
Abstract
Chronic obstructive pulmonary disease (COPD) and cardiovascular diseases often coexist. The mechanistic links between these two diseases are complex, multifactorial and not entirely understood, but they can influence the therapeutic approach. Therapy can be primarily directed towards treating the respiratory symptoms and reducing lung inflammation. Smoking cessation, bronchodilators and inhaled corticosteroids are central to this therapeutic approach. The underlying pathophysiological mechanisms that are responsible for the increased cardiovascular risk in COPD remain unclear, but might include arterial stiffness, inflammation and endothelial dysfunction as a consequence of systemic exposure to chemicals in cigarette smoke or airborne pollution. Therefore, it is plausible that treatment of cardiovascular co-morbidities might reduce morbidity and mortality in patients with COPD and, consequently, therapy of COPD should be shifted to the treatment of cardiovascular diseases and systemic inflammation. In support of this approach, early data suggest that patients with COPD treated with angiotensin-converting enzyme inhibitors, angiotensin II type 1 receptor blockers, statins, anti-platelet drugs or β-adrenoceptor blockers may have improved survival and reduced hospitalisation from acute exacerbations of COPD. In this review, the potential impact of traditional therapies for COPD that are centred on treating the lungs and newer strategies potentially able to affect and mitigate cardiovascular risks in patients with COPD are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chronic obstructive pulmonary disease (COPD) and cardiovascular diseases (CVDs) often coexist. While increasing evidence suggests that the systemic inflammatory pathway provides the common link between COPD and CVD, the mechanisms by which the systemic component arises are unclear. |
We still do not know whether the successful treatment of the co-morbid diseases associated with COPD, mainly CVD, also positively influences the course of the lung disease because, to date, there are few definite data documenting that treatment of COPD co-morbidities will reduce morbidity and mortality rates in these patients. |
Currently, no COPD guideline recommends a core cardiovascular evaluation for patients with COPD, and no CVD guideline recommends a core pulmonary evaluation for patients with CVD. This is the likely reason why there is little specific guideline guidance for treating COPD in patients with CVD, or vice versa. There is clearly a need for COPD and CVD to be considered in an integrated way. |
1 Coexistence of Chronic Obstructive Pulmonary Disease and Cardiovascular Diseases
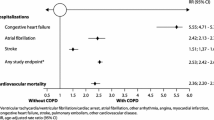
Chronic obstructive pulmonary disease (COPD) and cardiovascular diseases (CVDs) often coexist [1]. An Italian population-based retrospective study provided solid evidence that patients with the clinical diagnosis of COPD are highly associated with clinical diagnoses of CVDs [2]. Among patients with the diagnosis of COPD, there were no differences in sex for association with all CVDs except for angina, coronary disease and myocardial infarction, for which the odds ratios were higher in women compared with men. The age clusters between 35 and 54 years were those with the highest odds ratio for the simultaneous presence of the diagnoses of CVD and COPD. A progressive significant reduction in odds ratio values in older age clusters was also observed [2]. The largest and perhaps most conclusive systematic review with a meta-analysis from large international studies has recently documented that the risk of major CVD types (ischaemic heart disease, cardiac dysrhythmia, heart failure, diseases of the pulmonary circulation and artery diseases) is increased by up to five times in patients with COPD [3].
In this review, we examine the potential impact of traditional therapy of COPD centred on treating the lungs (mainly inhaled therapy) and the different strategies potentially able to affect and mitigate cardiovascular (CV) risks in patients with COPD (mainly systemic therapy). References for this review were identified through searches of PubMed, Scopus and Google Scholar for articles published from January 1971 to February 2017, by use of the terms ‘Chronic obstructive pulmonary disease (COPD)’, ‘cardiovascular disease (CVD)’, ‘bronchodilators’, ‘inhaled corticosteroids’, ‘ACE inhibitors’, ‘AT1 receptor blockers’, ‘statins’, ‘antiplatelet agents’ and ‘β-adrenoceptor blockers’. Articles resulting from these searches, published in English, French, German and Italian, as well as relevant references cited in those articles were reviewed.
2 Mechanistic Links between Chronic Obstructive Pulmonary Disease and Cardiovascular Diseases
The mechanistic links between COPD and CVD are complex, multifactorial and not entirely understood [4]. COPD and CVD share recognised risk factors, mainly smoking but also older age and unhealthy lifestyle choices play a role. However, we still do not know whether COPD and CVD are linked beyond these risk factors. COPD is characterised by persistent lung and systemic inflammation [5]. Simultaneously elevated levels of C-reactive protein (CRP), fibrinogen and leukocyte count are associated with a two- to four-fold increased risk of CVD in COPD [5].
While increasing evidence suggests that the systemic inflammatory pathway provides the common link between COPD and CVD, the mechanisms by which the systemic component arises are unclear [6]. It has been suggested that inflammation in the lung spills into the systemic circulation causing systemic inflammation [7]. This systemic inflammatory response, in turn, is related to acute endothelial dysfunction of systemic blood vessels [7]. However, it has also been proposed that COPD could be considered as part of a “chronic systemic inflammatory syndrome” and the systemic inflammation in COPD represents a systemic component of the disease that develops in parallel with, or prior to, pulmonary inflammation [8].
The two different views relating the observed associations between COPD and CVD have important therapeutic consequences and may not be mutually exclusive [9]. In the spill-over theory, the aims of therapy are primarily centred on treating the lungs. In the ‘systemic’ inflammatory state with multiple organs compromised, the centre of therapy should be shifted to the treatment of systemic inflammation.
3 Traditional Therapy of Chronic Obstructive Pulmonary Disease Centred on the Lungs
Smoking cessation and therapy with bronchodilators and inhaled corticosteroids (ICSs) are currently the cornerstone of treatment for COPD [10].
3.1 Smoking Cessation
Whether smoking cessation might reduce systemic inflammation is still unknown. This seems to be questionable because elevated CRP levels have been found in ex-smokers with COPD [11]. The factors that drive inflammation in COPD after smoking cessation have not been clearly established, although autoimmunity, embedded particles/heavy metals from smoking and chronic bacterial infection have all been proposed to have a role [12].
3.2 Bronchodilators
Using the Manitoba Health database to examine the relationship between use of inhaled respiratory drugs in people with COPD and CVD hospitalisations from 1996 through 2000, Macie and colleagues [13] observed that bronchodilators were associated with hospitalisations for CVD. These agents apparently imposed a larger relative risk in people with unrecognised CVD than in people recognised as having this condition.
3.2.1 β2-Adrenoceptor Agonists
There is evidence that even in healthy subjects, inhalation of a therapeutic dose of salbutamol results in significant haemodynamic changes, which are accompanied by a shift in CV autonomic tone towards increased sympathetic outflow in the absence of baroreceptor activation [14]. These changes in cardiac autonomic function may contribute to an increased cardiac risk associated with inhaled β2-adrenoceptor agonist (β2-agonist) treatment. In effect, using the computerised health administrative databases of the province of Quebec, Wilchesky and colleagues documented that initiation of treatment with both short- and long-acting β-agonists (LABAs) may be associated with a modest increase in the risk of cardiac arrhythmia in patients with COPD [15].
β-Agonists induce an increase in heart rate and a worsening of arrhythmia, mainly in a patient with COPD experiencing pre-existing cardiac arrhythmias and hypoxemia, but this has also been observed in normal subjects. The magnitude of these effects seems to be dependent on the dose of the agent used and whether it is a full or a partial agonist [16].
A case-control study within the Group Health Cooperative of Puget Sound showed that new use of β-agonists among patients with CVD was associated with an increased risk of myocardial infarction, although it was difficult to determine if the association is causal [17]. In fact, it was impossible to exclude that β-agonists were prescribed for nonspecific respiratory symptoms or chest discomfort that represents undiagnosed angina, which is a major risk factor for myocardial infarction. It was also reported that patients hospitalised for myocardial infarction or unstable angina were significantly more likely than control subjects to have received a β-agonist during the 3 months immediately prior to their index date [18]. A dose-response relationship was observed that did not appear to be confounded by tobacco use, COPD history, CVD or CV risk factors. A recent meta-analysis of 24 randomised controlled trials aimed to assess the CV safety of inhaled LABAs in COPD and indicated that these bronchodilators do not increase the risk of fatal CV events in COPD; in subgroup analyses, inhaled LABAs were able to significantly decrease fatal CV events in long-term trials, but there was no significant difference between inhaled LABAs and placebo in the short-term trials [19].
Although Au and colleagues demonstrated that β2-agonist use is associated with excess hospitalisations from heart failure, as well as an increased risk of all-cause mortality in patients with left-ventricular dysfunction [20], a large-scale study with a >3700 patient-years follow-up suggested that inhaled β2-agonists are not associated with increased mortality in community-managed heart failure patients when adjusted for the plasma B-type natriuretic peptide (BNP) level. This may provide an important method for assessing ventricular function, as well as other clinical, demographic and medication variables [21]. Pulmonary function abnormalities, especially diffusion impairment and airway obstruction, are highly prevalent in patients with chronic heart failure, even in a stable and non-congested condition, but without a chronic airway disorder [22]. It is not surprising that inhaled bronchodilators may have an additional role in the management of patients with chronic heart failure because of their potential to improve pulmonary function, especially in those with airway obstruction [23]. In patients with COPD experiencing an exacerbation, the prevalence of left-ventricular systolic dysfunction is common [24] and, consequently, plasma levels of BNP are elevated [25]. We have recently documented that both the short-acting β2-agonist salbutamol and the ultra-LABA indacaterol induce a rapid reduction in BNP levels in patients admitted to the emergency department for an acute exacerbation of COPD (AECOPD). This is likely because both β2-agonists attenuate air trapping, leading to a reduction in intrathoracic pressure, including pressure on the whole heart, and, consequently, to an improvement of right-ventricular overload and left-ventricular diastolic dysfunction [26].
3.2.2 Muscarinic Antagonists
Despite the concerns raised about an excess risk of CV adverse events with inhaled muscarinic antagonists [27], the results of a recent, very large pooled analysis of adverse event data of the use of tiotropium bromide, delivered by either the HandiHaler® (18 μg) or the Respimat® (5 μg) device in randomised clinical trials in patients with COPD (treatment duration ≥4 weeks) totalling 14,909 patient-years of tiotropium exposure, showed that tiotropium bromide was not associated with an increased risk of cardiac or vascular adverse events. Further, even more importantly, there was no evidence of an increased risk for major adverse CV events or fatal major adverse CV events for the tiotropium bromide-treated group [28].
However, because a meta-analysis with the fixed-effect model has suggested that there might be a significantly increased risk of death with tiotropium bromide delivered using the Respimat® device vs. placebo, the risk being more evident for CV death [29], we conducted a safety evaluation of tiotropium bromide delivered with the Handihaler® (18 µg) device vs. tiotropium bromide using the Respimat® (5 and 2.5 µg) device [30]. We performed a systematic review and network meta-analysis of the currently available clinical evidence, including data from the massive Tiotropium Safety and Performance in Respimat (TIOSPIR) trial [31]. This meta-analysis demonstrated that the safety profile of tiotropium bromide delivered by the Handihaler® device seems to be superior to that of the Respimat® device, although no statistical difference was detected between these two devices. However, the Surface Under the Cumulative Ranking analysis favoured tiotropium bromide delivered by the Respimat® device with regard to serious adverse events. It is unlikely that using the Respimat® device rather than the HandiHaler® device exposes patients to higher risks of real adverse CV events. It is rather more probable that there may be a different CV response to muscarinic receptor antagonism in individual patients.
3.2.3 Dual Bronchodilators
Recently, there has been the introduction of a number of inhaled medicines containing both a β2-agonist and a muscarinic receptor antagonist in the same device, which is being actively promoted as the cornerstone of therapy for patients with COPD. In a nested case-control study with the use of the nationwide insurance claims database of the Korean Health Insurance Review and Assessment Service, long-acting muscarinic antagonists (LAMAs) without LABAs, and LABAs without LAMAs showed a similar impact on the development of tachyarrhythmia; however, there were no significant multiplicative or additive interactions between LABAs and LAMAs from a safety standpoint [32].
Additionally, a systematic review with a meta-analysis of dual bronchodilation treatment with a LAMA/LABA combination for the treatment of stable COPD indicated that approved doses of LAMA/LABA fixed-dose combinations did not show any significant difference concerning the cardiac safety profile compared with treatment with the monocomponents [33]. Intriguingly, when compared with respective monocomponents, the umeclidinium/vilanterol combination provided a signal (p = 0.057) of possible protection against severe CV adverse events, whereas the glycopyrronium/indacaterol combination significantly (p < 0.05) protected against these events.
3.3 Inhaled Corticosteroids
Therapy with ICSs might suppress systemic inflammation in patients with stable COPD [34]. Inhaled corticosteroids can down-regulate the inflammatory process in the lungs, which in turn, reduces the spill-over of inflammatory mediators from the lungs into the systemic circulation, dampening the systemic inflammatory process that is persistent in patients with obstructive airways disease. Because systemic inflammation is a co-factor in the genesis of endothelial dysfunction and progression of atherosclerosis, ICSs can enhance blood vessel function and reduce the risk of ischaemic events. Critical to this process are mediators such as interleukin-6, tumour necrosis factor-α, matrix metalloproteinases and nitric oxide, which may be modulated by ICSs.
Initially, it was highlighted that only very low doses of ICSs may be associated with a reduction in the risk of acute myocardial infarction [35]. The lack of benefit at higher doses might plausibly reflect counterbalancing adverse effects of other risk factors or the fact that patients with more severe disease, itself linked to CV morbidity, are prescribed higher doses. Nevertheless, a post-hoc analysis of the European Respiratory Society’s study on Chronic Obstructive Pulmonary Disease (EUROSCOP), a 3-year placebo-controlled study of an ICS (budesonide 800 µg day−1) in smokers with mild COPD, suggested that ischaemic cardiac events among these patients could be significantly reduced by inhaled budesonide at a rather high dosage [36].
In any case, a post-hoc analysis of the pivotal 3-year TOwards a Revolution in COPD Health (TORCH) study not only confirmed that the occurrence of new CV adverse events was no more frequent in patients treated with a LABA than in those treated with placebo, but also suggested that the combination of a LABA and an ICS might offer a degree of cardioprotection [37]. Furthermore, the Study to Understand Mortality and MorbidITy (SUMMIT), which enrolled more than 16,000 patients with concomitant moderate COPD and CVD, documented that combined ICS and LABA treatment had no effect on overall mortality or CV events [38].
4 Use of Therapy to Treat Systemic Inflammation and Co-Morbidities
As already highlighted, the underlying pathophysiological mechanisms that are responsible for the increased CV risk in COPD remain unclear [1], but might include arterial stiffness, inflammation and endothelial dysfunction as a consequence of systemic exposure to chemicals in cigarette smoke or airborne pollution [4]. Therefore, it seems logical to think that treatment of CVD might reduce morbidity and mortality in patients with COPD and it is therefore perhaps not surprising that patients with COPD treated with angiotensin-converting enzyme (ACE) inhibitors, angiotensin II type 1 (AT1) receptor blockers, statins, anti-platelet drugs or β-adrenoceptor blockers (β-blockers) may have improved survival and reduced hospitalisation from AECOPDs [1, 4]. Mancini and colleagues were the first to suggest that the cardioprotective effects of ACE inhibitors and statins, and potentially those of AT1 receptor blockers, extend also to patients with COPD regardless of their concomitant CV risk profile [39].
4.1 Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Type 1 Receptor Blockers
The renin-angiotensin system is potentially implicated in the pathogenesis of COPD through its involvement in inducing proinflammatory mediators in the lung [9]. Angiotensin II stimulates the release of cytokines including interleukin-6, tumour necrosis factor-α and monocyte chemotactic protein-1, and also has an immunomodulatory effect on T-cell responses, which mediate the lung tissue injury associated with COPD. Furthermore, the AT1 receptor/AT2 receptor ratio increases noticeably in regions of marked fibrosis surrounding bronchioles, which correlated with the reduction in forced expiratory volume in 1 s (FEV1). These actions support a role for angiotensin II in inducing bronchoconstriction via the AT1 receptor. The renin-angiotensin system can also generate reactive oxygen species via the AT1 receptor, promoting mitochondrial dysfunction, which contributes to the oxidative stress and impaired redox signalling, observed in patients with COPD. ACE inhibitors and AT1 receptor antagonists also block the proinflammatory effect of angiotensin II [9]. Figure 1 illustrates the protective role of ACE inhibitors and AT1 receptor blockers against inflammation, vasoconstriction and small airways fibrosis induced by activation of the renin-angiotensin system. In effect, in an established murine model, antagonism of TGF-β signalling with angiotensin receptor blockade normalises histology and reduces oxidative stress, cell death, inflammation and, importantly, metalloprotease activation associated with chronic cigarette smoke exposure [40].

Impact of angiotensin-converting enzyme (ACE) inhibitors and angiotensin II type 1 receptor blockers (ARBs) on oxidative stress, proinflammatory signalling and proliferative effects induced by renin-angiotensin system activation. AT1 R angiotensin II type 1 receptor, eNOS endothelial nitric oxide synthase, IL interleukin, MCP-1 monocyte chemoattractant protein-1, NADPH reduced form of nicotinamide adenine dinucleotide phosphate, NF-kB transcription factor nuclear factor-kB, NO nitric oxide, RANTES regulated on activation normal T cell expressed and secreted, ROS reactive oxygen species, TGF-β1 transforming growth factor β1, TNF-α tumour necrosis factor-α
It is not surprising, therefore, that by using the New Mexico-based longitudinal Lovelace Smokers Cohort, it was shown that ACE inhibitors, but not other CV drugs may be protective against rapid FEV1 decline in smokers [41]. This protective effect was stronger on the decline in FEV1 than on the decline in forced vital capacity, suggesting that ACE inhibitors were more likely to be effective in preventing the development of obstructive rather than nonobstructive lung diseases among smokers. Furthermore, Mortensen and colleagues found that the use of ACE inhibitors prior to admission reduced the mortality of patients hospitalised with AECOPDs [42] and the study of Kim and colleagues revealed that the use of ACE inhibitors and angiotensin II receptor blockers was associated with reducing the risk of pneumonia in patients with COPD [43].
There is also documentation that use of the ACE-inhibitor enalapril alongside a programme of pulmonary rehabilitation, in patients without an established indication for ACE inhibition, reduced the peak work rate response to exercise training in patients with COPD [44]. However, this finding contrasts with the results of a previous study showing that ACE inhibition, using fosinopril for 3 months, did not improve quadriceps function or exercise performance in patients with COPD with quadriceps weakness [45].
4.2 Statins
Changes in high-sensitivity (hs)-CRP over time were associated with corresponding changes in exercise tolerance under statin therapy in COPD [46]. Improvement of exercise tolerance was greater in those with a greater decrease of hs-CRP levels and higher baseline CRP levels. Because of its significant correlation of the changes between exercise time and hs-CRP levels, it is likely that the involved mechanism could be directly to inhibit hs-CRP by pravastatin.
Statins have several pharmacological actions that might be beneficial in the treatment of patients with COPD, including antioxidant, anti-inflammatory and immunomodulatory effects [9]. The mechanism by which statins could be used for the treatment of patients with COPD, involving the inhibition of the mevalonate pathway, seems to be the same as that observed for cholesterol lowering [9]. Inflammatory cells involved in lung and systemic inflammation including eosinophils, neutrophils, macrophages, mast cells, T cells and dendritic cells are all affected by 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity, and mevalonate pathway metabolites and/or GTPases, and this evidence collectively indicates a fundamental role for the mevalonate pathway in respiratory health and disease [9]. Figure 2 shows the impact of statins on the mevalonate pathway, leading to anti-inflammatory effects and protection against bronchial hyperresponsiveness.

Mevalonate pathway and mechanism of action of statins. BHR bronchial hyperresponsiveness, CoA coenzyme A, FPP farnesyl-pyrophosphate, FTase farnesyl protein transferase, GGPP geranylgeranyl-pyrophosphate, GGTase geranylgeranyl protein transferase, HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A, IPP isopentenyl-pyrophosphate, SQase squalene synthase
Inhibition of the mevalonate pathway, which triggers the prenylation of small GTPases, by statins prevents the detrimental effects of lipopolysaccharide in isolated human airways [47]. This suggests a potential role for statins in preventing AECOPDs. In effect, the use of statins prior to admission is associated with decreased mortality in subjects hospitalised with an AECOPD [42]. This information has been generated by a retrospective national cohort study using Veterans Affairs administrative data including subjects ≥65 years of age hospitalised with an AECOPD.
However, in the Prospective Randomized Placebo-Controlled Trial of Simvastatin in the Prevention of COPD Exacerbations (STATCOPE), a large randomised multicentre trial that enrolled 885 participants with COPD who were at high risk for exacerbations and were treated from 12 to 36 months, the mean number of exacerbations per person-year was similar in the simvastatin and placebo groups [48]. There were no significant between-group differences in the time to the first exacerbation.
Furthermore, in the Cohort of Mortality and Inflammation in COPD (COMIC) study that used a large prospective cohort with patients with well-defined COPD, statin use was not associated with time until first hospitalisation for an AECOPD or time until first community-acquired pneumonia and even with a reduced risk of all-cause mortality in patients with COPD [49]. Furthermore, a post-hoc analysis of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico Heart Failure (GISSI-HF) trial, which investigated the efficacy of rosuvastatin at the dose of 10 mg daily in a population with co-existing heart failure and COPD, failed to show any beneficial effects on the analysed endpoints of all cause mortality, CV and non-CV mortality, and hospitalisation [50]. Nevertheless, the authors of this post-hoc analysis suggested that some sub-populations, for instance the “systemic inflammation novel phenotype”, might receive some beneficial effects from regular statin use, also in the absence of high-cholesterol levels [50]. In effect, in the Rosuvastatin therapy on peripheral vasoDilator function, inflammatory markers and pulmonary function in patients with stablE chronic Obstructive pulmonary disease (RODEO) study, which enrolled patients with stable COPD without the standard indications for statin therapy, but with evidence of systemic inflammation, rosuvastatin 10 mg once daily for 12 weeks was associated with a significant attenuation of systemic inflammation and improvement in endothelial-dependent vascular function [51].
4.3 β-Adrenoceptor Blockers
In the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme, which randomised 7599 patients with symptomatic heart failure, survival was significantly better in patients with heart failure receiving β-blockers, irrespective of concurrent bronchodilator therapy [52]. Use of β-blockade in patients with COPD and concomitant heart failure has a solid pharmacological basis. It has been documented for a long time that an enhanced and sustained cardiac adrenergic drive occurs in heart failure and also in patients with COPD [53]. The increased levels of plasma adrenaline stimulate desensitisation of the β2-adrenoceptors because of G protein-coupled receptor kinase 2-mediated phosphorylation that leads to β2-arrestin binding and receptor internalisation [54].
Rinaldi and colleagues showed that in a murine model of heart failure, these increased levels of plasma adrenaline result in a significant attenuation of relaxation of the airways, an effect that is reversed by treatment with metoprolol [54]. The same group has also documented that the co-administration of a β2-agonist and a β1-blocker can influence cardiac remodelling [55]. They showed that treatment with either indacaterol or metoprolol significantly reduced the infarct size in heart failure rats compared with the untreated heart failure group. The combination of indacaterol and metoprolol reduced the infarct size even further, reduced both blood pressure and heart rate, reversed the decrease in ejection fraction, and normalised left-ventricular systolic and diastolic internal diameters. Furthermore, the combination of indacaterol and metoprolol normalised the decreased β1-adrenoceptor messenger RNA expression as well as cardiac cAMP levels and reduced cardiac G protein-coupled receptor kinase 2 expression, catecholamine, ANP, BNP and collagen 1 messenger RNA values compared with the untreated heart failure group.
These findings fit very well with the hypothesis of Khan and colleagues [56], according to which synergistic β2-adrenoceptor stimulation and β1-adrenoceptor blockade together may be efficacious for the expansion of cardiac progenitor cells in the failing heart, potentially paving the way for a novel therapeutic approach to treat congestive heart failure. However, it has been suggested that reducing the sympathetic tone and up-regulation of β2-adrenoceptors in the lungs could be possible mechanisms by which β-blockers exhibit pulmonary beneficial effects in the long term in COPD [57]. Long-term use of β-blockers can up-regulate β2-adrenoceptors in the lungs and thus reduce the need for β2-agonists [58]. Figure 3 shows the pharmacological rationale for combining β-blockers and β2-agonists in patients with both COPD and heart failure.

Pharmacological rationale for combining β-adrenoreceptor (AR) blockers and β2-AR agonists in patients with chronic obstructive pulmonary disease (COPD) and concomitant heart failure. ASM airway smooth muscle, GPRK2 G protein-coupled receptor kinase 2
Whatever the case may be, a meta-analysis of 15 pooled cohort studies on the use of β-blockers in patients with COPD demonstrated that β-blockers produced a significant reduction in mortality and AECOPDs that was far greater than seen with any inhaled medicine [59]. This effect seemed to be greater in patients with pre-existing heart disease. However, a prospective national study of patients starting long-term oxygen therapy for COPD in Sweden suggested that β-blockers decrease survival in patients with severe oxygen-dependent COPD [60]. This finding has been challenged by the results of a prospective follow-up of the COPDGene cohort, a multicentre observational cohort of current and former smokers, which showed that β-blockers were associated with a significant reduction in AECOPDs also in subjects with severe COPD and on home oxygen [61]. The benefits of β-blockers in patients with COPD and coexistent CVD are less evident in those subjects that are older or who have more severe disease [62]. In any case, patients with COPD discharged from hospital with β-blockers after a myocardial infarction had a lower all-cause mortality compared with patients not prescribed β-blockers [63].
The question is still open as to which pharmacological properties of β-blockers make them effective in patients with concomitant COPD and CVD and indeed whether some of the benefit is the result of some of the β-blockers actually behaving as inverse agonists [53]. β-Blockers can be broadly classified into (1) nonselective, those producing a competitive blockade of both β1-adrenoceptors and β2-adrenoceptors and (2) those with much higher affinity for the β1-adrenoceptors than for the β2-adrenoceptors, usually called β1-adrenoceptor-selective (cardioselective) blockers. Although it has been suggested that there is no significant difference in AECOPDs based on β-blocker cardioselectivity [64], there is evidence that bisoprolol, a typical highly cardioselective β-blocker, reduced the incidence of chronic heart failure and/or AECOPDs compared with carvedilol, a typical non-selective β-adrenergic and α1-adrenoceptor blocker [65]. Apparently, intrinsic sympathomimetic activity (ISA) seems to be at least as important as β1-adrenoceptor selectivity in reducing the increase in airway resistance that results from β-blockade both at rest and during exertion [53]. Treatment with β-blockers that have ISA is associated with downregulation of β2-adrenoceptors. This finding is consistent with data showing that β-blockers with ISA did not produce the increase in β2-agonist response that was seen with β-blockers without ISA [53]. Because agents with ISA offer less cardioprotection than do β-blockers without this ancillary property, the use of these drugs should be severely restricted [53].
4.4 Anti-Platelet Drugs
Platelet activation is increased in patients with stable and acute exacerbation of COPD [66]. Actually, there is evidence that the mean platelet volume, as an index of platelet activation that is correlated with a higher risk of developing atherosclerotic thrombotic CV events, is higher in patients with COPD than in controls and even higher in patients with more severe COPD and during acute exacerbation [67]. Moreover, in elderly male patients with COPD, the mean platelet volume was negatively correlated with the level of FEV1 and left-ventricular ejection fraction [68]. It has been suggested that platelets may have an important inflammatory role in COPD, possibly related to hypoxia [69]. Platelets should no longer be considered just cell fragments that contribute to plugging of injured blood vessels, but as inflammatory cells in their own right that play a key role in the recruitment and activation of inflammatory cells in diseases such as COPD [70].
Nevertheless, the impact of antiplatelet drug administration in patients with COPD is still controversial. Ekström and colleagues [60] showed that antiplatelet drugs improve survival in patients with severe oxygen-dependent COPD. Furthermore, in patients admitted to hospital for AECOPD, antiplatelet therapy was associated with a reduced 1-year mortality [69]. A recent systematic review and meta-analysis suggested that antiplatelet therapy might significantly contribute to reduce all-cause mortality in patients with COPD, independently of the presence or not of ischaemic heart disease [71]. Regrettably, other studies came to the opposite conclusion [72, 73]. In particular, in patients with COPD taking dual antiplatelet therapy (aspirin plus clopidogrel), a lower drug responsiveness compared with patients without COPD was observed [74]. It has been speculated that the proinflammatory nature of COPD could be involved, influencing both platelet function and drug responsiveness.
5 Discussion
Currently, no COPD guideline recommends a core CV evaluation for patients with COPD and no CVD guideline recommends a core pulmonary evaluation for patients with CVD. This is the likely reason why there is little specific guideline guidance for treating COPD in patients with CVD, or vice versa. There is clearly a need for COPD and CVD to be considered in an integrated way [1].
The 2017 updated version of the Global Initiative for Chronic Obstructive Lung Disease Strategy recommends that treatment for any co-morbidity, including CVD, should follow that disease-specific guideline [10]. This is likely because treatment of CVD might reduce morbidity and mortality in patients with COPD and reduces hospitalisation from AECOPDs [4]. Unfortunately, to date, it is still unclear whether treatment of CVD alters the natural history of COPD or whether it has a beneficial effect in patients with COPD by treating the underlying disease or merely by reducing CV complications [9].
This is clearly an issue that needs resolving, as there is no direct evidence that this respiratory disease should be treated differently in the presence of CVD [75–77]. Despite this, regardless of any scientific speculation, we believe that, while waiting for studies specifically designed to understand which drugs are effective and safe in patients with coexisting COPD and CVD, a degree of caution in the choice of therapy is necessary, being aware that it is absolutely appropriate to consider the two diseases concomitantly.
We have already highlighted that β2-agonists may induce tachycardia or precipitate cardiac rhythm disorders in susceptible patients, without increasing the risk of CV events [16]. Therefore, concomitant CVD does not justify their avoidance. Nonetheless, because no distinction is made in the Global Initiative for Chronic Obstructive Lung Disease Strategy as to which class of bronchodilators should be considered first, but only recommends the use of long-acting bronchodilator agents [10], a patient requiring regular long-acting bronchodilators should be switched to a LAMA according to current CV safety data [28]. However, we recognise that there is a limitation in the existing LAMA CV safety database, and that there is a paucity of information on the safety of these drugs in high-risk patients, such as those with coronary artery disease, heart failure, cardiac arrhythmia, hypoxemia requiring daytime oxygen therapy and creatinine levels >2 mg/dL, because such patients were excluded from phase III clinical trials [78]. These patients may be at increased risk of drug-related cardiac events in a real-world setting. Therefore, adequately designed randomised trials pre-specified for CV outcomes to confirm CV safety of LAMAs are now mandatory.
In patients with COPD with concomitant chronic heart failure, concurrent administration of a LABA and a β-blocker seems an attractive option. However, the efficacy of concomitant β-blockers to offset the adverse CV effects of β2-agonists in patients suffering from COPD with coexistent heart failure and reduced ejection fraction has not yet been assessed in clinical trials [76]. The addition of an ICS does not seem to increase the risk of adverse CV events [38], while regarding the use of dual bronchodilators (LABA/LAMA FDCs) we must repeat the comments that we have made for LAMAs.
There is no specific contraindication to use ACE inhibitors, AT1 receptor blockers and statins in patients with COPD, although between 5 and 20% of subjects treated with ACE inhibitors develop a cough and, consequently, in these patients it is preferable to prescribe AT1 receptor blockers because they induce fewer cough events than ACE inhibitors [79]. Quite the reverse, the prescription of β-blockers has traditionally been contraindicated in COPD, mainly because of anecdotal evidence and case reports citing acute bronchospasm especially in patients with more severe COPD or increased airway hyperresponsiveness after their administration [80]. Actually, the use of β-blockers is significantly lower in patients with heart failure and COPD than in patients with heart failure alone, irrespective of the severity of COPD [81]. Nonetheless, the literature is reassuring [53, 80] and the use of β-blockers is recommended in international management guidelines for heart failure [82], coronary artery disease [83] and hypertension [84] and also for patients with concomitant COPD. Cardioselective β1-blockers are preferred to the nonselective β-blockers as they are less likely to produce bronchoconstriction in COPD [53].
Intriguingly, cardioselective β1-blockers had larger beneficial effects on mortality than nonselective ones but similar effects on the risk of AECOPDs [85]. Apparently, only β-blockers that are β2-adrenoceptor inverse agonists such as nadolol, which could also inactivate the spontaneously active β2-adrenoceptors, exert their beneficial effects on airway epithelial cells and immune cells by inhibiting constitutive proinflammatory signalling through non-canonical β-arrestin-2-mediated signalling, with a real potential to affect AECOPDs [80]. Therefore, it will be important to verify if the specific frequent exacerbator COPD phenotype is particularly sensitive to β2-adrenoceptor inverse agonists [80]. We are confident that a positive result of a randomised clinical trial with an inverse agonist on AECOPDs would cause a paradigm shift comparable to β-blocker use in patients with heart failure in the 1990s.
References
Cazzola M, Rogliani P, Matera MG. Cardiovascular disease in patients with COPD. Lancet Respir Med. 2015;3(8):593–5. doi:10.1016/S2213-2600(15)00279-9.
Cazzola M, Calzetta L, Bettoncelli G, et al. Cardiovascular disease in asthma and COPD: a population-based retrospective cross-sectional study. Respir Med. 2012;106(2):249–56. doi:10.1016/j.rmed.2011.07.021.
Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Respir Med. 2015;3(8):631–9. doi:10.1016/S2213-2600(15)00241-6.
Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1(1):73–83. doi:10.1016/S2213-2600(12)70060-7.
Thomsen M, Dahl M, Lange P, et al. Inflammatory biomarkers and comorbidities in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(10):982–8. doi:10.1164/rccm.201206-1113OC.
MacNee W. Systemic inflammatory biomarkers and co-morbidities of chronic obstructive pulmonary disease. Ann Med. 2013;45(3):291–300. doi:10.3109/07853890.2012.732703.
van Eeden S, Leipsic J, Man SP, Sin DD. The relationship between lung inflammation and cardiovascular disease. Am J Respir Crit Care Med. 2012;186(1):11–6. doi:10.1164/rccm.201203-0455PP.
Fabbri LM, Rabe KF. From COPD to chronic systemic inflammatory syndrome? Lancet. 2007;370(9589):797–9. doi:10.1016/S0140-6736(07)61383-X.
Matera MG, Calzetta L, Rinaldi B, Cazzola M. Treatment of COPD: moving beyond the lungs. Curr Opin Pharmacol. 2012;12(3):315–22. doi:10.1016/j.coph.2012.04.001.
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for diagnosis, management, and prevention of COPD: 2017 report. http://goldcopd.org/. Accessed 28 Dec 2016.
de Torres JP, Cordoba-Lanus E, Lopez-Aguilar C, et al. C-reactive protein levels and clinically important predictive outcomes in stable COPD patients. Eur Respir J. 2006;27(5):902–7. doi:10.1183/09031936.06.00109605.
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–54. doi:10.1056/NEJMra0804752.
Macie C, Wooldrage K, Manfreda J, Anthonisen N. Cardiovascular morbidity and the use of inhaled bronchodilators. Int J Chron Obstruct Pulmon Dis. 2008;3(1):163–9.
Cekici L, Valipour A, Kohansal R, Burghuber OC. Short-term effects of inhaled salbutamol on autonomic cardiovascular control in healthy subjects: a placebo-controlled study. Br J Clin Pharmacol. 2009;67(4):394–402. doi:10.1111/j.1365-2125.2009.03377.x.
Wilchesky M, Ernst P, Brophy JM, et al. Bronchodilator use and the risk of arrhythmia in COPD: part 2: reassessment in the larger Quebec cohort. Chest. 2012;142(2):305–11. doi:10.1378/chest.11-1597.
Cazzola M, Imperatore F, Salzillo A, et al. Cardiac effects of formoterol and salmeterol in patients suffering from COPD with preexisting cardiac arrhythmias and hypoxemia. Chest. 1998;114(2):411–5.
Au DH, Lemaitre RN, Curtis JR, et al. The risk of myocardial infarction associated with inhaled β-adrenoceptor agonists. Am J Respir Crit Care Med. 2000;161(3 Pt 1):827–30. doi:10.1164/ajrccm.161.3.9904006.
Au DH, Curtis JR, Every NR, et al. Association between inhaled beta-agonists and the risk of unstable angina and myocardial infarction. Chest. 2002;121(3):846–51.
Xia N, Wang H, Nie X. Inhaled long-acting β2-agonists do not increase fatal cardiovascular adverse events in COPD: a meta-analysis. PLoS One. 2015;10(9):e0137904. doi:10.1371/journal.pone.0137904.
Au DH, Udris EM, Curtis JR, et al. ACQUIP Investigators. Association between chronic heart failure and inhaled β-2-adrenoceptor agonists. Am Heart J. 2004;148(5):915–20. doi:10.1016/j.ahj.2004.03.048.
Bermingham M, O’Callaghan E, Dawkins I, et al. Are beta2-agonists responsible for increased mortality in heart failure? Eur J Heart Fail. 2011;13(8):885–91. doi:10.1093/eurjhf/hfr063.
Minasian AG, van den Elshout FJJ, Dekhuijzen PNR, et al. Using the lower limit of normal instead of the conventional cutoff values to define predictors of pulmonary function impairment in subjects with chronic heart failure. Respir Care. 2016;61(2):173–83. doi:10.4187/respcare.04101.
Minasian AG, van den Elshout FJ, Dekhuijzen PN, et al. Bronchodilator responsiveness in patients with chronic heart failure. Heart Lung. 2013;42(3):208–14. doi:10.1016/j.hrtlng.2012.11.007.
Rutten FH, Cramer MJ, Lammers JW, et al. Heart failure and chronic obstructive pulmonary disease: an ignored combination? Eur J Heart Fail. 2006;8(7):706–11. doi:10.1016/j.ejheart.2006.01.010.
Gariani K, Delabays A, Perneger TV, Agoritsas T. Use of brain natriuretic peptide to detect previously unknown left ventricular dysfunction in patients with acute exacerbation of chronic obstructive pulmonary disease. Swiss Med Wkly. 2011;141(13):13298–300. doi:10.4414/smw.2011.13298.
Segreti A, Fiori E, Calzetta L, et al. The effect of indacaterol during an acute exacerbation of COPD. Pulm Pharmacol Ther. 2013;26(6):630–4. doi:10.1016/j.pupt.2013.03.020.
Singh S, Loke YK, Furberg CD. Inhaled anticholinergics and risk of major adverse cardiovascular events in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. JAMA. 2008;300(12):1439–50. doi:10.1001/jama.300.12.1439.
Halpin DMG, Dahl R, Hallmann C, et al. Tiotropium HandiHaler® and Respimat® in COPD: a pooled safety analysis. Int J Chron Obstruct Pulmon Dis. 2015;10:239–59. doi:10.2147/COPD.S75146.
Dong YH, Lin HH, Shau WY, et al. Comparative safety of inhaled medications in patients with chronic obstructive pulmonary disease: systematic review and mixed treatment comparison meta-analysis of randomised controlled trials. Thorax. 2013;68(1):48–56. doi:10.1136/thoraxjnl-2012-201926.
Cazzola M, Calzetta L, Rogliani P, Matera MG. Tiotropium formulations and safety: a network meta-analysis. Ther Adv Drug Saf. 2017;8(1):17–30. doi:10.1177/2042098616667304.
Wise RA, Anzueto A, Cotton D, et al. Tiotropium Respimat inhaler and the risk of death in COPD. N Engl J Med. 2013;369(16):1491–501. doi:10.1056/NEJMoa1303342.
Lee CH, Choi S, Jang EJ, et al. Inhaled bronchodilators and the risk of tachyarrhythmias. Int J Cardiol. 2015;190:133–9. doi:10.1016/j.ijcard.2015.04.129.
Calzetta L, Rogliani P, Matera MG, Cazzola M. A systematic review with meta-analysis of dual bronchodilation with LAMA/LABA for the treatment of stable COPD. Chest. 2016;149(5):1181–96. doi:10.1016/j.chest.2016.02.646.
Sin DD, Van Eeden SF, Man SF. Slaying the CVD dragon with steroids. Eur Respir J. 2010;36(3):466–8. doi:10.1183/09031936.00082510.
Huiart L, Ernst P, Ranouil X, Suissa S. Low-dose inhaled corticosteroids and the risk of acute myocardial infarction in COPD. Eur Respir J. 2005;25(4):634–9. doi:10.1183/09031936.05.00079004.
Lofdahl CG, Postma DS, Pride NB, et al. Possible protection by inhaled budesonide against ischaemic cardiac events in mild COPD. Eur Respir J. 2007;29(6):1115–9. doi:10.1183/09031936.00128806.
Calverley PM, Anderson JA, Celli B, et al. Cardiovascular events in patients with COPD: TORCH study results. Thorax. 2010;65(8):719–25. doi:10.1136/thx.2010.136077.
Vestbo J, Anderson JA, Brook RD, et al. Fluticasone furoate and vilanterol and survival in chronic obstructive pulmonary disease with heightened cardiovascular risk (SUMMIT): a double-blind randomised controlled trial. Lancet. 2016;387(10030):1817–26. doi:10.1016/S0140-6736(16)30069-1.
Mancini GB, Etminan M, Zhang B, et al. Reduction of morbidity and mortality by statins, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers in patients with chronic obstructive pulmonary disease. J Am Coll Cardiol. 2006;47(12):2554–60. doi:10.1016/j.jacc.2006.04.039.
Podowski M, Calvi C, Metzger S, et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest. 2012;122(1):229–40. doi:10.1172/JCI46215.
Petersen H, Sood A, Meek PM, et al. Rapid lung function decline in smokers is a risk factor for COPD and is attenuated by angiotensin-converting enzyme inhibitor use. Chest. 2014;145(4):695–703. doi:10.1378/chest.13-0799.
Mortensen EM, Copeland LA, Pugh MJ, et al. Impact of statins and ACE inhibitors on mortality after COPD exacerbations. Respir Res. 2009;10:45. doi:10.1186/1465-9921-10-45.
Kim J, Lee JK, Heo EY, et al. The association of renin-angiotensin system blockades and pneumonia requiring admission in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2159–66. doi:10.2147/COPD.S104097.
Curtis KJ, Meyrick VM, Mehta B, et al. Angiotensin-converting enzyme inhibition as an adjunct to pulmonary rehabilitation in COPD. Am J Respir Crit Care Med. 2016;194(11):1349–57. doi:10.1164/rccm.201601-0094OC.
Shrikrishna D, Tanner RJ, Lee JY, et al. A randomized controlled trial of angiotensin-converting enzyme inhibition for skeletal muscle dysfunction in COPD. Chest. 2014;146(4):932–40. doi:10.1378/chest.13-2483.
Lee T-M, Lin M-S, Chang N-C. Usefulness of C-reactive protein and interleukin-6 as predictors of outcomes in patients with chronic obstructive pulmonary disease receiving pravastatin. Am J Cardiol. 2008;101(4):530–5. doi:10.1016/j.amjcard.2007.09.102.
Cazzola M, Calzetta L, Page CP, et al. Protein prenylation contributes to the effects of LPS on EFS-induced responses in human isolated bronchi. Am J Respir Cell Mol Biol. 2011;45(4):704–10. doi:10.1165/rcmb.2010-0306OC.
Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med. 2014;370(23):2201–10. doi:10.1056/NEJMoa1403086.
Citgez E, van der Palen J, Koehorst-Ter Huurne K, et al. Statins and morbidity and mortality in COPD in the COMIC study: a prospective COPD cohort study. BMJ Open Respir Res. 2016;3(1):e000142. doi:10.1136/bmjresp-2016-000142.
Rossi A, Inciardi RM, Rossi A, et al. Prognostic effects of rosuvastatin in patients with co-existing chronic obstructive pulmonary disease and chronic heart failure: a sub-analysis of GISSI-HF trial. Pulm Pharmacol Ther. 2017. doi:10.1016/j.pupt.2017.03.001 (Epub ahead of print).
Neukamm A, Hoiseth AD, Einvik G, et al. Rosuvastatin treatment in stable chronic obstructive pulmonary disease (RODEO): a randomized controlled trial. J Intern Med. 2015;278(1):59–67. doi:10.1111/joim.12337.
Hawkins NM, Wang D, Petrie MC, et al. Baseline characteristics and outcomes of patients with heart failure receiving bronchodilators in the CHARM programme. Eur J Heart Fail. 2010;12(6):557–65. doi:10.1093/eurjhf/hfq040.
Matera MG, Martuscelli E, Cazzola M. Pharmacological modulation of β-adrenoceptor function in patients with coexisting chronic obstructive pulmonary disease and chronic heart failure. Pulm Pharmacol Ther. 2010;23(1):1–8. doi:10.1016/j.pupt.2009.10.001.
Rinaldi B, Capuano A, Gritti G, et al. Effects of chronic administration of β-blockers on airway responsiveness in a murine model of heart failure. Pulm Pharmacol Ther. 2014;28(2):109–13. doi:10.1016/j.pupt.2014.04.005.
Rinaldi B, Donniacuo M, Sodano L, et al. Effects of chronic treatment with the new ultra-long-acting β2-adrenoceptor agonist indacaterol alone or in combination with the β1-adrenoceptor blocker metoprolol on cardiac remodelling. Br J Pharmacol. 2015;172(14):3627–37. doi:10.1111/bph.13148.
Khan M, Mohsin S, Avitabile D, et al. β-Adrenergic regulation of cardiac progenitor cell death versus survival and proliferation. Circ Res. 2013;112(3):476–86. doi:10.1161/CIRCRESAHA.112.280735.
Rutten FH, Hoes AW. Chronic obstructive pulmonary disease: a slowly progressive cardiovascular disease masked by its pulmonary effects? Eur J Heart Fail. 2012;14(4):348–50. doi:10.1093/eurjhf/hfs022.
Lin R, Peng H, Nguyen LP, et al. Changes in β2-adrenoceptor and other signaling proteins produced by chronic administration of ‘β-blockers’ in a murine asthma model. Pulm Pharmacol Ther. 2008;21(1):115–24. doi:10.1016/j.pupt.2007.06.003.
Du Q, Sun Y, Ding N, et al. Beta-blockers reduced the risk of mortality and exacerbation in patients with COPD: a meta-analysis of observational studies. PLoS One. 2014;9(11):e113048. doi:10.1371/journal.pone.0113048.
Ekström MP, Hermansson AB, Strom KE. Effects of cardiovascular drugs on mortality in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(7):715–20. doi:10.1164/rccm.201208-1565OC.
Bhatt SP, Wells JM, Kinney GL, et al. β-Blockers are associated with a reduction in COPD exacerbations. Thorax. 2016;71(1):8–14. doi:10.1136/thoraxjnl-2015-207251.
Puente-Maestu L, Álvarez-Sala LA, de Miguel-Díez J. β-blockers in patients with chronic obstructive disease and coexistent cardiac illnesses. COPD Res Pract. 2015;1:11. doi:10.1186/s40749-015-0013-y.
Andell P, Erlinge D, Smith JG, et al. β-Blocker use and mortality in COPD patients after myocardial infarction: a Swedish nationwide observational study. J Am Heart Assoc. 2015;4(4):e001611. doi:10.1161/JAHA.114.001611.
Farland MZ, Peters CJ, Williams JD, et al. β-Blocker use and incidence of chronic obstructive pulmonary disease exacerbations. Ann Pharmacother. 2013;47(5):651–6. doi:10.1345/aph.1R600.
Kubota Y, Asai K, Furuse E, et al. Impact of β-blocker selectivity on long-term outcomes in congestive heart failure patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2015;10:515–23. doi:10.2147/COPD.S79942.
Maclay JD, McAllister DA, Johnston S, et al. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax. 2011;66(9):769–74. doi:10.1136/thx.2010.157529.
Malerba M, Olivini A, Radaeli A, et al. Platelet activation and cardiovascular comorbidities in patients with chronic obstructive pulmonary disease. Curr Med Res Opin. 2016;32(5):885–91. doi:10.1185/03007995.2016.1149054.
Cui H, Liu L, Wei Z, et al. Clinical value of mean platelet volume for impaired cardiopulmonary function in very old male patients with chronic obstructive pulmonary disease. Arch Gerontol Geriatr. 2012;54(2):e109–12. doi:10.1016/j.archger.2011.09.013.
Harrison MT, Short P, Williamson PA, et al. Thrombocytosis is associated with increased short and long term mortality after exacerbation of chronic obstructive pulmonary disease: a role for antiplatelet therapy? Thorax. 2014;69(7):609–15. doi:10.1136/thoraxjnl-2013-203996.
Idzko M, Pitchford S, Page C. Role of platelets in allergic airway inflammation. J Allergy Clin Immunol. 2015;135(6):1416–23. doi:10.1016/j.jaci.2015.04.028.
Pavasini R, Biscaglia S, d’Ascenzo F, et al. Antiplatelet treatment reduces all-cause mortality in COPD patients: a systematic review and meta-analysis. COPD. 2016;13(4):509–14. doi:10.3109/15412555.2015.1099620.
Søyseth V, Brekke PH, Smith P, Omland T. Statin use is associated with reduced mortality in COPD. Eur Respir J. 2007;29(2):279–83. doi:10.1183/09031936.00106406.
Sheng X, Murphy MJ, MacDonald TM, et al. Effect of statins on total cholesterol concentrations, cardiovascular morbidity, and all-cause mortality in chronic obstructive pulmonary disease: a population based cohort study. Clin Ther. 2012;34(2):374–84. doi:10.1016/j.clinthera.2011.12.014.
Campo G, Pavasini R, Pollina A, et al. On-treatment platelet reactivity in patients with chronic obstructive pulmonary disease undergoing percutaneous coronary intervention. Thorax. 2014;69(1):80–1. doi:10.1136/thoraxjnl-2013-203608.
Roversi S, Fabbri LM, Sin DD, et al. Chronic obstructive pulmonary disease and cardiac diseases: an urgent need for integrated care. Am J Respir Crit Care Med. 2016;194(11):1319–36. doi:10.1164/rccm.201604-0690SO.
Jaiswal A, Chichra A, Nguyen VQ, et al. Challenges in the management of patients with chronic obstructive pulmonary disease and heart failure with reduced ejection fraction. Curr Heart Fail Rep. 2016;13(1):30–6. doi:10.1007/s11897-016-0278-8.
Campo G, Pavasini R, Malagu M, et al. Chronic obstructive pulmonary disease and ischemic heart disease comorbidity: overview of mechanisms and clinical management. Cardiovasc Drugs Ther. 2015;29(2):147–57. doi:10.1007/s10557-014-6569-y.
Cazzola M, Calzetta L, Matera MG. The cardiovascular risk of tiotropium: is it real? Expert Opin Drug Saf. 2010;9(5):783–92. doi:10.1517/14740338.2010.500611.
Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs. 2012;12(4):263–77. doi:10.2165/11599990-000000000-00000.
Matera MG, Calzetta L, Cazzola M. β-Adrenoceptor modulation in chronic obstructive pulmonary disease: present and future perspectives. Drugs. 2013;73(15):1653–63. doi:10.1007/s40265-013-0120-5.
Lipworth B, Skinner D, Devereux G, et al. Underuse of β-blockers in heart failure and chronic obstructive pulmonary disease. Heart. 2016;102(23):1909–14. doi:10.1136/heartjnl-2016-309458.
McMurray JJ, Adamopoulos S, Anker SD, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: 2012. The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14(8):803–69. doi:10.1093/eurjhf/hfs105.
Smith SC Jr, Allen J, Blair SN, et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update: endorsed by the National Heart, Lung, and Blood Institute. Circulation. 2006;113(19):2363–72. doi:10.1161/CIRCULATIONAHA.106.174516.
Mancia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC practice guidelines for the management of arterial hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens. 2007;25(9):1751–62. doi:10.1097/HJH.0b013e3282f0580f.
Rutten FH, Zuithoff NP, Hak E, et al. β-Blockers may reduce mortality and risk of exacerbations in patients with chronic obstructive pulmonary disease. Arch Intern Med. 2010;170(10):880–7. doi:10.1001/archinternmed.2010.112.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflict of interest
Mario Cazzola, Luigino Calzetta, Barbara Rinaldi, Clive Page, Giuseppe Rosano, Paola Rogliani and Maria Gabriella Matera report no relationships that could be construed as a conflict of interest with regard to the subject matter of this review.
Rights and permissions
About this article

Cite this article
Cazzola, M., Calzetta, L., Rinaldi, B. et al. Management of Chronic Obstructive Pulmonary Disease in Patients with Cardiovascular Diseases. Drugs 77, 721–732 (2017). https://doi.org/10.1007/s40265-017-0731-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0731-3