MIC) by 2- to 128-fold] depending on the presence or absence of β-lactamase enzymes. Against Pseudomonas aeruginosa, the addition of relebactam also improves the activity of imipenem (MIC reduced eightfold). Based on the data available, the addition of relebactam does not improve the activity of imipenem against Acinetobacter baumannii, Stenotrophomonas maltophilia and most anaerobes. Similar to imipenem–relebactam, the addition of vaborbactam significantly (2- to > 1024-fold MIC reduction) improves the activity of meropenem against most species of Enterobacteriaceae depending on the presence or absence of β-lactamase enzymes. Limited data suggest that the addition of vaborbactam does not improve the activity of meropenem against A. baumannii, P. aeruginosa, or S. maltophilia. The pharmacokinetics of both relebactam and vaborbactam are described by a two-compartment, linear model and do not appear to be altered by the co-administration of imipenem and meropenem, respectively. Relebactam’s approximate volume of distribution (V d) and elimination half-life (t ½) of ~ 18 L and 1.2–2.1 h, respectively, are similar to imipenem. Likewise, vaborbactam’s V d and t½ of ~ 18 L and 1.3–2.0 h, respectively, are comparable to meropenem. Like imipenem and meropenem, relebactam and vaborbactam are both primarily renally excreted, and clearance correlates with creatinine clearance. In vitro and in vivo pharmacodynamic studies have reported bactericidal activity for imipenem–relebactam and meropenem–vaborbactam against various Gram-negative β-lactamase-producing bacilli that are not inhibited by their respective carbapenems alone. These data also suggest that pharmacokinetic–pharmacodynamic parameters correlating with efficacy include time above the MIC for the carbapenems and overall exposure for their companion β-lactamase inhibitors. Phase II clinical trials to date have reported that imipenem–relebactam is as effective as imipenem alone for treatment of complicated intra-abdominal infections and complicated urinary tract infections, including acute pyelonephritis. Imipenem–relebactam is currently in two phase III clinical trials for the treatment of imipenem-resistant bacterial infections, as well as hospital-associated bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP). A phase III clinical trial has reported superiority of meropenem–vaborbactam over piperacillin–tazobactam for the treatment of complicated urinary tract infections, including acute pyelonephritis. Meropenem–vaborbactam has recently demonstrated higher clinical cure rates versus best available therapy for the treatment of carbapenem-resistant Enterobacteriaceae (CRE), as well as for HABP and VABP. The safety and tolerability of imipenem–relebactam and meropenem–vaborbactam has been reported in various phase I pharmacokinetic studies and phase II and III clinical trials. Both combinations appear to be well tolerated in healthy subjects and hospitalized patients, with few serious drug-related treatment-emergent adverse events reported to date. In conclusion, relebactam and vaborbactam serve to broaden the spectrum of imipenem and meropenem, respectively, against β-lactamase-producing Gram-negative bacilli. The exact roles for imipenem–relebactam and meropenem–vaborbactam will be defined by efficacy and safety data from further clinical trials. Potential roles in therapy for these agents include the treatment of suspected or documented infections caused by resistant Gram-negative bacilli-producing ESBL, KPC, and/or AmpC β-lactamases. The usage of these agents in patients with CRE infections will likely become the standard of care. Finally, increased activity of imipenem–relebactam against P. aeruginosa may be of clinical benefit to patients with suspected or documented P. aeruginosa infections.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Carbapenems, which are increasing in usage worldwide, have the broadest spectrum of activity of all β-lactam antimicrobials and therefore are often reserved for severe, complicated, and multidrug-resistant (MDR) infections [1, 2]. Resistance to carbapenems has emerged in Enterobacteriaceae, P. aeruginosa, and A. baumannii [3]. The World Health Organization ranks MDR and carbapenem-resistant Enterobacteriaceae (CRE), particularly K. pneumoniae and Escherichia coli, as well as carbapenem-resistant P. aeruginosa and A. baumannii as the three most critical antimicrobial-resistant bacteria on their global priority list to guide research, discovery, and development of new antimicrobials [4]. For infections where effective antimicrobial agents are limited, older agents with well-defined toxicities such as polymyxins, aminoglycosides, and tigecycline may be necessary to treat MDR infections [5, 6]. There is a critical need for new antimicrobials that have increased efficacy and/or decreased toxicity to treat patients with carbapenem-resistant and MDR infections.
Resistance to carbapenems arises from increased antimicrobial efflux, decreased permeability, and most importantly the production of β-lactamases, including carbapenemases [6]. In Enterobacteriaceae, carbapenem resistance is frequently attributable to the production of carbapenemases such as K. pneumoniae carbapenemases (KPCs) as well as to the production of class B metallo-β-lactamases (e.g., NDM, IMP, VIM), and class D β-lactamases (OXA-type) [6, 7]. In P. aeruginosa, the carbapenem resistance is often a combination of decreased expression of OprD (a porin protein), increased efflux, and increased production of AmpC [7].
Older β-lactamase inhibitors such as tazobactam, clavulanic acid, and sulbactam are effective in potentiating the activities of β-lactam antimicrobials against some class A β-lactamase-producing Enterobacteriaceae [8]. However, these older β-lactamase inhibitors have no activity against carbapenemases and limited to no effect versus some class C enzymes, including AmpC [8]. The novel β-lactamase inhibitors relebactam (formerly known as MK-7655) and vaborbactam (formerly known as RPX7009) are being developed to address the need for agents with activity against carbapenem-resistant Gram-negative bacilli.
Relebactam and vaborbactam are the first β-lactamase inhibitors developed for use in combination with carbapenems, specifically with imipenem (relebactam) and meropenem (vaborbactam). Both relebactam and vaborbactam have displayed in vitro activity against class A β-lactamases (e.g. KPC) and class C β-lactamases (e.g. AmpC) [5]. In vitro, both novel β-lactamase inhibitors have restored the activity of carbapenems against various phenotypes and genotypes of CRE, especially in KPC-producing isolates [7, 9]. Relebactam has also been shown to be effective in potentiating imipenem activity against P. aeruginosa [6, 7].
Vaborbactam in combination with meropenem (proprietary name, Vabomere) was the first of these two combination agents to complete a randomized, comparative Phase III clinical trial (TANGO I). In the TANGO I trial, meropenem–vaborbactam was assessed for the treatment of complicated urinary tract infections (cUTI), including acute pyelonephritis (AP) (http://clinicaltrials.gov, identifier NCT02166476) and was shown to provide superior efficacy compared to piperacillin-tazobactam and to be safe [10]. In a phase III randomized, comparative trial (TANGO II), meropenem–vaborbactam has recently demonstrated higher clinical cure rates versus best available therapy for the treatment of patients with CRE infections, including patients with hospital-associated bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP) (NCT02168946 and NCT03006679). Relebactam is currently in phase III clinical trials in combination with imipenem and the renal dehydropeptidase-1 inhibitor cilastatin, for the treatment of imipenem-resistant bacterial infections, HABP, and VABP (NCT02452047 and NCT02493764). Whenever imipenem is referred to in the review, unless otherwise stipulated, it refers to imipenem/cilastatin. Two completed Phase II clinical trials have reported efficacy and safety of imipenem–relebactam in the treatment of complicated intra-abdominal infections (cIAI) and cUTI/AP compared to imipenem treatment alone (NCT01506271 and NCT01505634).
This review considered all published data for imipenem–relebactam and meropenem–vaborbactam, including chemistry, mechanisms of action, mechanisms of resistance, microbiology, pharmacokinetics, pharmacodynamics, and efficacy and safety results from animal studies and clinical trials. Literature was obtained via a standard comprehensive search (up to October 2017) of PubMed for all materials including the terms “imipenem” and “relebactam” or “MK-7655”, “meropenem” and “vaborbactam” or “RPX7009”, and “Carbavance”. All results were reviewed by at least two authors and supplemented with abstracts and posters from scientific meetings as well as citations from relevant review articles.
Vaborbactam has been previously studied with biapenem, a carbapenem marketed in Japan. Compared to meropenem, biapenem demonstrates stability against certain Ambler class B and D β-lactamases (e.g. NDM-1 and OXA-48) and is more resistant to efflux by P. aeruginosa [8, 11, 12]. However, meropenem’s safety record and ultimately its registration status lead to further clinical development in combination with vaborbactam [11]. As biapenem is only currently approved for use in Japan and is no longer being studied in combination with vaborbactam, these studies will not be discussed in this review [13, 14].
2 Chemistry
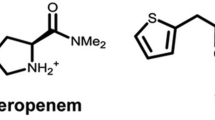
Carbapenems originated from the compound thienamycin, which is an antimicrobial produced by the soil bacterium Streptomyces cattleya [2]. However, due to thienamycin’s chemical instability, carbapenems with increased stability such as imipenem and meropenem were developed and approved for use in the USA in 1987 and 1996, respectively [1]. Carbapenems (imipenem and meropenem) differ from penicillins by having a carbon in place of the sulfur atom at position 1 and an unsaturated double bond between carbons 2 and 3 in their thiazolidine ring structure (Fig. 1) [2]. Carbapenems are well known for their intrinsic resistance to β-lactamases, including class A extended-spectrum β-lactamases (ESBLs) and class C β-lactamases (AmpCs) [2, 6]. The stability of carbapenems to β-lactamases arises from the trans-1α-hydroxyethyl substituent at carbon 6 [2]. The trans configuration at carbon 6 is characteristic of carbapenems compared to the cis side chains of penicillins and cephalosporins [1].

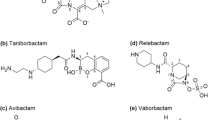
Chemical structures of imipenem (a), relebactam (b), avibactam (c), meropenem (d), and vaborbactam (e)
Even though imipenem demonstrated increased chemical stability compared to thienamycin, it is still susceptible to degradation by dehydropeptidase-1 (DHP-1) in the proximal renal tubules of mammals [1]. Therefore, imipenem must be administered with cilastatin, an inhibitor of DHP-1. Unlike imipenem, meropenem is intrinsically resistant to DHP-1 degradation due to the addition of a 1-β-methyl group on carbon 1 located in the carbapenem core [2]. Structurally, meropenem also differs from imipenem with a pyrrolidinyl substituent at position 2, which presumably increases its activity against Gram-negative bacteria, including P. aeruginosa [1]. With regard to the MexAB-OprM efflux system in P. aeruginosa, meropenem acts as a substrate due to the presence of a hydrophobic side chain at position 2 versus imipenem, which possesses a hydrophilic side chain making it a poor substrate for the MexAB-OprM efflux system [15, 16].
2.1 Imipenem–Relebactam
Relebactam is a potent non-β-lactam, bicyclic diazabicyclooctane, β-lactamase inhibitor (Fig. 1) [17]. It is structurally related to avibactam (a β-lactamase inhibitor currently approved for use in combination witsh ceftazidime) with the main distinguishing feature being the addition of a piperidine ring to the 2-position carbonyl group (Fig. 1) [17, 18]. Relebactam is highly reactive due to its highly strained bicyclic urea core and electron withdrawing aminoxy sulfate moiety [19]. The high reactivity results in limited stability in the presence of base or nucleophiles; however, this is also the same property that makes this compound a potent β-lactamase inhibitor [19]. Stability is only achieved at pH 4–8 in aqueous solution [19]. The positive charge on the piperidine side chain at physiological pH was shown to be essential in preventing efflux of relebactam from bacterial cells [18].
2.2 Meropenem–Vaborbactam
Vaborbactam is a non-β-lactam, cyclic boronic acid with high affinity to serine β-lactamases (Fig. 1) [11, 17]. Vaborbactam’s boronic ester ring was designed with the intention of constraining the inhibitor into a preferred conformation to increase potency [11]. The boron atom in vaborbactam acts as an electrophile and forms a reversible covalent bond with the catalytic serine of specific β-lactamases [11, 17]. As boronic acids are potent serine protease inhibitors, there was concern vaborbactam would potentially inhibit other important mammalian serine proteases in vivo [5, 11]. However, Hecker et al showed that vaborbactam had IC50 values ≥ 1000 μM for inhibition of 11 common mammalian serine proteases, confirming selective inhibition exclusively to serine β-lactamases [11]. Vaborbactam also has a 2-thienylacetyl side chain similar to the side chains of cephalothin and cefoxitin [11, 20].
3 Mechanisms of Action
Carbapenems, like other β-lactam antimicrobials, inhibit cross-linking of peptidoglycan, the structural component in bacterial cell walls by binding to penicillin binding proteins (PBPs) [1]. The inability of bacterial cells to form an intact cell wall ultimately leads to cell death, which classifies β-lactams as bactericidal antimicrobials [1]. Carbapenems enter the periplasmic space of Gram-negative bacteria by passing through outer membrane proteins (OMPs) [2]. Important OMPs include OprD in P. aeruginosa and OmpK in K. pneumoniae, as decreased expression can limit uptake and consequently decrease the activity of antimicrobials [2].
Increased potency of meropenem compared to imipenem against Gram-negative bacilli is partially explained by differing affinities to various PBPs [1]. Imipenem has weak affinity for PBP3 but preferentially binds to PBP2, followed by PBPs 1a and 1b [1]. Alternatively, meropenem preferentially binds to PBP2, followed by PBPs 3, 1a, and 1b [1]. The preferable affinity to PBPs is unlike aminopenicillins and cephalosporins, which primarily bind PBP3 [1, 2]. The low affinity of carbapenems to PBP3 is predicted to be responsible for their ability to achieve cell lysis without filamentation, which allows for a less significant increase in cell mass before lysis and less lipopolysaccharide (endotoxin) release [1, 2, 21].
Mechanistically both relebactam and vaborbactam exhibit their effects by inhibiting β-lactamase enzymes produced by Gram-negative bacilli. Available published data displaying the activities of relebactam and vaborbactam against different β-lactamases can be found in Tables 1 and 2, respectively. The current prevalence of the various β-lactamases per individual country is beyond the scope of this review; however, β-lactamases, including ESBL, and carbapenemases, such as KPC and NDM continue to spread worldwide The studies summarized in Tables 1 and 2 used assays that measured the ability of each β-lactamase inhibitor to prevent degradation of nitrocefin [11, 18, 22,23,24]. Ki and IC50 values from various studies are not directly comparable due to small variations in assays; however, Tables 1 and 2 are useful for a general comparison of relebactam and vaborbactam to other β-lactamase inhibitors currently used in clinical practice. Table 3 provides a comparative summary of activities for various β-lactamase inhibitors including relebactam, vaborbactam, avibactam, clavulanic acid, sulbactam, and tazobactam, compiled from available studies and publications [5, 24,25,26,27].
β-lactamase enzymes are either classified based on genetic similarly (amino acid sequence), as described by Ambler et al which groups enzymes into classes A, B, C, and D, or based on functional similarity as described by Bush et al [28,29,30]. For simplicity, only the Ambler classification system will be referred to in this review. Vaborbactam and relebactam demonstrate potent activity against class A β-lactamases, especially KPCs, and class C β-lactamases (Tables 1 and 2). Of interest, it was recently shown that vaborbactam demonstrates activity against the newly discovered class A carbapenemases BKC-1 and FRI-1 found in clinical isolates of K. pneumoniae and E. cloacae, respectively (Table 2) [22].
As demonstrated in Table 1, relebactam has no activity against OXA class D β-lactamases produced by A. baumannii [18]. However, relebactam has been shown to demonstrate variable potentiation of imipenem in vitro against OXA-48 producing Enterobacteriaceae [6, 7]. Neither relebactam nor vaborbactam has consistently demonstrated activity against class B metallo-β-lactamases (e.g. NDM, IMP, VIM), which utilize a zinc atom in their active site or class D β-lactamases (e.g. OXA) [5, 31]. Winkler et al identified reduced activity of relebactam against the class A carbapenemase, GES-2 (Ki app, 19 ± 2 μM), potentially revealing a limitation to class A carbapenemase inhibition by relebactam [32].
3.1 Imipenem–Relebactam
The mechanism of action of relebactam is not well described in the literature; however, relebactam is predicted to have a mechanism of action similar to avibactam because of their similar chemical structures [12, 17, 31]. Avibactam rapidly acylates β-lactamases and slowly reversibly de-acylates from these enzymes, producing a regenerated enzyme and an active inhibitor capable of rebinding [33]. Acylation occurs between the active site serine residue of the β-lactamase and the carbonyl at position 7 in the cyclic urea core of avibactam [24]. The recyclization of the 5-membered ring is unlike other β-lactamase inhibitors such as clavulanic acid, which have 4-membered rings and require much more energy to reform their ring structures [31].
Barnes et al determined that relebactam has more potent activity against PER-2, a β-lactamase produced by P. aeruginosa, compared to avibactam (Ki app 5.8 ± 0.6 and 29 ± 3 μM, respectively) [34]. However, when comparing imipenem–relebactam to ceftazidime–avibactam, ceftazidime is hydrolyzed by PER unlike imipenem [34]. In a similar study, both relebactam and avibactam were determined to have activity against PDC-3, an ESBL produced by P. aeruginosa (Ki app 3.4 ± 0.4 and 2.5 ± 0.3 μM, respectively) [35].
3.2 Meropenem–Vaborbactam
Crystallography studies have shown that the catalytic serine residue of both CTM-M-15 and AmpC covalently bound to the boron atom of vaborbactam [11]. These studies also demonstrated insight into the coordination of vaborbactam in the active site of these β-lactamase enzymes [11]. The boron atom in vaborbactam mimics the carbonyl carbon of the β-lactam ring, which forms an association with the serine residue in β-lactamases ultimately mimicking the tetrahedral transition state of β-lactam hydrolysis [11, 17]. Even though vaborbactam forms a covalent bond with theses enzymes, this association is reversible, and therefore vaborbactam serves as a competitive inhibitor and is not hydrolyzed [5, 31].
4 Mechanisms of Resistance
Carbapenem resistance can arise due to various mechanisms such as decreased permeability via reduced expression of OMPs, expression of efflux systems, alteration of PBPs thus decreasing affinity of β-lactams, and most importantly the production of carbapenemases [2]. In 2011, the Clinical and Laboratory Standards Institute (CLSI) reduced the carbapenem MIC (minimum inhibitory concentration) breakpoints four-fold against Enterobacteriaceae, including imipenem and meropenem [2]. This change was made because of resistance mechanisms that were undetected at higher MIC breakpoints [2].
The addition of the β-lactamase inhibitors relebactam and vaborbactam prevent degradation of imipenem and meropenem, respectively, by certain β-lactamases. These novel β-lactamase inhibitors have displayed synergy in potentiating carbapenems against Ambler class A and C β-lactamases, especially KPCs and AmpCs; however, they have not been proven to significantly inhibit class B (e.g. IMP, VIM, NDM) or D enzymes (e.g. OXA) [6, 7, 9, 36]. Therefore, organisms that produce specific carbapenemases outside the spectrum of inhibition of relebactam and vaborbactam will continue to be resistant to imipenem and meropenem, respectively.
Generally, pathogens that remain resistant to imipenem–relebactam and meropenem–vaborbactam include S. maltophilia, Elizabethkingia meningoseptica, and Aeromonas spp., which produce class B metallo-β-lactamases that are chromosomally encoded and utilize a zinc atom at their active site [1, 2, 37]. Other resistant organisms include A. baumannii and some isolates of K. pneumoniae, which produce OXA class D β-lactamases [2, 37, 38].
Some pathogens have intrinsic resistance to carbapenems due to mechanisms other than β-lactamase production and therefore the addition of β-lactamase inhibitors provides no benefit. One example is pathogens that produce altered PBPs with low affinities to β-lactam antimicrobials. These organisms include MRSA (methicillin-resistant Staphylococcus aureus) due to production of PBP2a as well as Enterococcus faecium, which produces PBP5 [1, 2]. Another example of a resistance mechanism other than β-lactamase production is overexpression of multi-drug efflux pumps such as the MexA-MexB-OprM efflux system in P. aeruginosa [1, 2]. In particular, meropenem is susceptible to P. aeruginosa efflux compared to imipenem, which is unaffected by this mechanism of resistance [39]. Likewise, relebactam is unaffected by P. aeruginosa efflux, which makes this compound an optimal inhibitor to be used in combination with imipenem [6, 7].
P. aeruginosa resistance to carbapenems is a significant and growing problem [1, 2]. The most common mechanism of imipenem resistance in P. aeruginosa is a combination of overproduction of AmpC β-lactamase and decreased expression of OprD, an OMP specific to the uptake of imipenem [6, 39]. However, relebactam in combination with imipenem helps to prevent resistance via this mechanism by inhibiting AmpC; this effect has not been described with meropenem–vaborbactam [7, 9, 36]. Similarly, the decreased expression of various OmpK porin proteins in K. pneumoniae and OmpC and OmpF in Enterobacter spp. is another mechanism of resistance [2, 37]. Down-regulation of OmpK36 or major mutations in this porin have been shown to increase imipenem–relebactam MICs [36, 40].
Haidar et al. described that variant KPC-3 enzymes, which confer resistance to ceftazidime-avibactam, do not affect the activity of imipenem–relebactam as this mutation causes the enzyme to no longer function as a carbapenemase, but rather as an ESBL [40]. Similarly, Lomovskaya and Tsivkovski determined that Asp179Tyr amino acid substitutions in KPC-2 and KPC-3 do not affect activity of meropenem–vaborbactam in contrast to causing resistance to ceftazidime-avibactam [41].
Two recent studies explored enzyme-inhibitor interactions of vaborbactam and KPC-2, which provide insight on different interactions that allow resistance to form against β-lactamase inhibitors such as avibactam and clavulanic acid but not against vaborbactam [23, 42]. Tsivkovski and Lomovskaya determined that substitutions in Trp105 (an amino acid residue in KPC-2, which is important for recognition of substrates) that confer resistance to ampicillin-clavulanic acid, had no effect on vaborbactam’s whole cell potency in combination with antimicrobials [42]. The authors determined this effect may be due to very slow K off rates of vaborbactam, which reserves its potency [42]. Tsivkovski et al determined that inhibition of KPC-2 by vaborbactam does not involve S130, which is important for inhibition by avibactam [23]. This was a significant finding as a point mutation, KPC-2-S130G, caused approximately a 6000-fold decrease in K i for avibactam but had no significant effect on the K i of vaborbactam [23].
Sun et al. identified exposure levels of meropenem and vaborbactam in order to prevent resistance development using sub-optimal exposures of both drugs in seven meropenem-resistant strains of K. pneumoniae [43]. The authors concluded that concentrations of 8 mg/L meropenem along with 8 mg/L vaborbactam were associated with a resistance frequency < 10−9 [43]. The mutants that were selected for in isolates incubated at sub-inhibitory concentrations had a resistant phenotype involving inactivation of OmpK36 due to various insertions, deletions, stop mutations, or substitutions [43]. This study provided target exposures for clinical use in order to prevent the development of resistance due to changes in OmpK35 and OmpK36 [43].
Interestingly, in another study by Sun et al, loss of resistance was observed in an in vitro hollow fiber infection model (HFIM) with 6 KPC-producing isolates of K. pneumoniae when treated with meropenem–vaborbactam [44]. Approximately 5–10% of cells that survived exposure to meropenem–vaborbactam became susceptible to carbapenems as well as other β-lactam antimicrobials due to the loss of bla KPC [44]. It is predicted that the loss of the bla KPC gene was due to loss of a plasmid rather than genetic recombination to remove the gene in the isolates studied [44]. In summary, resistance developing upon administration of imipenem–relebactam and meropenem–vaborbactam is currently limited. As the usage of these agents increases, novel resistance mechanisms may arise and will require investigation.
5 Microbiology
The in vitro activities of imipenem–relebactam compared with imipenem alone against various Gram-negative aerobes and anaerobes, including drug-resistant and specific β-lactamase-producing isolates are presented in Tables 4, 5, and 6 [6, 7, 32, 36, 40, 45,46,47,48,49,50,51,52,53,54,55,56,57]. The MIC values presented in these tables are modal MIC values derived from a review of available in vitro studies conducted with similar methods. Comparative data for imipenem alone was also pooled from these studies and included when data were available. Susceptible and resistant phenotypes are described using the current CLSI breakpoints for imipenem against Enterobacteriaceae (susceptible ≤ 1 mg/L, resistant ≥ 4 mg/L), P. aeruginosa and Acinetobacter spp. (susceptible ≤ 2 mg/L, resistant ≥ 8 mg/L), and anaerobes and other non-Enterobacteriaceae (susceptible ≤ 4 mg/L, resistant ≥ 16 mg/L) [58].
5.1 Imipenem–Relebactam
Table 4 shows the activities of imipenem–relebactam and imipenem alone against common Gram-negative aerobes [6, 7, 36, 40, 45, 47,48,49,50,51,52,53,54, 57]. Also included in this table are various resistant phenotypes such as imipenem non-susceptible isolates, ESBL, KPC, and serine carbapenemase producers and MDR isolates. The activity of imipenem against Gram-negative bacteria is either retained or enhanced with the addition of relebactam. Significant increases in activity of imipenem with the addition of relebactam are observed against imipenem non-susceptible and β-lactamase (ESBL, KPC, and serine carbapenemase) producing Enterobacteriaceae (2- to 128-fold MIC reductions) and against P. aeruginosa (eightfold MIC reduction). The addition of relebactam has little impact on the activity of imipenem against A. baumannii, Chryseobacteria and S. maltophilia.
Table 5 shows in vitro activity of imipenem–relebactam compared to imipenem alone against Enterobacteriaceae and P. aeruginosa expressing specific β-lactamase enzymes [7, 32, 36, 45, 46]. Overall, relebactam significantly improved the activity of imipenem against Enterobacteriaceae isolates producing Ambler class A ESBLs (2- to 16-fold MIC reduction) and KPC carbapenemases (32- to 128-fold MIC reduction) (Tables 4 and 5). The addition of relebactam showed significant benefit in potentiating the activity of imipenem against imipenem non-susceptible P. aeruginosa with AmpC production and OprD porin loss [7, 36, 46]. The addition of relebactam did not potentiate imipenem against A. baumannii isolates producing OXA-23 and had minimal impact on the activity of imipenem against Gram-negative isolates producing OXA-48. As expected based on relebactam’s spectrum of activity, the addition of relebactam did not potentiate the activity of imipenem against organisms producing Ambler class B metallo-β-lactamases, including IMP, VIM, and NDM. Overall, the majority of isolates with elevated imipenem–relebactam will likely contain multiple resistance mechanisms including production of β-lactamases not inhibited by relebactam (most Ambler class B and D β-lactamases), porin alterations, and overexpression of efflux pumps.
Table 6 shows the activities of imipenem–relebactam and relebactam against anaerobic bacteria [55, 56]. Overall, imipenem–relebactam demonstrated little or no improvement compared to imipenem alone; however, imipenem alone demonstrated excellent activity against most anaerobes presented in Table 6. Two exceptions were Bilophila wadsworthia and Fusobacterium varium with MIC90 values that dropped from a CLSI resistant MIC of 16 mg/L to a susceptible MIC of 4 mg/L (fourfold reduction). There was also a fourfold reduction in imipenem MIC90 values for Fusobacterium necrophorum with the addition of relebactam, which dropped from 2 to 0.5 mg/L (both CLSI susceptible values). Bacteriodes spp. with decreased imipenem susceptibility showed no benefit with the addition of relebactam, the majority of isolates continued to remain above the CLSI breakpoint for imipenem (MIC50, 8 mg/L; MIC90, > 32 mg/L). Limited data have been published on the activity of imipenem–relebactam versus Gram-positive bacteria.
5.2 Meropenem–Relebactam
The in vitro activities of meropenem–vaborbactam compared with meropenem alone against various Gram-negative aerobes, including antimicrobial-resistant and specific β-lactamase-producing isolates are presented in Tables 7 and 8 [9, 59,60,61,62,63,64,65,66,67,68,69,70,71,72,73]. The MIC values presented in these tables are modal MIC values derived from a review of available in vitro studies conducted with similar methods. Comparative data on meropenem alone was also pooled from these studies and included when data were available. Susceptible and resistant phenotypes are described using the current CLSI breakpoints for meropenem, which are the same as those for imipenem, as stated above.
Table 7 shows the activities of meropenem–vaborbactam and meropenem alone against common Gram-negative aerobes [9, 59,60,61,62,63,64,65,66,67,68]. Also included in this table are various resistant phenotypes such as meropenem non-susceptible isolates, β-lactamase-producers, MDR and XDR (extensively drug resistant) isolates and CRE. The activity of meropenem against Gram-negative bacteria is either retained or enhanced with the addition of vaborbactam. Significant increases in activity of meropenem with the addition of vaborbactam are observed against meropenem non-susceptible, β-lactamase- (ESBL, KPC and other carbapenemase) producing, and MDR Enterobacteriaceae including CRE (32 to ≥ 256-fold MIC reductions). The addition of vaborbactam had little impact on the activity of meropenem against A. baumannii and P. aeruginosa. Meropenem–vaborbactam MIC90 values for Acinetobacter spp., S. maltophilia, non-KPC-producing CRE, and metallo-β-lactamase-producing and XDR Enterobacteriaceae continued to remain above the CLSI breakpoint for meropenem.
Table 8 shows in vitro activity of meropenem–vaborbactam compared to meropenem alone against Enterobacteriaceae expressing specific β-lactamase enzymes [9, 59, 64, 69,70,71,72,73]. Vaborbactam significantly improved the activity of imipenem against Enterobacteriaceae isolates producing β-lactamases from Ambler class A and C (4- to ≥ 1024-fold MIC reduction), including KPC carbapenemases (> 32- to ≥ 128-fold reduction). The addition of vaborbactam did not potentiate the activity of meropenem against Enterobacteriaceae producing OXA-48-like β-lactamases. Similar to imipenem–relebactam, the majority of isolates with elevated meropenem–vaborbactam MICs will likely contain multiple resistance mechanisms including production of β-lactamases not inhibited by relebactam (most Ambler class B and D β-lactamases), porin alterations, and overexpression of efflux pumps.
No data have been published on the activity of meropenem–vaborbactam versus anaerobic bacteria or Gram-positive bacteria. Of note, neither relebactam nor vaborbactam have been shown to have antimicrobial activity alone [46, 60].
6 Pharmacokinetics
The pharmacokinetics of intravenous imipenem and meropenem have been well established. Imipenem, when administered with cilastatin, has an approximate volume of distribution (V d) of 0.23 to 0.31 L/kg with 20% serum protein binding, 60–70% renal clearance, and an elimination half-life (t ½) of 1 h [1]. Meropenem has a V d of 0.23–0.35 L/kg with 2% protein binding, 70% renal elimination, and half-life (t ½) of 1 h [1].
6.1 Imipenem–Relebactam
A population pharmacokinetic model of relebactam in combination with imipenem was constructed using data from three phase I trials in healthy adults and one phase II study in patients with cIAIs [74]. Relebactam pharmacokinetics were described by a two-compartment, linear model with first order elimination. Co-administration of imipenem and relebactam had no effect on the pharmacokinetics of either agent. The population estimates for the central volume, peripheral volume, and total clearance of relebactam were 12.1 L [relative standard error (RSE), 5.4%], 5.9 L (RSE, 5.8%), and 8.0 L/h (RSE, 2.7%), respectively.
Results from four Phase I pharmacokinetic studies of relebactam are summarized in Table 9 [75,76,77,78]. The phase I studies in healthy individuals describe a total V d of 11.9–12.3 L in females, 14.0–17.7 L in males, and 22.8 L in hemodialysis patients [75,76,77,78]. Relebactam is approximately 20% bound to plasma proteins [74, 77]. A phase I study by Rhee et al evaluated the intrapulmonary pharmacokinetics of relebactam and imipenem after 5 doses of imipenem/cilastatin-relebactam 500/500/250 mg administered q6 h [77]. Epithelial lining fluid (ELF) penetration was determined from the ratio of area under the concentration-time curve (AUC) in ELF to free AUC (fAUC) in plasma. The relative exposures in ELF versus plasma were 53 and 54% for imipenem and relebactam, respectively.
Relebactam is at least 90% renally eliminated as unchanged parent, with a total CL and t ½ ranging from 5.3 to 9.1 L/h and 1.2 to 2.1 h, respectively [74,75,76,77]. The AUC achieved with 125 mg and 250 mg of relebactam ranged from 14.9 to 17.1 mg·h/L and 28.6 to 30.0 mg·h/L, respectively, with higher exposures in renal impairment and elderly subjects [75,76,77]. A study by Rizk et al. of relebactam pharmacokinetics in subjects with varying degree of renal function showed reduced CL with decreasing renal function [78]. The relative increases in AUC compared to healthy matched controls are detailed in Table 9. The study also reported that approximately half of the administered doses of imipenem, cilastatin, and relebactam were removed by hemodialysis. The authors suggested recommending dosing adjustments for imipenem with renal impairment can be maintained with the addition of relebactam.
6.2 Meropenem-Relebactam
The population pharmacokinetics of vaborbactam in combination with meropenem were studied using data from two phase I trials in healthy subjects and two phase II trials in patients with cUTIs or other CRE infections [79]. Vaborbactam pharmacokinetics were described by a two-compartment model with first order elimination. The mean population estimates for the non-renal clearance, maximum renal clearance, V c, and V p were 0.169 L/h [standard error of the mean (SEM), 12.5%], 9.34 L/h (SEM, 3.3%), 16.9 L (SEM, 3.9%), and 1.41 L (SEM, 27.2%), respectively.
Results from three phase I pharmacokinetics studies of vaborbactam are summarized in Table 10 [80,81,82]. Griffith et al determined that co-administration of meropenem and vaborbactam did not alter the pharmacokinetics of either agent [80]. The total V d for vaborbactam from phase I studies ranged from 17.5 to 21.8 L in healthy adults [80,81,82]. Vaborbactam is approximately 33% protein bound in plasma [81]. A Phase I study by Wenzler et al. determined ELF penetration following three doses of meropenem–vaborbactam 2000/2000 mg administered q8 h [82]. ELF penetration according to the ratio of AUC in ELF to fAUC in plasma, was 65 and 79% for meropenem and vaborbactam, respectively.
Vaborbactam pharmacokinetic studies demonstrated 80 to 90% renal elimination with total CL and t ½ of 10.1–14.0 L/h and 1.3–2.0 h, respectively [80,81,82]. The AUC achieved with 2000 mg of vaborbactam ranged from 145.0 to 204 mg·h/L [80,81,82]. The population pharmacokinetic study by Trang et al. characterized relationships between glomerular filtration rate and drug CL, which were similar for meropenem and vaborbactam. As such, dose adjustments based upon estimated glomerular filtration rate (eGFR) thresholds for meropenem are expected to be appropriate for vaborbactam [79].
7 Pharmacodynamics
7.1 Imipenem–Relebactam
The pharmacodynamics of relebactam were studied by Mavridou et al. in a neutropenic murine thigh model of β-lactamase-producing isolates of K. pneumoniae (n = 2) and P. aeruginosa (n = 4) [46]. Treatment included imipenem at various doses every 2 h with or without relebactam at various doses and intervals over 24 h. Antimicrobial activity correlated with relebactam exposure (i.e. fAUC24h), where bacteriostasis was associated with a mean fAUC24h of 26.0 mg·h/L and range of 6.3–45.2 mg·h/L depending on imipenem dose and MIC.
The in vivo activity of imipenem–relebactam was evaluated by Powles et al in a neutropenic murine model of pulmonary infection with imipenem-resistant P. aeruginosa and systemic infection with imipenem-resistant P. aeruginosa and K. pneumoniae [83]. Treatment was initiated 15 min post-infection in both models, and also delayed 16.5 h in the pulmonary model. Relebactam doses from 10 to 80 mg/kg were studied in combination with imipenem/cilastatin [5/50 mg/kg q6 h, with a 1-h infusion time (t’)] for 24 h. Bactericidal activity (≥ 3-log10 bacterial kill) was observed with immediate treatment with regimens containing relebactam 20 mg/kg for systemic P. aeruginosa infection, whereas 40 mg/kg was required for bactericidal activity in P. aeruginosa pulmonary and K. pneumoniae systemic infections. Delayed treatment with relebactam at a dose of 20 mg/kg was bacteriostatic in the P. aeruginosa pulmonary model. The authors characterized an association between bacteriostatic activity and a relebactam AUC24h of 52.2 mg·h/L equivalent to a fAUC24h of 41.8 mg·h/L (assuming 20% protein binding).
Bhagunde et al. investigated a pharmacodynamics index for imipenem–relebactam described as the percentage of time that imipenem concentrations exceed the instantaneous MIC (%T > MICi), representing the reducing imipenem susceptibility with decreasing relebactam concentrations over time [84]. Their mathematical model simulated various relebactam doses in combination with imipenem (500 mg q6 h, t’ 30 min). Antimicrobial activity was then tested in a (HFIM using a KPC-2-producing K. pneumoniae strain with an imipenem MIC of 64 mg/L. Bacteriostasis was observed over 48 h with an imipenem %T > MICi of 69% in combination with relebactam doses that achieved AUC24h values of either 51.9 or 86.6 mg·h/L.
In another HFIM study by Wu et al. the activity of relebactam at doses of 125 and 250 mg in combination with imipenem (500 mg q6 h, t’ 30 min) was tested against imipenem-resistant isolates of P. aeruginosa (n = 5) and K. pneumoniae (n = 1) [85]. Sustained bactericidal activity was observed within 10 h for both relebactam doses against all isolates except one, P. aeruginosa (imipenem MIC = 64 mg/L), which required the higher dose of 250 mg for bactericidal activity. Using a pharmacodynamics parameter (%T > MICDynamic) similar to the %T > MICi defined by Bhagunde et al. data for two P. aeruginosa isolates suggested maximum antimicrobial activity with an imipenem %T > MICDynamic of 40–50% [84, 85].
Finally, using a population pharmacokinetic model derived from a phase II and three phase I studies, Lucasti et al. simulated the probability of target attainment for imipenem/cilastatin-relebactam (500/500/250 mg q6 h, t’ 30 min) against P. aeruginosa and KPC-producing isolates [86]. The distribution of MICs was obtained from the Study for Monitoring Antimicrobial Resistance Trends (SMART) 2011 global surveillance study. Pharmacodynamic targets were defined as an imipenem fAUC24h/MIC of ≥ 30% and relebactam AUC24h ≥ 150 μM·h (52.2 mg·h/L). The authors concluded that target attainment was achieved in at least 90% of simulated cases with imipenem–relebactam MICs at or below 2 mg/L.
7.2 Meropenem–Relebactam
The pharmacodynamics of meropenem–vaborbactam (2000/2000 mg q8 h, t’ 3 h) were studied by Tarazi et al. in an in vitro HFIM against various carbapenem-resistant isolates over 32 h [71, 87]. Concentration profiles in the model were adapted from the pharmacokinetic data from phase I studies [71, 87]. Sustained bactericidal activity was observed within 8 h against seven β-lactamase and KPC-producing K. pneumoniae, three E. cloacae, one E. coli, and two meropenem non-susceptible P. aeruginosa [71, 87].
In another study by Tarazi et al. higher exposures of vaborbactam were also tested based on the pharmacokinetics of infected patients in phase III trials (AUC24h 547 vs 343 mg·h/L), which unlike lower exposures, was able to suppress regrowth of one highly-resistant K. pneumoniae isolate (meropenem MIC ≥ 64 mg/L, meropenem–vaborbactam MIC = 16 mg/L) [70]. Sustained bactericidal activity was observed within 10 h against the other four isolates of KPC-producing and carbapenem-resistant K. pneumoniae and one isolate of E. cloaceae.
Griffith et al studied 17 KPC-producing CRE isolates in an HFIM and 5 isolates in a neutropenic murine thigh infection model (meropenem MICs of 8 to > 64 mg/L and meropenem–vaborbactam MICs ≤ 0.06–64 mg/L) [73]. A clinical dose equivalent to 2000 mg q8 h (t’ 3 h) of meropenem was simulated in combination with vaborbactam exposures (i.e. AUC24h) of 192, 300, and 550 mg·h/L over 32 h in the HFIM and from 0 to 725 mg·h/L over 24 h in the animal model. The ratio of vaborbactam fAUC to meropenem–vaborbactam MIC (fAUC24h:MIC) was best correlated with antimicrobial activity in both models (r 2 = 0.81 and 0.70 in the HFIM and animal model, respectively) [73]. In the HFIM, fAUC24h:MIC values of 12 and 36 were associated with bacteriostatic and bactericidal activity, respectively. In the thigh infection model, fAUC24h:MIC values of 9 and 220 were associated with bacteriostasis and 2-log10 bacterial kill, respectively. For isolates with elevated meropenem–vaborbactam MICs of 8 to 16 mg/L (meropenem MICs > 64 mg/L), a fAUC24h:MIC above 24 was required to prevent the emergence of resistance in the HFIM.
Finally, Bhavnani et al investigated the pharmacodynamics target attainment of meropenem–vaborbactam (2000/2000 mg q8 h, t’ 3 h) using data from two phase III studies of cUTI, which included 175 microbiologically evaluable patients [88]. Clinical and microbiological responses ranged from 93 to 100% and 76.3 to 100%, respectively. The predicted target attainment for a meropenem %fT > MIC of ≥ 45% was 96.6%, with over 90% of patients achieving a %f T > MIC of 100%.
8 Animal Studies
The in vivo antimicrobial efficacy of imipenem–relebactam and meropenem–vaborbactam for the treatment of resistant Gram-negative infections has been evaluated in various animal models. Imipenem–relebactam has been studied in murine systemic, pulmonary, and thigh infection models, while meropenem–vaborbactam has been studied in murine thigh and urinary tract infection models. These studies are summarized in Table 11 and represent available data to date [46, 69, 83, 89]. MIC values of isolates tested in each model are included in Table 11.
8.1 Imipenem–Relebactam
As mentioned previously, Powles et al. evaluated the in vivo efficacy of imipenem–relebactam using pulmonary and systemic infection in neutropenic mice caused by imipenem-resistant P. aeruginosa and K. pneumoniae [83]. In the systemic P. aeruginosa and K. pneumoniae infection models, mice (n = 5 for each regimen) were intraperitoneally (IP) inoculated with 2.2 × 106 colony forming units (CFU) and 5.5 × 105 CFU, respectively. Treatment was initiated intravenously 15 min post-infection with imipenem/cilastatin at 5/50 mg/kg alone or in combination with 10–80 mg/kg relebactam q6 h for 24 h (t’ 1 h). Spleen bacterial density was determined at the end of treatment and compared to untreated controls sacrificed at the same point in time. In the P. aeruginosa systemic model, a log10 reduction in CFU of 0.45 was observed for imipenem treatment alone, compared to 1.72, 3.13, and 3.73 log10 reductions in CFU with the addition of 10, 20, and 40 mg/kg relebactam, respectively. In the K. pneumoniae systemic model, a log10 increase in CFU of 0.52 was observed for imipenem treatment alone, compared to 2.29, 3.06, and 2.36 log10 reductions in CFU with the addition of 20, 40, and 80 mg/kg relebactam, respectively.
In the P. aeruginosa pulmonary infection models by Powles et al, mice (n = 3–5 for each regimen) were intranasally inoculated with 1.4 × 105 CFU and 1.8 × 104 CFU in the immediate and delayed treatment models, respectively [83]. Treatment was initiated intravenously 15 min post-infection and 16.5 h post-infection in the immediate and delayed treatment models, respectively. Imipenem/cilastatin at 5/50 mg/kg was administered alone or in combination with relebactam 20, 40, and 80 mg/kg q6 h for 24 h (t’ 1 h). Lung bacterial density was determined at the end of treatment and compared to untreated controls sacrificed at the same point in time. For immediate treatment in the P. aeruginosa pulmonary model, a log10 increase in CFU of 0.11 was observed for imipenem treatment alone, compared to 2.37, 3.59, and 4.59 log10 reductions in CFU with the addition of 20, 40, and 80 mg/kg relebactam, respectively. For delayed treatment in the P. aeruginosa pulmonary model, a log10 reduction in CFU of 0.78 was observed for imipenem treatment alone, compared to 2.94, 2.06, and 2.12 log10 reductions in CFU with the addition of 20, 40, and 80 mg/kg relebactam, respectively. Data comparing lung bacterial density of treated mice and controls sacrificed at the beginning of treatment (16.5 h post-infection) for the delayed treatment P. aeruginosa pulmonary model are also represented in Table 11.
Also mentioned previously, Mavridou et al. used a neutropenic murine thigh model of imipenem non-susceptible and β-lactamase-producing isolates of K. pneumoniae (n = 2) and P. aeruginosa (n = 4) to determine the dose of relebactam in combination with imipenem required to achieve bacteriostasis [46]. Treatment was initiated 2 h after mice (n ≥ 2 for each regimen) were infected with approximately 5 × 106 CFU in each thigh. Imipenem/cilastatin 2/2 to 15.9/15.9 mg/kg was administered IP q2 h for 24 h with various doses and intervals of relebactam. Thigh bacterial density was compared to untreated controls sacrificed at the start of treatment (2 h post-infection). Table 11 shows the various total daily doses of relebactam needed to achieve bacteriostasis against each of the six isolates tested as well as the fAUC24h achieved with each regimen. Total daily doses to achieve bacteriostasis ranged from 11.8 to 85.1 mg/kg/day (mean 48.9 mg/kg/day) with fAUC24h from 6.3 to 45.2 mg·h/L (mean 26.0 mg·h/L), which were highly dependent on the imipenem dose and MIC of each isolate.
8.2 Meropenem–Relebactam
Sabet et al. evaluated the in vivo efficacy of meropenem–vaborbactam against meropenem-resistant and β-lactamase producing Enterobacteriaceae (three E. cloacae, one E. coli, and six K. pneumoniae) in a neutropenic murine thigh model [89]. Mice were infected intramuscularly with approximately 1 × 106 CFU in each thigh. Treatment was initiated 2 h post-infection with either meropenem 300 mg/kg alone or in combination with 50 mg/kg vaborbactam, administered IP q2 h for 24 h. Dosing regimens were humanized to provide exposures equivalent to meropenem 2000 mg alone or in combination with vaborbactam 2000 mg q8 h (t’ 3 h). Thigh bacterial density was compared to untreated controls sacrificed at the start of treatment (2 h post-infection). Changes in bacterial density compared to controls for meropenem treatment alone and in combination with vaborbactam against various Enterobacteriaceae are summarized in Table 11. Antimicrobial efficacy of meropenem was greatly improved with the addition of vaborbactam.
Finally, Weiss et al. evaluated the in vivo efficacy of meropenem–vaborbactam in a neutropenic murine urinary tract infection model against meropenem-resistant and KPC-producing E. coli and K. pneumoniae [69]. Mice (n = 5–10) were transurethrally infected, 4–5-day post-infection mean kidney bacterial densities were 4.2 × 106–1.2 × 107 log10 CFU and 3.4 to 6.2 × 106 CFU, respectively. Treatment was initiated four days post-infection with meropenem 50–300 mg/kg alone or in combination with vaborbactam 25–50 mg/kg IP q2 h for 24 h. Kidney bacterial density was compared to untreated controls sacrificed at the start of treatment on day 4. Reductions in bacterial density compared to controls are summarized in Table 11. Overall, the addition of vaborbactam increased the antimicrobial efficacy of meropenem in both models, achieving an additional 1.37–1.98 and 0.67–2.25 log10 CFU reduction in kidney bacterial density compared to meropenem alone, in the E. coli and K. pneumoniae infection models, respectively. Data comparing kidney bacterial density of treated mice and controls sacrificed at the end of treatment on day 5 are also represented in Table 11.
9 Clinical Trials
Clinical trials completed to date are summarized in Table 12 [10, 86, 90,91,92], including two imipenem–relebactam phase II clinical trials for the treatment of cIAI and cUTI/AP (NCT01506271 and NCT01505634) and one meropenem–vaborbactam phase III clinical trial for the treatment of cUTI/AP (NCT02166476).
9.1 Imipenem–Relebactam
The efficacy, tolerability, and safety of imipenem–relebactam has been studied for the treatment of cIAI in a global, double-blind, randomized, phase II, non-inferiority trial (NCT01506271) (Table 12) [86]. Imipenem/cilastatin-relebactam (500/500/250 mg and 500/500/125 mg) were compared to imipenem/cilastatin alone (500/500 mg), administered IV (t’ 30 min) q6 h (q8 h for renal insufficiency) for 4–14 days. The dose of imipenem was adjusted for renal insufficiency and/or low body weight according to the approved label, and the dose of relebactam was adjusted proportionally. In this study, 351 patients were randomized (1:1:1) with stratification for disease severity (APACHE II score of ≤ 15 or > 15). Inclusion criteria were patients aged ≥ 18 years with an eligible diagnosis of cIAI requiring hospitalization and treatment with IV antimicrobial therapy. Exclusion criteria were: an APACHE II score > 30; patients who received antimicrobial therapy effective against the identified causative pathogen(s) after culture collection and prior to study therapy initiation; patients who received antimicrobial therapy for > 24 h within the preceding 72 h, effective against presumed/documented pathogen(s); renal dysfunction [creatinine clearance (CLCR) < 50 mL/min]; and hepatic dysfunction (ALT or AST > 3 times the upper limit of normal [× ULN]).
The primary outcome was a favorable clinical response in the microbiologically evaluable (ME) population at discontinuation of IV therapy (DCIV). Clinical response was evaluated based on resolution of presenting clinical signs/symptoms including evidence of a systemic inflammatory response (fever, elevated white blood cell count, decreased blood pressure, increased pulse/respiratory rate, hypoxemia, and/or altered mental status) and physical findings associated with IAI (such as abdominal pain and/or tenderness, abdominal wall rigidity, abdominal mass, or ileus). The ME population was defined as subjects with an eligible diagnosis of cIAI, a prestudy/postoperative culture growing at least one Gram-negative enteric and/or anaerobic pathogen, and no significant protocol deviations, and patients who received at least 4 days of IV study therapy. The ME population included 72.6% of randomized patients, of whom 52.5 and 16.5% had a diagnosis of complicated appendicitis and complicated cholecystitis, respectively. A non-inferiority margin was set at ≥ 15% for the lower bound of the 95% confidence interval (CI) with a one-sided p value (α = 0.025) for between-treatment differences, with 80% power to determine non-inferiority of imipenem–relebactam compared to imipenem alone. Clinical response in the ME population at DCIV (primary outcome) for the relebactam 250 mg arm was 96.3 versus 95.2% for imipenem alone, a difference of 1.1% (p < 0.001, 95% CI − 6.2 to 8.6), while response for the relebactam 125 mg arm was 98.8 versus 95.2% for imipenem alone, a difference of 3.7% (p < 0.001, 95% CI − 2.0 to 10.8). Therefore, both regimens were non-inferior to imipenem alone for the primary outcome. A sensitivity analysis showed similar results in the microbiological intention-to-treat (MITT) population (defined as patients who received at least one dose of IV study therapy and a prestudy/postoperative culture growing at least one Gram-negative enteric and/or anaerobic pathogen). Clinical response in the MITT population at DCIV for the relebactam 250 mg arm was 89.9 versus 90.2% for imipenem alone, a difference of − 0.3% (p < 0.002, 95% CI − 9.6 to 8.9), while response for the relebactam 125 mg arm was 91.7 versus 90.2% for imipenem alone, a difference of 1.4% (p < 0.001, 95% CI − 7.2 to 10.3). Neither regimen was superior to imipenem alone for the primary outcome.
The most common pathogens isolated at baseline included E. coli (n = 171), K. pneumoniae (n = 38), and P. aeruginosa (n = 37). Per-pathogen clinical response in the ME population at DCIV was similar among treatment groups for nearly all pathogens. At baseline, 40 imipenem non-susceptible organisms were isolated from 34 patients in the ME population. All 34 patients had a favorable clinical and microbiological response at DCIV. In vitro, 7 of the 40 isolates were non-susceptible to imipenem alone (one isolate of Alcaligenes xylosoxidans, one E. coli, two Proteus mirabilis, and three P. aeruginosa), while the remaining 33 isolates were non-susceptible to imipenem alone and imipenem–relebactam (19 isolates of Proteus spp., five M. morganii, four Acinetobacter spp., three S. maltophilia, and two P. aeruginosa).
Secondary endpoints included clinical response at early follow-up (EFU) 5–9 days after DCIV and late follow-up (LFU) 28–42 days after DCIV, microbiological response, and global response. Microbiologic response was imputed from clinical response if follow-up cultures from the site of infection were not available. Global response (i.e. cure), was defined as resolution of presenting signs/symptoms of IAI, survival, no unplanned percutaneous or surgical procedures for IAI, no antimicrobial therapy needed for initial or emergent IAI, and no other event related to the initial or emergent IAI that resulted in clinical instability or worsening. Global response was measured 28 days after randomization in the MITT population. Results for secondary endpoints were similar across all three treatment groups (Table 12). Clinical response in the ME population at EFU and LFU was similar to the response seen at DCIV, and was similar across all three treatment groups (Table 12). Adverse events observed in this study will be discussed in Sect. 10.
The efficacy, tolerability, and safety of imipenem–relebactam has been studied for the treatment of cUTI and AP in a global, double-blind, randomized, phase II, non-inferiority trial (NCT01505634) (Table 12) [90]. Imipenem/cilastatin-relebactam (500/500/250 mg and 500/500/125 mg) was compared to imipenem/cilastatin alone (500/500 mg), administered IV (t’ 30 min) q6 h with optional oral step-down to ciprofloxacin after at least 4 days of IV therapy. The total treatment duration was a maximum of 14 days. The dose of imipenem was adjusted for renal insufficiency and/or low body weight according to the approved label, and the dose of relebactam was adjusted proportionally. In this study, 302 patients were block randomized 1:1:1. Inclusion criteria were patients ≥ 18 years of age with an eligible diagnosis of cUTI or AP requiring hospitalization and IV antimicrobial therapy. An eligible diagnosis of cUTI required at least two specific signs and symptoms of UTI (e.g. dysuria, urinary urgency or frequency, fever) and at least one risk factor for cUTI (e.g. indwelling catheter, obstructive uropathy, history of urinary retention, etc.). An eligible diagnosis of AP included patients with a normal urinary tract anatomy and presence of a systemic ascending UTI with at least two specific signs and symptoms (e.g. fever, flank pain, nausea and/or vomiting, costovertebral angle tenderness). Patients also needed to have pyuria and a positive urine culture within 48 h of enrollment. Exclusion criteria were: uncomplicated UTI, patients who received antimicrobial therapy effective against the identified causative pathogen(s) after culture collection and prior to study therapy initiation; patients who received antimicrobial therapy for > 24 h within the preceding 72 h, effective against presumed/documented causative pathogen(s); complete obstruction of any urinary tract portion, ileal loop, intractable vesicoureteral reflux, or temporary indwelling urinary catheter that could not be removed; perinephric or intrarenal abscess, or known/suspected prostatitis; recent pelvic or urinary tract trauma; need for non-study systemic antimicrobial agents; renal dysfunction (CLCR ≤ 5 mL/min and/or need for dialysis); and hepatic dysfunction (ALT or AST > 3× ULN, total bilirubin > 2× ULN, or ALT/AST > 2× ULN with total bilirubin > ULN).
The primary outcome was a favorable microbiological response in the ME population at DCIV. Microbiological response was determined based on urine culture results on follow-up relative to the pathogen(s) isolated at baseline. The ME population was defined as subjects with an eligible diagnosis of cUTI or AP, a prestudy culture growing at least one Gram-negative enteric and/or anaerobic pathogen at a sufficient quantity, and no significant protocol deviations, who received at least 4 days of IV study therapy. The ME population at DCIV included 77.2% of randomized patients. A non-inferiority margin was set at ≥ 15% for the lower bound of the 95% CI, with 87% power to determine non-inferiority of imipenem–relebactam compared to imipenem alone. Microbiological response in the ME population at DCIV (primary outcome) for the relebactam arm 250 mg was 95.5 versus 98.7% for imipenem alone, a difference of − 3.1% (95% CI − 11.3 to 3.2), while response for the relebactam 125 mg arm was 98.6 versus 98.7% for imipenem alone (95% CI − 6.4 to 5.9). Therefore, both regimens were non-inferior to imipenem alone for the primary outcome. All patients in the ME population with an unfavorable microbiological response at DCIV had imipenem-susceptible pathogens at baseline. A sensitivity analysis showed similar results in the microbiological intention-to-treat (MITT) population (defined as patients who received at least one dose of IV study therapy and a prestudy culture growing at least one Gram-negative enteric and/or anaerobic pathogen at any quantity). Microbiological response in the MITT population at DCIV for the relebactam 250 mg arm was 87.8 versus 92.6% for imipenem alone, a difference of − 4.8% (95% CI − 15.1 to 4.9), while response for the relebactam 125 mg arm was 87.8 versus 92.6% for imipenem alone, a difference of − 4.8% (95% CI − 14.7 to 4.7). Thus, the two regimens were deemed non-inferior.
The most common pathogens isolated at baseline in the ME population included E. coli (n = 143), K. pneumoniae (n = 34), and P. aeruginosa (n = 16). At baseline, 25 out of 220 patients (11.4%) had a Gram-negative pathogen that was non-susceptible to imipenem, and 15 of these patients had isolates that were also non-susceptible to imipenem–relebactam (five isolates of P. mirabilis, four A. baumannii, four M. morganii, one Providencia rettgeri, and one P. aeruginosa).
Secondary endpoints were assessed in the ME population and included microbiological response at EFU (5–9 days after completion of all study therapy) and LFU (28–42 days after completion of all study therapy), microbiological response at DCIV in patients with imipenem non-susceptible pathogens, and clinical response at DCIV, EFU, and LFU. A composite of clinical and microbiological response in the ME population at EFU was an additional exploratory endpoint. Clinical response was determined by comparing cUTI signs/symptoms at follow-up with those at baseline. After DCIV, microbiological responses decreased at EFU and LFU with similar reductions across all three treatment groups (Table 12). Clinical responses at DCIV were similar across all three treatment groups, with similar decreases in response at EFU and LFU (Table 12). Of the 25 patients mentioned above with a Gram-negative isolate non-susceptible to imipenem, 23 were ME and had favorable microbiological responses at DCIV (Table 12). The composite for clinical and microbiological response in the ME population at EFU was similar among treatment groups with a response rate of 54.1, 59.8, and 61.7% for the relebactam 250 mg arm, the relebactam 125 mg arm, and imipenem alone, respectively. Adverse events observed in this study will be discussed in Sect. 10.
9.2 Meropenem–Relebactam
The efficacy, tolerability, and safety of meropenem–vaborbactam for the treatment of cUTI and AP have been studied in a multicenter, double-blind, randomized, phase III, non-inferiority trial (TANGO I, NCT02166476) (Table 12) [10, 91, 92]. Meropenem–vaborbactam (2000/2000 mg) IV (t’ 3 h) q8 h was compared to piperacillin-tazobactam (4000/500 mg) IV (t’ 30 min) q8 h. Oral step-down to levofloxacin after at least 5 days of IV therapy was permitted in both treatment arms. The total treatment duration was 10 days. In this study, 550 patients were randomized 1:1, stratified by geographic region and type of infection (AP, cUTI with removable source of infection [cUTI-R], and cUTI with a non-removable source [cUTI-NR]). Inclusion criteria were patients ≥ 18 years of age with an eligible diagnosis of cUTI or AP that required at least 5 days of IV antimicrobial therapy, and any indwelling urinary catheter or instrumentation removed or replaced not longer than 12 h after randomization. Key exclusion criteria were renal impairment (CLCR < 30 mL/min), and patients who received any potentially therapeutic antimicrobial agent within 48 h before randomization. Exceptions to the exclusion criteria were patients with signs/symptoms of cUTI or AP while on antimicrobials for another indication, clear clinical evidence of treatment failure (worsening signs/symptoms), and those who received a single dose of a short-acting oral or IV antimicrobial (no more than 25% of subjects were to be enrolled who met this criterion).
The primary outcome was overall success in the microbiologic modified intention-to-treat (m-MITT) population at DCIV. Overall success included clinical cure or improvement and microbiological eradication defined as the baseline pathogen being reduced to < 104 CFU/mL (FDA criteria). The m-MITT population included 68% of randomized patients who had at least one baseline pathogen at ≥ 105 CFU/mL. In the m-MITT population, 59.1% of patients were diagnosed with AP and 40.9% with cUTI. Overall success in the m-MITT population at DCIV (primary outcome) was 98.4 and 94.0% for meropenem–vaborbactam and piperacillin-tazobactam, respectively, a difference of 4.5% (95% CI 0.7–9.1). Meropenem–vaborbactam was non-inferior to piperacillin-tazobactam for the primary outcome. Overall success in the m-MITT population at DCIV was 97.5, 100, and 100% for patients with AP, cUTI-R, and cUTI-NR treated with meropenem–vaborbactam, while treatment with piperacillin-tazobactam resulted in overall success in 94.1, 92.1, and 95.3%, respectively.
The most common pathogens isolated at baseline in the m-MITT population were Enterobacteriaceae (n = 333) and P. aeruginosa (n = 15). Approximately 29% of Enterobacteriaceae at baseline had an ESBL phenotype (ceftazidime or aztreonam MIC ≥ 2 mg/L). Per-pathogen clinical outcome and microbiological eradication rates at DCIV were similar among treatment groups for nearly all pathogens.
Secondary endpoints were assessed in the m-MITT and ME populations and included microbiological eradication at test of cure [(TOC), day 15–19]. Here microbiological eradication was defined as the baseline pathogen being reduced to < 103 CFU/mL (EMA criteria). The microbiological eradication rate in the m-MITT population at TOC was 66.7 and 57.7% for meropenem–vaborbactam and piperacillin-tazobactam, respectively, a difference of 9.0% (95% CI − 0.9 to 18.7). Results were similar in the ME population (Table 12). Adverse events observed in this study will be discussed in Sect. 10.
9.3 Imipenem–Relebactam
Imipenem–relebactam is currently being studied in two Phase III clinical trials, RESTORE-IMI 1 and RESTORE-IMI 2, with estimated primary completion dates of September 2017 and May 2019, respectively (NCT02452047 and NCT02493764). In the first study, RESTORE-IMI 1, imipenem–relebactam is compared with colistimethate sodium plus imipenem for the treatment of imipenem-non-susceptible bacterial infections (including HABP, VABP, cIAI, and cUTI). In the second study, RESTORE-IMI 2, imipenem–relebactam is compared with piperacillin-tazobactam, with empiric linezolid administered in both treatment arms, for the treatment of HABP/VABP.
9.4 Meropenem–Relebactam
Meropenem–vaborbactam is currently being studied in two phase III clinical trials, TANGO II and TANGO III (NCT02168946 and NCT03006679). Preliminary data with TANGO II were presented at ID week 2017 and will be discussed below. Estimated completion of TANGO III is expected June 2020. In the first study, TANGO II, a randomized, multi-national open-label trial studied patients with complicated urinary tract infection (cUTI), acute pyelonephritis (AP), hospital-acquired or ventilator-associated bacterial pneumonia (HABP/VABP), bacteremia, or complicated intra-abdominal infection (cIAI), due to known or suspected carbapenem-resistant Enterobacteriaceae (CRE) [93, 94]. Patients were randomized in a 2:1 fashion to meropenem–vaborbactam ((2000/2000 mg) IV (t’ 3 h) q8 h or best available therapy (BAT) for 7–14 days [93]. BAT included alone or in combination: carbapenems, aminoglycosides, polymyxin B, colistin, tigecycline or ceftazidime-avibactam. Clinical cure was defined as a complete resolution of signs or symptoms such that no further antimicrobial therapy as required. Seventy-two patients were enrolled; 50 (69.4%) had a Gram-negative baseline organism (m-MITT), and 43 (59.7%) had a baseline CRE (mCRE- MITT) [93]. Within mCRE-MITT, 20 patients had bacteremia, 15 had cUTI/AP, 5 had HABP/VABP, and 3 had cIAI. The most common baseline CRE pathogens were K. pneumoniae (86%) and E. coli (7%). There was no consensus BAT regimen; however, combination therapy was used in 66.7% of cases. Treatment duration was similar across arms (mean 8.5 days for meropenem–vaborbactam and 8.1 days for BAT). Although the patient numbers were relatively small (n = 50 in m-MITT and n = 43 in mCRE-MITT groups, respectively), meropenem–vaborbactam was associated with higher clinical cure rates in the mCRE-MITT group both at EOT (64.3 vs 40.0%, respectively) and TOC (57.1 vs 26.7%, respectively), as well as in the m-MITT group at both EOT (67.7 vs 42.1%, respectively) and TOC (58.0 vs 31.6%, respectively), than BAT across all indications [93]. These superior outcomes in the meropenem–vaborbactam group versus the BAT group occurred even in patients who were immunocompromised [94]. Due to the superiority of meropenem–vaborbactam compared to BAT, this study was terminated prematurely.
In the second study, TANGO III, meropenem–vaborbactam is compared with piperacillin-tazobactam for the treatment of HABP/VABP.
Both imipenem–relebactam and meropenem–vaborbactam are undergoing phase I trials in pediatric populations. The phase I pharmacokinetic study of imipenem–relebactam in serious pediatric Gram-negative infections is set to be completed in August 2020 (NCT03230916). The Phase I pharmacokinetic study of meropenem–vaborbactam for the treatment of serious bacterial infections in pediatric patients is set to be completed in August 2019 (NCT02687906).
Currently, for adult patients without renal dysfunction, imipenem–relebactam is being dosed at 500/500/250 mg IV (t’ 30 min) q6 h, while meropenem–vaborbactam is being dosed at 2000/2000 mg IV (t’ 3 h) q8 h.
10 Adverse Events
The safety and tolerability of relebactam has been reported in one Phase I pharmacokinetic study and imipenem–relebactam in three phase I pharmacokinetic studies and two phase II clinical trials [75,76,77,78, 86, 90]. The safety and tolerability of vaborbactam has been reported in one phase I pharmacokinetic study and meropenem–vaborbactam in one phase III clinical trial [10, 81].
10.1 Imipenem–Relebactam
In a phase I pharmacokinetic study by Butterton et al. 16 healthy males received single doses of relebactam ranging from 0 tSo 1150 mg [75]. No subjects discontinued due to an adverse event and no serious adverse events were reported. Adverse events reported by three or more subjects included; headache, coryza, and somnolence.
In a pharmacokinetic study, Jumes et al. reported safety and tolerability of relebactam 50–625 mg with imipenem/cilastatin 500/500 mg IV q6 h for 7–14 days in a cohort of 90 healthy adult males, and a single dose of imipenem/cilastatin-relebactam 500/500/125 mg in a cohort of 24 healthy elderly subjects and adult females [76]. In the cohort of 90 adult men, 6 discontinued the study due to an adverse event including; vasovagal reaction, vomiting, pain at cannula site, skin rash, and elevated transaminases. Both vomiting and vasovagal episodes were considered severe adverse events while all other reported events were mild-to-moderate. Adverse events reported by three or more subjects included; erythema, tenderness, pain or swelling at the infusion or cannula site, diarrhea, yellow discoloration of the tongue, abdominal discomfort, headache, vasovagal episodes, presyncope, light-headedness, dysgeusia, and nausea. For subjects dosed q6 h for seven days with imipenem–relebactam, 8 of 42 subjects (19%) experienced elevations of ALT and/or AST above ULN. For subjects dosed q6 h for 14 days, 4 of 24 subjects (17%) administered imipenem–relebactam experienced ALT elevated above ULN, compared to 2 of 8 subjects (25%) administered imipenem-cilastatin. All increases in transaminases were reversible. For subjects dosed q6 h for 14 days, on day 10 and 11 two patients developed a generalized erythematous maculopapular pruritic rash and were discontinued from study therapy [1 of 24 (4%) administered imipenem–relebactam, 1 of 8 (13%) administered imipenem]. In this same group, 9 of 24 subjects (38%) on imipenem–relebactam and 3 of 8 subjects (38%) on imipenem reported yellow staining of tongue or teeth. In the cohort of 24 healthy elderly subjects and adult females, no severe adverse events were reported, only headache and dizziness were reported, which were considered mild drug-related adverse events.
In a pharmacokinetic study, Rizk et al. reported safety and tolerability of single doses of imipenem–relebactam 250/125 mg in 24 healthy subjects with varying renal function [78]. No severe adverse events were seen, and no subjects discontinued the study due to an adverse event. Drug-related adverse events included headache and left arm muscle irritation.
In an intrapulmonary pharmacokinetic study by Rhee et al, safety and tolerability of five doses of imipenem/cilastatin-relebactam 500/500/250 mg IV q6 h, in 16 healthy adult subjects was reported [77]. No serious adverse events were seen. Drug-related adverse events included fatigue, increased creatinine, nausea, emesis, and diarrhea. The subject who experienced mild diarrhea, and moderate nausea and emesis was discontinued from the study.
In a phase II study comparing the treatment of cIAI, drug-related adverse events occurred in 13.7, 13.8, and 9.6% of patients for treatment with imipenem/cilastatin-relebactam 500/500/250 mg, imipenem/cilastatin-relebactam 500/500/125 mg, and imipenem/cilastatin 500/500 mg, respectively [86]. The most common treatment-emergent adverse events included diarrhea, nausea, and vomiting, which were relatively similar in all three treatment groups (range of 2.6–7.8%). Three deaths occurred in the relebactam 125 mg treatment group due to septic shock, ventricular fibrillation, and intestinal infraction, all determined to be non-drug related. One serious drug-related adverse event, severe thrombocytosis, occurred in the imipenem alone treatment group and this patient consequently discontinued therapy. Drug-related adverse events leading to discontinuation occurred in four patients, 0 (0%), 1 (0.9%), and 3 (2.6%) in the three treatment groups, respectively, due to decreased creatinine clearance, thrombocytosis, nausea, and increased ALT. Four patients experienced AST or ALT ≥ 5× ULN, two receiving relebactam 250 mg and two receiving imipenem alone. One patient experienced AST or ALT ≥ 3× ULN with total bilirubin ≥ 2× ULN and alkaline phosphatase < 2× ULN in the relebactam 250 mg treatment group; however, this was determined to be non-drug related.
In a Phase II study comparing the treatment of cUTI and AP, drug-related adverse events occurred in 10.1, 9.1, and 9% of patients for treatment with imipenem/cilastatin-relebactam 500/500/250 mg, imipenem/cilastatin-relebactam 500/500/125 mg, and imipenem/cilastatin 500/500 mg, respectively [90]. No deaths occurred; however, serious treatment-emergent adverse events were reported in 3.0, 1.0, and 3.0%, respectively. The most common treatment-emergent adverse events included nausea, headache and diarrhea, which were relatively similar in all three treatment groups (range of 2.0–7.1%). Treatment-related adverse events leading to discontinuation occurred in four patients, 2 (2%), 1 (1%), and 1 (1%) in the three treatment groups, respectively, due to diarrhea, rash, nausea, and diarrhea. One patient in the relebactam 250 mg group experienced AST elevations ≥ 5× ULN, determined to be drug related.
10.2 Meropenem–Relebactam
In a phase I pharmacokinetic study by Griffith et al, 88 healthy males received single or multiple doses of vaborbactam ranging from 0 to 2000 mg, multiple doses were administered q8 h for 7 days [81]. No deaths or severe adverse events occurred in this study. For single doses of vaborbactam, the most common treatment-emergent event was headache; however, there was no correlation with an increasing dose, and rates were similar to placebo [19% (8/42) versus 21% (3/14) for placebo]. Ten adverse events were classified as mild, and four moderate including myalgia (placebo group), musculoskeletal (buttock) pain (vaborbactam 750 mg), pain in extremity (thigh) (vaborbactam 750 mg), and infusion site thrombosis (vaborbactam 2000 mg). For multiple doses, adverse events occurring in at least five subjects included infusion site phlebitis [42% (10/24) vs 50% (4/8) for placebo], headache [29% (7/24) vs 13% (1/8) for placebo], lethargy [21% (5/24) vs 0% (0/8) placebo], and dermatitis contact [13% (3/24) vs 0% (0/8) placebo]. Mild lethargy was most commonly observed in the highest dose (2000 mg) group for vaborbactam (4 of 6 patients in this group, accounting for 4 of 5 events). Drug-related adverse events occurred in 58% of patients, 27 events were mild, and three moderate including infusion-site phlebitis (vaborbactam 250 and 2000 mg) and infusion site erythema (placebo).
In a phase III trial comparing the treatment for cUTI and AP, drug-related adverse events occurred in 15.1% (41/272) and 12.8% (35/273) of patients treated with meropenem–vaborbactam and piperacillin-tazobactam, respectively [10]. Two deaths occurred in each treatment group. Serious adverse events occurred in 4% (11/272) and 4.4% (12/273) of patients treated with meropenem–vaborbactam and piperacillin-tazobactam, respectively. Discontinuation of therapy occurred due to an adverse event in 2.6% (7/272) and 5.1% (14/273) of patients treated with meropenem–vaborbactam and piperacillin-tazobactam, respectively. Specific adverse events reported in this study were not available.
To date, both imipenem–relebactam and meropenem–vaborbactam appear to be well tolerated in healthy subjects as well as patients with infectious diseases, with few serious drug-related treatment-emergent adverse events reported.
11 Drug Interactions
Limited information is currently available regarding specific drug interactions involving co-administration with imipenem–relebactam or meropenem–vaborbactam. However, drug interactions have been well described for imipenem and meropenem without the additions of relebactam and vaborbactam.
Co-administration of probenecid causes a decrease in renal excretion of imipenem or meropenem, therefore increasing plasma concentrations and prolonging t ½ [1, 95, 96]. Probenecid causes increases in meropenems t½ by approximately 33%; alternatively, imipenem plasma concentrations are minimally affected as non-renal clearance mechanisms increase to compensate for a 30% decrease in renal clearance of imipenem [1]. Co-administration is not recommended [95].
Both imipenem and meropenem have been reported to decrease serum concentrations of valproic acid [95, 96]. Meropenem has been reported to decreased valproic acid serum concentrations by 60–100% in approximately two days [96]. Co-administration is not recommended due to the risk of valproic acid serum concentrations falling below a therapeutic range [95, 96].
Generalized seizures have been reported for co-administration of imipenem with ganciclovir [1, 95]. Co-administration is not recommended [95].
The potential of relebactam to be a substrate or inhibitor of human renal uptake and efflux transporters to determine potential drug interactions was investigated (MSD unpublished data). Relebactam was determined to be a substrate of tubular transporters hOAT3, hOAT4, MATE1 and MATE2K, but not a substrate of other common human renal uptake and efflux transporters (hOAT1, hOCT2, MDR1 P-gp, MRP2, MRP4, or BCRP). It was also determined that relebactam was not an inhibitor of common human renal uptake and efflux transporters (MDR1 P-gp, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, MATE2 K, BCRP, or BSEP). Probenecid inhibited hOAT3-mediated relebactam uptake in vitro (IC50 = 1.9 ± 0.4 μM in MDCKII-hOAT3 cells). Other common antimicrobials (piperacillin-tazobactam, ciprofloxacin, fluconazole, ampicillin, levofloxacin, metronidazole, vancomycin, linezolid, daptomycin, and cefazolin) did not inhibit hOAT3-mediated relebactam uptake in vitro (IC50 > 50 μM in MDCKII-hOAT3 cells). Overall, given that active secretion accounts for only ~ 35% of the total renal clearance of relebactam, the extent of drug interactions due to inhibition of these tubular transporters is expected to be of limited clinical significance.
12 Place of Imipenem–Relebactam and Meropenem–Vaborbactam in Therapy
The addition of relebactam and vaborbactam restore the activity of imipenem and meropenem, respectively, against resistant Gram-negative bacilli that produce Ambler class A and C β-lactamases, including ESBLs and serine carbapenemases. Available safety and pharmacokinetic data currently suggest no additional considerations need to be taken into consideration when administering imipenem and meropenem with the additions of relebactam and vaborbactam, respectively.
Imipenem–relebactam has demonstrated clinical efficacy similar to that of imipenem alone in phase II studies of cIAI and cUTI/AP. Completion of Phase III clinical trials for the treatment of resistant bacterial infections (including HABP/VABP, cIAI, and cUTI) and for the treatment of HABP/VABP will help to define the efficacy and safety of this novel antimicrobial combination in serious and resistant infections. The excellent ELF penetration of imipenem–relebactam combined with potent activity against CRE and P. aeruginosa could potentially make this novel antimicrobial combination an important treatment option in HABP and VABP.
In phase III clinical trials, meropenem–vaborbactam has demonstrated clinical efficacy compared to piperacillin-tazobactam in the treatment of cUTI/AP. Recent clinical trial data demonstrated that meropenem–vaborbactam was associated with higher clinical cure rates than “best available therapy” across a variety of infections caused by CRE (including cUTI/AP, HABP/VABP, and bacteremia), including in immunocompromised patients, providing compelling data that meropenem–vaborbactam is a new option for the treatment of CRE infections in seriously ill patients. As with imipenem–relebactam, the excellent ELF penetration of meropenem–vaborbactam combined with potent activity against CRE could also potentially make this novel antimicrobial combination an important treatment option in HABP and VABP.
Potential roles in therapy for imipenem–relebactam and meropenem–vaborbactam include treatment of suspected or documented infections caused by resistant Gram-negative bacilli-producing ESBL, KPC, and/or AmpC β-lactamases. The usage of these agents in patients with CRE infections will likely become the standard of care. The increased activity of imipenem–relebactam against P. aeruginosa may also be clinically beneficial in patients with suspected or documented P. aeruginosa infections. Although more clinical efficacy and safety data are required, imipenem–relebactam and meropenem–vaborbactam provide clinicians with an alternative option for the empiric treatment of serious infections caused by resistant Gram-negative bacilli, although the lack of activity against isolates producing metallo-β-lactamases remains a limitation.
Change history
10 May 2018
Section heading 5.2, which currently reads 5.2 Meropenem–Relebactam.
References
Zhanel GG, Wiebe R, Dilay L, Thomson K, Rubinstein E, Hoban DJ, et al. Comparative review of the carbapenems. Drugs. 2007;67(7):1027–52.
Doi Y, Chambers HF. Other β-lactam antibiotics. In: Mandell G, Bennett J, Dolin R, editors. Principles and practice of infectious diseases. 8th ed. Philadelphia: Saunders Elsevier; 2015. p. 293–7.
Cerceo E, Deitelzweig SB, Sherman BM, Amin AN. Multidrug-resistant Gram-negative bacterial infections in the hospital setting: overview, implications for clinical practice, and emerging treatment options. Microb Drug Resist. 2016;22(5):412–31.
Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics [Internet]: World Health Organization [published 27 February 2017; cited 23 May 2017]. http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/. Accessed 17 May 2017.
Wong D, van Duin D. Novel beta-lactamase inhibitors: unlocking their potential in therapy. Drugs. 2017;77(6):615–28.
Lob SH, Hackel MA, Kazmierczak KM, Hoban DJ, Young K, Motyl MR, et al. In vitro activity of imipenem–relebactam against gram-negative bacilli isolated from patients with lower respiratory tract infections in the United States in 2015–Results from the SMART global surveillance program. Diagn Microbiol Infect Dis. 2017;88(2):171–6.
Livermore DM, Warner M, Mushtaq S. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J Antimicrob Chemother. 2013;68(10):2286–90.
Toussaint KA, Gallagher JC. β-lactam/β-lactamase inhibitor combinations: from then to now. Ann Pharmacother. 2015;49(1):86–98.
Lapuebla A, Abdallah M, Olafisoye O, Cortes C, Urban C, Quale J, et al. Activity of meropenem combined with RPX7009, a novel β-lactamase inhibitor, against Gram-negative clinical isolates in New York City. Antimicrob Agents Chemother. 2015;59(8):4856–60.
The Medicines Company TANGO-1 analyst call, Carbavance TANGO-1 phase III trial results [Internet]: The Medicines Company [published 27 June 2016; cited 22 May 2017]. http://www.themedicinescompany.com/investors/event/medicines-company-tango-1-analyst-call. Accessed 16 May 2017.
Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, Griffith DC, et al. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem. 2015;58(9):3682–92.
Lin S, Huang C, Ko W, Chen Y, Hsueh P. Recent developments in antibiotic agents for the treatment of complicated intra-abdominal infections. Expert Opin Pharmacother. 2016;17(3):339–54.
Goldstein EJC, Citron DM, Tyrrell KL, Merriam CV. In vitro activity of biapenem plus RPX7009, a carbapenem combined with a serine β-lactamase inhibitor, against anaerobic bacteria. Antimicrob Agents Chemother. 2013;57(6):2620–30.
Livermore DM, Mushtaq S. Activity of biapenem (RPX2003) combined with the boronate β-lactamase inhibitor RPX7009 against carbapenem-resistant Enterobacteriaceae. J Antimicrob Chemother. 2013;68(8):1825–31.
Li XZ, Ma D, Livermore DM, Nikaido H. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: active efflux as a contributing factor to β-lactam resistance. Antimicrob Agents Chemother. 1994;38(8):1742–52.
Köhler T, Michea-Hamzehpour M, Epp SF, Pechere JC. Carbapenem activities against Pseudomonas aeruginosa: respective contributions of OprD and efflux systems. Antimicrob Agents Chemother. 1999;43(2):424–7.
Olsen I. New promising β-lactamase inhibitors for clinical use. Eur J Clin Microbiol Infect Dis. 2015;34(7):1303–8.
Blizzard TA, Chen H, Kim S, Wu J, Bodner R, Gude C, et al. Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin®. Bioorg Med Chem Lett. 2014;24(3):780–5.
Mangion IK, Ruck RT, Rivera N, Huffman MA, Shevlin M. A concise synthesis of a β-lactamase inhibitor. Org Lett. 2011;13(20):5480–3.
Morandi S, Morandi F, Caselli E, Shoichet BK, Prati F. Structure-based optimization of cephalothin-analogue boronic acids as beta-lactamase inhibitors. Bioorg Med Chem. 2008;16(3):1195–205.
Jackson JJ, Kropp H. β-lactam antibiotic-induced release of free endotoxin: in vitro comparison of penicillin-binding protein (PBP) 2-specific imipenem and PBP 3-specific ceftazidime. J Infect Dis. 1992;165(6):1033–41.
Lomovskaya O, Tsivkovski R. Vaborbactam (RPX7009) plus meropenem is active against the newly discovered BKC-1 and FRI-1 carbapenemases [abstract no. P1289 plus poster]. In: 26th European Congress of Clinical Microbiology and Infectious Diseases; 2016; Netherlands.
Tsivkovski R, Totrov M, Lomovskaya O. Inhibition of KPC-2 by vaborbactam (VAB; formerly Rpx7009) does not involve Ser130 (S130) that is important for its inhibition by avibactam (AVI) [presentation abstract]. In: ASM Microbe; 2016; Boston.
Zhanel GG, Lawson CD, Adam H, Schweizer F, Zelenitsky S, Lagacé-Wiens PRS, et al. Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs. 2013;73(2):159–77.
Jacoby GA, Munoz-Price LS. The new β-lactamases. N Engl J Med. 2005;352(4):380–91.
Shahid M, Sobia F, Singh A, Malik A, Khan HM, Jonas D, et al. Beta-lactams and beta-lactamase-inhibitors in current- or potential-clinical practice: a comprehensive update. Crit Rev Microbiol. 2009;35(2):81–108.
Drawz SM, Bonomo RA. Three decades of β-lactamase inhibitors. Clin Microbiol Rev. 2010;23(1):160–201.
Ambler RP. The structure of β-lactamases. Philos Trans R Soc Lond. 1980;289:321–31.
Bush K, Jacoby GA. Updated functional classification of β-lactamases. Antimicrob Agents Chemother. 2010;54(3):969–76.
Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 1995;39(6):1211–33.
Drawz SM, Papp-Wallace KM, Bonomo RA. New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother. 2014;58(4):1835–46.
Winkler M, Hujer AM, Bethel CR, Domitrovic TN, Young K, Donomo RA. Imipenem-ciastatin-relebactam (IMI/REL): an analysis of resistance in Pseudomonas aerugoinsa (Pa) isolates [abstract no. C-147 plus poster]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2015; San Diego.
Shlaes DM. New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann N Y Acad Sci. 2013;1277(1):105–14.
Barnes MD, Papp-Wallace KM, Alsop J, Domitrovic TN, Becka SA, Hujer AM, et al. Determining resistance mechanisms in Pseudomonas aeruginosa clinical isolates that demonstrate altered susceptibility profiles to β-lactam-relebactam (REL) vs. β-lactam-avibactam (AVI) combinations [abstract no. P0235 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Barnes MD, Bethel CR, Alsop J, Becka SA, Rutter JD, Papp-Wallace KM, et al. Relebactam (REL) inhibits the PDC-3 β-lactamase and restores the susceptibility of imipenem (IMI) against Pseudomonas aeruginosa [abstract no. 2780 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Lapuebla A, Abdallah M, Olafisoye O, Cortes C, Urban C, Landman D, et al. Activity of imipenem with relebactam against Gram-negative pathogens from New York City. Antimicrob Agents Chemother. 2015;59(8):5029–31.
Thaden JT, Pogue JM, Kaye KS. Role of newer and re-emerging older agents in the treatment of infections caused by carbapenem-resistant Enterobacteriaceae. Virulence. 2017;8(4):403–16.
Bush K. A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int J Antimicrob Agents. 2015;46(5):483–93.
El Amin N, Giske CG, Jalal S, Keijser B, Kronvall G, Wretlind B. Carbapenem resistance mechanisms in Pseudomonas aeruginosa: alterations of porin OprD and efflux proteins do not fully explain resistance patterns observed in clinical isolates. APMIS. 2005;113(3):187–96.
Haidar G, Clancy CJ, Chen L, Samanta P, Shields RK, Kreiswirth BN, et al. Identifying spectra of activity and therapeutic niches for ceftazidime-avibactam and imipenem–relebactam against carbapenem-resistant Enterobacteriaceae. Antimicrob Agents Chemother. 2017;AAC.00642-17 (in press).
Lomovskaya O, Tsivkovski R. Vaborbactam (VAB) is not affected by KPC-2 and KPC-3 variants containing Asp179Tyr amino acid substitution that are resistant to ceftazidime (CAZ) potentiation with avibactam [abstract no. 4169 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Tsivkovski R, Lomovskaya O. The effect of Trp105 substitutions in KPC on interactions with the novel beta-lactamase inhibitor RPX7009 [presentation abstract no. C-1194]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2014; Washington, D.C.
Sun D, Rubio-Aparicio D, Dudley MN, Lomovskaya O. Characterization of mutants selected in vitro using sub-optimal exposures of meropenem alone and with RPX7009 [abstract no. C-103 plus poster]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2014; Washington, D.C.
Sun D, Rubio-Aparicio D, Griffith D, Dudley MN, Lomovskaya O. Losing my resistance: loss of KPC following exposure of KPC-producing strains of Klebsiella pneumonia to carbapenems in combination with RPX7009 [presentation abstract no. C-1193]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2014; Washington, D.C.
Lob S, Hackel M, Badal R, Young K, Motyl M, Sahm D. In vitro activity of imipenem–relebactam (MK-7655) against Enterobacteriaceae and Pseudomonas aeruginosa from Latin America-SMART 2015 [abstract no. 2857 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Mavridou E, Melchers RJB, van Mil AC, Mangin E, Motyl MR, Mouton JW. Pharmacodynamics of imipenem in combination with β-lactamase inhibitor MK7655 in a murine thigh model. Antimicrob Agents Chemother. 2015;59(2):790–5.
Lob SH, Hackel MA, Kazmierczak KM, Young K, Motyl MR, Karlowsky JA, et al. In vitro activity of imipenem–relebactam against Gram-negative ESKAPE pathogens isolated by clinical laboratories in the United States in 2015–Results from the SMART global surveillance program. Antimicrob. Agents Chemother. 2017;61(6):e02209–16.
Lob S, Young K, Motyl M, Hawser S, Morrissey I, Magnet S, et al. Activity of imipenem–relebactam against Enterobacteriaceae and Pseudomonas aeruginosa from respiratory tract infections in Europe - SMART 2015 [abstract no. P1283 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Lob S, Hackel M, Badal R, Young K, Motyl M, Sahm D. In vitro activity of imipenem–relebactam (MK-7655) against Enterobacteriaceae from United States ICU and Non-ICU wards-SMART 2015-2016 [abstract no. 2846 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Lob S, Hackel M, Badal R, Young K, Motyl M, Sahm D. In vitro activity of imipenem–relebactam (MK-7655) against P. aeruginosa from United States ICU and Non-ICU Wards-SMART 2015-2016 [abstract no. 2848 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Hackel M, Young K, Motyl M, Sahm D. In vitro activity of imipenem–relebactam (MK-7655) against Enterobacteriaceae and Pseudomonas aeruginosa from urinary tract infections in North America—SMART 2015 [abstract no. 1840]. Open Forum Infect Dis. 2016;3(Suppl. 1):S515. Plus poster presented at IDWeek; 2016; New Orleans.
Hackel M, Young K, Motyl M, Sahm D. In vitro activity of imipenem–relebactam (MK-7655) against Enterobacteriaceae and Pseudomonas aeruginosa from respiratory tract infections in North America—SMART 2015 [abstract no. 1839]. Open Forum Infect Dis. 2016;3(Suppl. 1):S515. Plus poster presented at IDWeek; 2016; New Orleans.
Hackel M, Young K, Motyl M, Sahm D. In vitro activity of imipenem–relebactam (MK-7655) against Enterobacteriaceae and Pseudomonas aeruginosa from intraabdominal infections in North America—SMART 2015 [abstract no. 1838]. Open Forum Infect Dis. 2016;3(Suppl. 1):S515. Plus poster presented at IDWeek; 2016; New Orleans.
Zhang Y, Carpenter J, Altalhi S, Bush K. In vitro susceptibility of imipenem–relebactam against recent carbapenemase-producing carbapenem-resistant Enterobacteriaceae (CP-CRE) isolates [abstract no. 2944 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Snydman DR, Jacobus NV, McDermott LA. In vitro evaluation of the activity of imipenem–relebactam against 451 recent clinical isolates of Bacteroides group and related species. Antimicrob Agents Chemother. 2016;60(10):6393–7.
Citron DM, Merriam CV, Tyrrell KL, Leoncio E, Goldstein EJC. The in vitro activity of relebactam, imipenem, and the combination of the two, plus six comparator antimicrobial agents against 432 strains of anaerobic bacteria including imipenem-resistant organisms [abstract no. 5138 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Papp-Wallace KM, Barnes MD, Alsop J, Taracila MA, Bethel CR, Becka SA, et al. Relebactam is a potent inhibitor of the KPC-2 β-lactamase and restores the susceptibility of imipenem against KPC-producing Enterobacteriaceae [abstract no. P1284 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Performance standards for antimicobial susceptibility testing. Clinical and Laboratory Standards Institute (CLSI). 2017. M100-S27. 2017.
Castanheira M, Huband MD, Mendes RE, Flamm RK. Meropenem–vaborbactam tested against contemporary Gram-negative isolates collected worldwide during 2014, including carbapenem-resistant, KPC-producing, multidrug-resistant, and extensively drug-resistant Enterobacteriaceae. Antimicrob Agents Chemother. 2017;AAC.00567-17 (in press).
Castanheira M, Rhomberg PR, Flamm RK, Jones RN. Effect of the β-lactamase inhibitor vaborbactam combined with meropenem against serine carbapenemase-producing Enterobacteriaceae. Antimicrob Agents Chemother. 2016;60(9):5454–8 (Plus supplemental material).
Castanheira M, Rhomberg PR, Watters AA, Jones RN. In vitro activity of meropenem/RPX7009, a carbapenem/β-lactamase inhibitor combination tested against contemporary populations of Enterobacteriaceae and KPC-producing strains [abstract no. 257]. Open Forum Infect Dis. 2014;1(Suppl. 1):S70. Plus poster presented at IDWeek; 2014; Philadelphia.
Huband MD, Flamm RK, Rhomberg PR, Jones RN, Castanheira M. In vitro antibacterial activity of meropenem/RPX7009, (a carbapenem/β-lactamase inhibitor combination) against contemporary Enterobacteriaceae isolated from intra-abdominal and urinary tract infections in the United States [abstract no. 781]. Open Forum Infect Dis. 2015;2(Suppl. 1):S149. Plus poster presented at IDWeek; 2015; San Diego.
Hackel M, Badal R, Sahm D. In vitro activity of meropenem–vaborbactam against isolates of KPC-producing Enterobacteriaceae collected worldwide in 2014–2015 [abstract no. 1830]. Open Forum Infect Dis. 2016;3(Suppl. 1):S515. Plus poster presented at IDWeek; 2016; New Orleans.
Hackel M, Badal R, Sahm D. In vitro activity of meropenem–vaborbactam against KPC-producing Enterobacteriaceae from Europe collected in 2014–2015 [abstract no. P1287 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Castanheira M, Mendes RE, Flamm RK, Jones RN. Activity of meropenem/RPX7009 and comparator agents tested against contemporary Enterobacteriaceae isolates collected from bloodstream infections in USA hospitals [abstract no C-152 plus poster]. In: 55th Interscience Conference of Antimicrobial Agents and Chemotherapy; 2015; San Diego.
Castanheira M, Woosley LN, Huband MD, Flamm RK. Meropenem–vaborbactam activity against Enterobacteriaceae isolates, including carbapenem-resistant and carbapenemase-producing isolates, collected in United States (US) hospitals during 2016. [abstract no. 2705 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Yang Q, Xu Y, Xu Z, Zhang G, Chen X. In vitro activity of meropenem combined with RPX7009 against Enterobacteriaceae producing KPC-type carbapenemases in China [abstract no. P1285 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Castanheira M, Huband MD, Flamm RK, Jones RN. Meropenem–vaborbactam (MER-VAB) tested against contemporary Enterobacteriaceae isolates from USA hospitals [abstract no. 452 plus poster]. In: ASM Microbe; 2016; Boston.
Weiss WJ, Pulse ME, Nguyen P, Peterson K, Silva J, Simecka JW, et al. Efficacy of Carbavance (meropenem + RPX7009) against carbapenem-resistant E. coli and K. pneumoniae in a murine UTI model [abstract no. B-078 plus poster]. In: 55th Interscience Conference of Antimicrobial Agents and Chemotherapy; 2015; San Diego.
Tarazi Z, Sabet M, Rubio-Aparicio D, Nolan T, Parkinson J, Dudley MN, et al. Meropenem–vaborbactam against highly carbapenem-resistant Enterobacteriaceae in an in vitro hollow fiber model using PK from phase 1 and phase 3 data in patients [abstract no. P1288 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Tarazi Z, Sabet M, Rubio-Aparicio D, Nolan T, Parkinson J, Lomovskaya O, et al. Efficacy of simulated human exposures of Carbavance (meropenem-RPX7009) against carbapenem-resistant Enterobacteriaceae in an in vitro hollow fiber model [abstract no. F-959 plus poster]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2014; Washington, D.C.
Lomovskaya O, Griffith DC, Loutit JS, Dudley MN. Rationale for dose selection for Carbavance (CVC; meropenem/RPX7009 in phase 3 trials [abstract no. 64E plus poster]. In: 19th Making A Difference in Infectious Diseases; 2016; Florida.
Griffith DC, Sabet M, Tarazi Z, Lomovskaya O, Dudley MN. Pharmacodynamics of vaborbactam when administered in combination with meropenem [abstract no. 5980 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Lala M, Brown M, Kantesaria B, Walker B, Paschke A, Rizk M. Population pharmacokinetic analysis of relebactam (REL) and imipenem in phase 1 healthy volunteers and phase 2 patients with complicated intra-abdominal infection (cIAI) [abstract no. A-040 plus poster]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2015; San Diego.
Butterton JR, Jumes P, Calder N, Rizk ML, Nefliu M, Sun P, et al. A phase I study evaluating the safety, tolerability, and pharmacokinetics of an intravenous beta-lactamase inhibitor in healthy male volunteers [abstract no. F1-1967 plus presentation]. In: 50th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2010; Boston.
Jumes P, Rizk ML, Calder N, Gutierrez M, Warrington S, Li X, et al. Phase I studies evaluating the safety, tolerability, and pharmacokinetics of multiple doses of an intravenous beta-lactamase inhibitor in healthy young males and single doses in healthy elderly male, elderly female and young female volunteers [abstract no. A-009 plus poster]. In: 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2012; San Francisco.
Rhee EG, Jumes PA, Gotfried MH, Rizk ML, Liu Y, Mangin E, et al. Intrapulmonary pharmacokinetics of MK-7655, a novel β-lactamase inhibitor, dosed in combination with imipenem/cilastatin in healthy subjects [abstract no. A-1028 plus poster]. In: 53th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2013; Denver.
Rizk ML, Jumes P, Lasseter K, Marbury T, Mangin E, Liu Y, et al. Pharmacokinetics of MK-7655, a novel β-lactamase inhibitor (BLI), in combination with imipenem/cilastatin (IPM/CIL) in subjects with impaired renal function [abstract no. A-010 plus poster]. In: 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2012; San Francisco.
Trang M, Griffith DC, Bhavnani SM, Loutit JS, Dudley MN, Ambrose PG, et al. Population pharmacokinetics of meropenem and vaborbactam in healthy volunteers and infected patients [abstract no. 2685 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Griffith DC, Rubino CM, Loutit JS, Morgan EE, White D, Dudley MN. A phase 1 study of the safety, tolerability, and pharmacokinetics of the beta-lactamase inhibitor RPX7009 alone, meropenem alone, and both in combination (Carbavance) TID for 7 days in healthy adult subjects [abstract no. 401]. Open Forum Infect Dis. 2014;1(Suppl. 1):S70. Plus poster presented at IDWeek; 2014; Philadephia.
Griffith DC, Loutit JS, Morgan EE, Durso S, Dudley MN. Phase 1 study of the safety, tolerability, and pharmacokinetics of the β-lactamase inhibitor vaborbactam (RPX7009) in healthy adult subjects. Antimicrob Agents Chemother. 2016;60(10):6326–32.
Wenzler E, Gotfried MH, Loutit JS, Durso S, Griffith DC, Dudley MN, et al. Meropenem-RPX7009 concentrations in plasma, epithelial lining fluid, and alveolar macrophages of healthy adult subjects. Antimicrob Agents Chemother. 2015;59(12):7232–9.
Powles MA, Galgoci A, Misura A, Liberator P, Hammond M. In vivo efficacy of the beta-lactamase inhibitor, MK-7655, in combination with imipenem in murine models of infection [abstract no. F1-2140 plus poster]. In: 50th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2010; Boston.
Bhagunde P, Chang K, Hirsch EB, Ledesma KR, Nikolaou M, Tam VH. Novel modeling framework to guide design of optimal dosing strategies for β-lactamase inhibitors. Antimicrob Agents Chemother. 2012;56(5):2237–40.
Wu J, Racine F, Rizk ML, Wismer MK, Harradine P, Young K, et al. Exploring PK/PD relationship of a novel beta-lactamase inhibitor MK-7655 in combination with imipenem in a hollow fiber infection model [abstract no. P1740 plus poster]. In: 24th European Congress of Clinical Microbiology and Infectious Diseases; 2014; Barcelona.
Lucasti C, Vasile L, Sandesc D, Venskutonis D, McLeroth P, Lala M, et al. Phase 2, dose-ranging study of relebactam with imipenem-cilastatin in subjects with complicated intra-abdominal infection. Antimicrob Agents Chemother. 2016;60(10):6234–43.
Tarazi Z, Sabet M, Rubio-Aparicio D, Sun D, Nolan T, Parkinson J, et al. Efficacy of simulated human exposures of meropenem compared to Carbavance (meropenem-RPX7009) against Pseudomonas aeruginosa in an in vitro hollow fiber model [abstract no. P1288 plus poster]. In: 25th European Congress of Clinical Microbiology and Infectious Diseases; 2015; Denmark.
Bhavnani SM, Hammel JP, Rubino CM, Trang M, Loutit JS, Griffith DC, et al. Meropenem–vaborbactam pharmacokinetic-pharmacodynamic analyses for efficacy based on data from patients enrolled in phase 3 studies [abstract no. 2834 plus poster]. In: 2nd ASM Microbe; 2017; New Orleans.
Sabet M, Tarazi Z, Nolan T, Parkinson J, Rubio-Aparicio D, Lomovskaya O, et al. In vivo efficacy of carbavance (meropenem/RPX7009) against KPC-producing Enterobacteriaceae [abstract no. F-958 plus poster]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; 2014; Washington, D.C.
Sims M, Mariyanovski V, McLeroth P, Akers W, Lee Y, Brown ML, et al. Prospective, randomized, double-blind, phase 2 dose-ranging study comparing efficacy and safety of imipenem/cilastatin plus relebactam with imipenem/cilastatin alone in patients with complicated urinary tract infections. J Antimicrob Chemother. 2017. (in press).
Bidair M, Zervos M, Sagan OS, Zaitsev V, Loutit J, Dudley M, et al. Clinical outcomes in adults with complicated urinary tract infections (cUTI), including acute pyelonephritis (AP) in TANGO 1, a phase 3 randomized, double-blind, double-dummy trial comparing meropenem–vaborbactam (M-V) with piperacillin-tazobactam (P-T) [abstract no. P1289 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Walsh TJ, Bhowmick T, Darouiche RO, Shorr A, Zaitsev VI, Perlin DS, et al. Per pathogen outcomes of meropenem–vaborbactam (M-V) versus piperacillin-tazobactam (P-T) in the treatment of adults with complicated urinary tract infections (cUTI), including acute pyelonephritis (AP), in TANGO 1, a phase 3 randomized, double-blind,. double-dummy trial [abstract no. P1290 plus poster]. In: 27th European Congress of Clinical Microbiology and Infectious Diseases; 2017; Vienna.
Kaye KS, Vazquez J, Mathers A, Daikos G, Alexander E, Loutit JS, et al. Clinical outcomes of serious infections due to carbapenem-resistant enterobacteriaceae (CRE) in TANGO II a phase 3 randomized multi-national open-label trial of meropenem–vaborbactam (M-V) versus best available therapy (BAT). [abstract no. 1862]. In: IDWeek; 2017; San Diego.
Paterson D, Kwak EJ, Bhowmick T, Alexander E, Loutit JS, et al. Meropenem–vaborbactam vs. best available therapy for carbapenem-resistant enterobacteriaceae infections in TANGO II: Outcomes in immunocompromised patients. [abstract no. 1868]. In: IDWeek; 2017; San Diego.
Primaxin® product monograph. New Jersey: Merck Sharp & Dohme Corporation, a subsidiary of Merck & Company, Incorporated, 2016.
Merrem® product monograph. Ontario: AstraZeneca Canada Incorporated, 2017.
Acknowledgements
The authors would like to thank Pia Graham from Merck and Dr. Niki Patel from The Medicines Company for their efforts obtaining published literature on imipenem–relebactam and meropenem–vaborbactam, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this manuscript.
Conflict of interest
George Zhanel and Daryl Hoban have both received research grant funding from Merck and The Medicines Company. Courtney Lawrence, Heather Adam, Frank Schweizer, Sheryl Zelenitsky, Michael Zhanel, Philippe Lagacé-Wiens, Andrew Walkty, Andrew Denisuik, Alyssa Golden, Alfred Gin, Joseph Lynch and James Karlowsky have no conflicts to declare.
Rights and permissions
About this article

Cite this article
Zhanel, G.G., Lawrence, C.K., Adam, H. et al. Imipenem–Relebactam and Meropenem–Vaborbactam: Two Novel Carbapenem-β-Lactamase Inhibitor Combinations. Drugs 78, 65–98 (2018). https://doi.org/10.1007/s40265-017-0851-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0851-9