
Abstract
Trifluridine/tipiracil (Lonsurf®) is a novel, orally active, antimetabolite agent comprised of trifluridine, a thymidine-based nucleoside analogue, and tipiracil, a potent thymidine phosphorylase inhibitor. Trifluridine is incorporated into DNA via phosphorylation, ultimately inhibiting cell proliferation. Tipiracil increases systemic exposure of trifluridine when coadministered. Trifluridine/tipiracil has recently been approved for the treatment of adult patients with metastatic colorectal cancer (mCRC) who are refractory to or are not considered candidates for, current standard chemotherapy and biological therapy in the EU and USA and in unresectable advanced or recurrent CRC in Japan. The approved regimen of oral twice-daily trifluridine/tipiracil (35 mg/m2 twice daily on days 1–5 and 8–12 of each 28-day cycle) significantly improved overall survival and progression-free survival and was associated with a significantly higher disease control rate than placebo when added to best supportive care in the multinational, pivotal phase III trial (RECOURSE) and a phase II Japanese trial. Trifluridine/tipiracil was associated with an acceptable tolerability profile, with adverse events generally being managed with dose reductions, temporary interruptions in treatment or administration of granulocyte-colony stimulating factor. The most common grade 3–4 adverse events (≥10 %) were anaemia, neutropenia, thrombocytopenia and leukopenia. In conclusion, trifluridine/tipiracil is a useful additional treatment option for the management of mCRC in patients who are refractory to, or are not considered candidates for, currently available therapies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
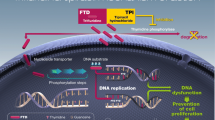
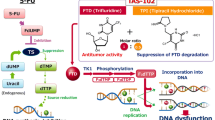
Tipiracil inhibits thymidine phosphorylase, enabling sufficient plasma concentrations of the cytotoxic trifluridine to be achieved |
Trifluridine is incorporated into DNA via phosphorylation and inhibits cell proliferation |
Significantly prolongs median OS and PFS compared with placebo in patients refractory to available therapies |
Myelosuppression is the most commonly reported treatment-emergent Grade 3–4 adverse event |
Adverse events are generally manageable with dose modification and/or supportive measures |
1 Introduction
Colorectal cancer (CRC) is the third most frequently diagnosed malignancy and the fourth most common cause of cancer-related death worldwide [1]. Approximately, 15–25 % of newly diagnosed patients present with metastatic CRC (mCRC) and ≈50–60 % of patients with CRC will ultimately develop metastases [2]. For many patients with mCRC, potentially curative surgery is not an option [1–4]. Therefore, treatment is palliative, and the goal is to prolong overall survival (OS) and maintain a good quality of life [1–4]. Chemotherapy agents (e.g. fluoropyrimidines, oxaliplatin and irinotecan), anti-vascular endothelial growth factor (VEGF) monoclonal antibodies (e.g. bevacizumab) and anti-epidermal growth factor receptor (EGFR) monoclonal antibodies (e.g. cetuximab, panitumumab) are considered standard therapies for the treatment of unresectable mCRC [3, 4].
Few effective options are available for patients with disease progression in the later lines of treatment [3]. Until recently, treatment options for refractory patients were limited to best supportive care (BSC), entering a clinical trial or the multikinase inhibitor regorafenib [3–5]. Trifluridine, a fluoropyrimidine initially synthesised over 50 years ago, had shown antitumour activity in an early clinical trial in breast and colon cancer, but the pharmacokinetic and toxicity profile was not considered feasible for long-term administration, and further drug development was halted [6]. More recently, combining trifluridine with tipiracil, a potent thymidine phosphorylase inhibitor, has been hypothesised to increase the bioavailability of trifluridine without increasing the toxicity [6]. This led to the development of a novel orally administered single, fixed-dose tablet combining trifluridine and tipiracil (Lonsurf®) [6–9], hereafter referred to as trifluridine/tipiracil.
Trifluridine/tipiracil is approved in the USA [7] and EU [8] for the treatment of mCRC in patients who have been previously treated with [7, 8], or are not considered candidates for [8], currently available therapies, including fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, anti-VEGF biological agents and anti-EGFR agents (if RAS wild-type) [7, 8]. In Japan, trifluridine/tipiracil is approved for the treatment of unresectable advanced or recurrent CRC [9]. This article reviews the pharmacological properties, clinical efficacy and tolerability of trifluridine/tipiracil in patients with mCRC who are refractory to or are not considered candidates for currently available therapies.
2 Pharmacodynamic Properties of Trifluridine/Tipiracil
Trifluridine/tipiracil is an orally administered combination of an antineoplastic, thymidine-based nucleoside analogue (trifluridine) and a thymidine phosphorylase inhibitor (tipiracil) [7, 8, 10]. Trifluridine is the active cytotoxic component, and tipiracil improves the bioavailability of trifluridine (Sect. 3.2) [11]. The optimal molar ratio was determined to be 1 M of trifluridine and 0.5 M of tipiracil [6].
In addition to its inhibition of thymidylate synthase (a rate-limiting enzyme of DNA synthesis), trifluridine/tipiracil antitumour activity is thought to be predominantly attributable to trifluridine incorporation into DNA; this provides an important additional mechanism of action and a point-of-difference to that of traditional fluoropyrimidines [12–15].
As seen with 5-fluorouracil (5-FU), trifluridine is rapidly phosphorylated by thymidine kinase to its active monophosphate form. However, unlike 5-FU, trifluorothymidine monophosphate reversibly binds to the active site of thymidylate synthase, with this short-term inhibition not considered sufficient enough to fully account for the cytotoxicity seen with trifluridine [13, 16]. Subsequent phosphorylation results in the production of trifluridine triphosphate, which is readily incorporated into the DNA of tumour cells (in the place of thymidine bases), interfering with DNA function and inhibiting tumour growth [12, 13, 15, 17]. Additionally, the amount of trifluridine incorporated into DNA was correlated with the antitumour activity of trifluridine/tipiracil [15], suggesting that this is its predominant route of cytotoxicity, which is distinct to that of 5-FU (primarily via thymidylate synthase inhibition). Importantly, incorporation into DNA via this route occurs with considerably greater efficacy with trifluridine than that seen with 5-FU [16].
In a preclinical study in human colorectal cancer lines, trifluridine/tipiracil induced cytotoxicity regardless of DNA mismatch repair (MMR) status, or 5-FU sensitivity/refractoriness. Conversely, sensitivity to 5-FU was reduced in a cell line that was MMR deficient [18].
The administration of trifluridine/tipiracil dose-dependently inhibited tumour growth and improved survival in human colorectal cancer xenograft models, irrespective of sensitivity or resistance to 5-FU and other fluoropyrimidines [6, 13–15, 19].
Trifluridine exposure is correlated with efficacy, and haematological toxicity [8, 10, 15, 20, 21]. Systemic exposure of trifluridine significantly correlated with trifluridine incorporation into tumour and white blood cell DNA, and with trifluridine/tipiracil antitumour activity and haematological toxicity in a colorectal tumour xenograft mouse model [15]. In mCRC patients who were administered trifluridine/tipiracil in a phase I trial, a reduction in neutrophil count was significantly (p < 0.001) associated with trifluridine systemic exposure [20]. In an expanded access program cohort study in patients with refractory mCRC (n = 149), trifluridine/tipiracil recipients who developed grade ≥2 neutropenia at 1 month had a better prognosis [significantly longer progression-free survival (PFS) and OS (p = 0.01 and p < 0.0001, respectively)] than those who did not [22].
A post hoc analysis of the phase III study in patients with mCRC (RECOURSE) [23] discussed in Sect. 4.2 suggests that occurrence of grade 3–4 neutropenia with trifluridine/tipiracil is correlated with an improved OS benefit [21]. In addition, in a subpopulation of patients from this trial for whom trifluridine/tipiracil AUC data were available, OS appeared more favourable in those with high trifluridine exposure (median OS of 9.3 vs. 8.1 months in low-exposure patients), while a higher frequency of grade ≥3 neutropenia was observed (47.8 vs. 30.4 %) [8, 10].
Trifluridine/tipiracil was not associated with clinically relevant effects on the QTc interval in a dedicated QTc study in patients with solid tumours [24].
3 Pharmacokinetic Properties
The oral bioavailability of trifluridine is low due to the first-pass effect by thymidine phosphorylase, which is highly expressed in the liver and gastrointestinal (GI) tract; therefore, monotherapy with trifluridine is not feasible [6]. Co-administration of trifluridine with tipiracil resulted in 38- and 22-fold increases in trifluridine AUC from time zero to the last measurable concentration (AUClast) and peak plasma concentration (Cmax), respectively [11]. This section provides a brief overview of the pharmacokinetics of oral trifluridine/tipiracil. All data have been obtained from studies in patients with cancer. Unless stated otherwise, a single dose of trifluridine/tipiracil was 35 mg/m2 and multiple-dose administration was 35 mg/m2 given twice daily on days 1–5 and 8–12 of each 28-day treatment cycle [20].
3.1 Absorption and Distribution
The pharmacokinetics of oral trifluridine and tipiracil were best described by one- and two-compartment models with transit and first-order absorptions, respectively [10, 25]. After twice-daily administration of trifluridine/tipiracil, systemic exposure of trifluridine increased more than dose proportionally over the dose range of 15–35 mg/m2, although oral clearance and apparent volume of distribution of trifluridine were generally constant over the dose range of 20–35 mg/m2 [7, 8]. Trifluridine is well absorbed (>57 % to almost complete absorption of administered dose) [10]. In contrast, tipiracil demonstrates moderate GI absorption (>27 but <50 % of administered dose) [10].
After multiple dose administration of trifluridine/tipiracil, the accumulation of trifluridine was ≈2-fold higher for Cmax and ≈3-fold higher for AUClast at steady-state (day 12 cycle 1) than after a single dose (day 1 cycle 1) [7, 8, 20]. There was no evidence of further accumulation of trifluridine during subsequent treatment cycles. No accumulation was observed for tipiracil [7, 8, 20].
A standardized high-fat, high-calorie meal decreased the Cmax of trifluridine and the Cmax and AUC of tipiracil by ≈40 %, but did not change the AUC of trifluridine, compared with the fasting state after administration of a single dose of trifluridine/tipiracil [26]. Since food consumption did not affect the exposure of trifluridine, it is unlikely to affect efficacy and safety of the drug; however, as the Cmax of trifluridine is correlated with decreased neutrophil counts (Sect. 2) [20], postprandial administration of trifluridine/tipiracil is recommended (Sect. 6) [7, 8].
Trifluridine and tipiracil are preferentially distributed to plasma rather than blood cells in humans [8, 10]. The in vitro protein binding of trifluridine in human plasma is high (96 %) and is not dependent on drug concentration over the range of 0.5–50 μg/mL or the presence of tipiracil. Trifluridine is mostly bound to human serum albumin. In contrast, over a concentration range 0.05–5 μg/mL, the plasma protein binding of tipiracil is low (<8 %) [8, 10].
3.2 Metabolism and Elimination
Trifluridine is extensively metabolised by thymidine phosphorylase into 5-(trifluoromethyl)uracil (major metabolite) which is pharmacologically inactive [7, 8]. In a mass balance study during which patients received an oral solution of 60 mg [14C]-trifluridine/[14C]-tipiracil, the major circulating moieties were trifluridine (53 %) and 5-(trifluoromethyl) uracil (33 %) [27]. Minor metabolites of trifluridine were detected at low or trace levels in plasma and urine (5-carboxyuracil and 5-carboxy-2′-deoxyuridine) [8, 10]. Neither trifluridine or tipiracil are metabolised by cytochrome P450 (CYP) enzymes in vitro [7, 8].
Approximately 60 % of the radioactivity was recovered over a 7-day period after oral administration of [14C]-trifluridine/[14C]-tipiracil, consisting of 55 % urinary excretion [mostly as 5-(trifluoromethyl)uracil (25 %) and trifluridine glucuronide isomers (18 %)], 3 % faecal excretion and 2 % expired CO2 [27]. The mean elimination half-life (t1/2) of trifluridine was 2.1 h on day 12 of cycle 1 (multiple doses) [8, 10]. After a single dose of trifluridine/tipiracil, the oral clearance for trifluridine was 10.5 L/h [10].
Tipiracil does not undergo significant first-pass metabolism in the liver and is mainly excreted in the urine in an unchanged form [7, 8, 10]. Approximately 77 % of the dose of [14C]-tipiracil was recovered over a 7-day period, with faecal excretion representing 50 % of total excretion and renal excretion accounted for 27 % [27]. The t1/2 for tipiracil was 2.4 h on day 12 of cycle 1 (multiple doses) [8, 10]. The oral clearance for tipiracil was 109 L/h following a single trifluridine/tipiracil dose [8, 10].
Renal impairment may increase exposure to trifluridine and tipiracil by reducing clearance of the drugs [10]. Consequently, patients with moderate renal impairment (CLCR 30–59 mL/min) should be frequently monitored and may require dose modifications for increased toxicity [7, 8, 10]. The pharmacokinetics of trifluridine/tipiracil have not been assessed in patients with moderate or severe hepatic impairment, or in patients with severe renal impairment or end-stage renal disease; therefore, trifluridine/tipiracil is not recommended in these patient populations [7, 8, 25]. Gender, age, ethnicity and mild renal or hepatic impairment have no clinically meaningful effects on the exposure to trifluridine/tipiracil [7, 8, 25]. No formal drug–drug interaction studies have been conducted in patients with mCRC receiving trifluridine/tipiracil [7, 8].
4 Therapeutic Efficacy of Trifluridine/Tipiracil
Key data showing the efficacy of oral trifluridine/tipiracil for the treatment of mCRC in patients who were refractory or intolerant to standard therapies are available from the supportive double-blind, placebo-controlled, multicenter, phase II trial in Japanese patients (n = 169) [28] and the pivotal randomized, double-blind, placebo-controlled, multinational phase III study (RECOURSE; n = 800) [23]. Some data are from abstracts/posters [29–33].
Eligible patients received twice-daily oral trifluridine/tipiracil or placebo in addition to BSC (see Table 1 for dosage details) [23, 28]. Treatment was continued until progressive disease, intolerable toxicity or withdrawal of consent; crossover was not allowed following disease progression [23, 28]. The primary endpoint was OS (defined as the time between randomization and death from any cause [23, 28] or the date of the last follow-up [28]) in the ITT population [23, 28].
4.1 Phase II Trial in Japanese Patients
The phase II trial evaluated the efficacy of oral trifluridine/tipiracil in Japanese patients aged ≥20 years with a confirmed diagnosis of unresectable mCRC, a treatment history of ≥2 regimens of standard chemotherapy and who were refractory to or intolerant of fluoropyrimidine, irinotecan and oxaliplatin [28]. Baseline characteristics were generally well matched, except twice as many patients in the trifluridine/tipiracil group had received adjuvant chemotherapy than patients in the placebo group. Overall, ≈52 % of patients had KRAS wild-type tumours. Of these patients 91 and 96 % in the trifluridine/tipiracil and placebo groups had received an anti-EGFR therapy. The cut-off date for the primary analysis was 4 February 2011 (123 deaths had occurred at this time), and the median follow-up was 11.3 months. The trifluridine/tipiracil dose intensity after the first dose was 147 mg/m2 per week and the relative dose intensity was 85.7 % [28]. The final cut-off date for the updated survival analysis was 19 January 2015 (167 deaths had occurred), and the median follow-up was 57.5 months [33].
In previously-treated Japanese patients with mCRC, the addition of trifluridine/tipiracil to BSC significantly prolonged median OS (primary endpoint) compared with placebo plus BSC (Table 1) [28]. Furthermore, trifluridine/tipiracil significantly improved median PFS as assessed by the independent review committee (Table 1) and by the investigators (hazard ratio [HR] 0.35; 95 % CI 0.25–0.50; p < 0.0001) relative to placebo [28].
The updated OS analysis at the final data cut-off date confirmed the statistically significant improvement in survival in patients receiving trifluridine/tipiracil compared with placebo [HR 0.63 (95 % CI 0.45–0.87); p = 0.0066]; the median OS was 9.0 months in the trifluridine/tipiracil arm compared with 6.6 months in the placebo arm [33].
Among patients evaluable for clinical response, one patient (1 %) in the trifluridine/tipiracil group achieved a partial response (for >225 days; continued response) and 48 (43 %) had stable disease [28]. Six patients (11 %) had stable disease in the placebo group. No cases of objective response were observed in the placebo group [28]. The proportion of patients who achieved disease control was significantly higher with trifluridine/tipiracil than with placebo (Table 1) [28]. Furthermore, trifluridine/tipiracil was associated with a significantly longer median time to treatment failure, assessed by the independent review committee, than placebo (Table 1) [28].
4.2 Phase III RECOURSE Trial
The RECOURSE trial evaluated the clinical efficacy of oral trifluridine/tipiracil in adults (aged ≥18 years) with mCRC who were refractory to, or are not considered candidates for, current standard therapies [23]. Eligible patients had histologically proven mCRC which had been previously treated with ≥2 chemotherapy regimens containing a fluoropyrimidine, oxaliplatin, irinotecan and bevacizumab, and an anti-EGFR therapy (cetuximab or panitumumab) in patients with KRAS wild-type tumours. Other eligibility criteria included an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and adequate renal, hepatic and haematopioetic function [23].
Patients were stratified according to KRAS status, time since diagnosis of first metastasis and geographical region (Japan vs. USA, EU and Australia), and then randomized to twice-daily oral trifluridine/tipiracil or placebo in addition to BSC (Table 1) [23]. Baseline characteristics were well matched between the two treatment arms [23]. The median patient age was 63 years, and the main primary site of disease was the colon (62 % of patients). All patients had received earlier treatment regimens containing a fluoropyrimidine, oxaliplatin, and irinotecan; all but one patient (in the placebo group) had received bevacizumab. The median number of prior lines of therapy for metastatic disease was three [10]. Approximately half of the study population had KRAS wild-type mCRC (49 %), and all except two of these patients (one in each treatment arm) had received prior panitumumab or cetuximab therapy. At baseline, 17 and 20 % of patients in the trifluridine/tipiracil and placebo groups had received regorafenib. Almost all patients (>90 %) had disease that was refractory to a fluoropyrimidine at the time of last exposure, and >50 % had disease refractory to a fluoropyrimidine administered as part of the most recent regimen prior to study entry. Overall, 67 % of patients were from the US, Europe and Australia, and 33 % were from Japan [23].
The clinical data cut-off for the primary analysis of the study was 24 January 2014 [after 574 (72 %) deaths], with a median follow-up of 11.8 months [23]. The cut-off date for the final survival data was 8 October 2014 [after 710 deaths (89 %) deaths] [30]. The duration of treatment during the double-blind period ranged from 0.1 to 78.0 (mean 12.7) weeks in the trifluridine/tipiracil arm and 0.1 to 63.7 (mean 6.8) weeks in the placebo arm [23]. Patients in the trifluridine/tipiracil and placebo groups received an average of 89 and 94 % of the planned dose, respectively, and the mean dose intensity was 155.1 and 165.3 mg/m2 per week [23].
In heavily-pretreated patients with mCRC who were refractory to or intolerant of standard chemotherapy, trifluridine/tipiracil plus BSC was associated with a clinically and statistically significant improvement in OS (primary endpoint) compared with placebo plus BSC (Table 1) [23]. This corresponded to a 32 % reduction in the risk of death. At 6 months, 58 and 44 % of trifluridine/tipiracil and placebo recipients, respectively, were alive. The corresponding survival rates at 12 months were 27 and 18 %. Trifluridine/tipiracil also significantly prolonged PFS and the median time to treatment failure compared with placebo at the primary cut-off date (Table 1) [23].
The updated OS analysis at the final data cut-off date confirmed the clinically and statistically significant improvement in survival in patients receiving trifluridine/tipiracil compared with placebo [HR 0.69 (95 % CI 0.59–0.81); p < 0.0001] [30]. The median OS was 7.2 months in the trifluridine/tipiracil arm compared with 5.2 months in the placebo arm; corresponding 12-month survival rates were 27.1 % (95 % CI 23.3–30.9) and 16.6 % (95 % CI 12.4–21.4). The clinical benefit of trifluridine/tipiracil was consistently maintained regardless of survival prognosis at randomization. Trifluridine/tipiracil also benefited patients’ functionality, since the median time to worsening in ECOG performance status (from 0–1 to ≥2) was significantly longer in the trifluridine/tipiracil arm compared with the placebo arm [6.2 vs. 4.4 months; HR 0.74 (95 % CI 0.64–0.87); p < 0.001] [30].
The significant OS and PFS benefits observed with trifluridine/tipiracil were generally consistent across the pre-specified subgroups, including KRAS status, time since diagnosis of first metastasis, number of metastatic sites, disease refractory to fluoropyrimidines (received as part of their last regimen), and prior treatment with regorafenib [23, 29, 31, 32].
In a multivariate Cox regression analysis, time since diagnosis of first metastasis, ECOG performance status and the number of metastatic sites were identified as prognostic factors; however, the trifluridine/tipiracil treatment benefits were still evident after adjusting for these three factors (HR 0.69; 95 % CI 0.58–0.81) [23]. No predictive factors were identified among the demographic and baseline characteristics.
Among patients in the trifluridine/tipiracil (n = 502) and placebo (n = 258) groups who were evaluable for tumour response, objective response rates were 1.6 % (eight patients had a partial response) and 0.4 % (one had a complete response), respectively [23]. Stable disease was seen in 42 % of trifluridine/tipiracil and 16 % of placebo recipients [10]. The median duration of response was 7.4 months in trifluridine/tipiracil recipients [10]. Treatment with trifluridine/tipiracil was also associated with a significant improvement in the disease control rate compared with placebo (Table 1) [23].
Trifluridine/tipiracil significantly delayed the worsening of ECOG performance status from baseline of 0–1 to ≥2 compared with placebo [5.7 vs. 4.0 months; HR 0.66 (95 % CI 0.56–0.78); p < 0.001] [23].
5 Tolerability of Trifluridine/Tipiracil
Trifluridine/tipiracil was associated with an acceptable tolerability profile during the clinical trials in patients with mCRC, with adverse events generally being manageable (by dose reductions, temporary interruptions or supportive care) [11, 20, 23, 28, 34–36]. In the RECOURSE trial, treatment-emergent adverse events (TEAEs) of any grade were reported in 98 % of trifluridine/tipiracil recipients and 93 % of placebo recipients [23] and serious TEAEs (any grade) were reported in 30 and 34 % of patients. Overall, 4 % of trifluridine/tipiracil recipients and 2 % of placebo recipients discontinued treatment due to adverse events [23]. TEAEs generally appeared more often during the first dosing cycle of trifluridine/tipiracil, with the incidence generally declining over subsequent dosing cycles (abstract presentation) [37].
The most common TEAEs in patients treated with trifluridine/tipiracil in RECOURSE were haematological or GI in nature; (Fig. 1) [23]. Grade ≥3 adverse events occurred more frequently in the trifluridine/tipiracil group than in the placebo group (69 vs. 52 %); however, most were haematological events (Sect. 5.1) and the incidence of non-haematological grade 3–4 TEAEs was low (<5 %). Typical fluoropyrimidine-associated adverse events, such as stomatitis, hand-foot syndrome and coronary spasm were infrequent (each <1 % for grade 3–4 events) with trifluridine/tipiracil [23].

Tolerability of trifluridine/tipiracil (FTD/TPI) in treatment-experienced patients with metastatic colorectal cancer in the RECOURSE trial. Treatment-emergent adverse events of a all grades (incidence ≥5 and ≥2 % higher in the FTD/TPI arm) and b grades 3 or 4 (incidence of ≥5 % and a between-group difference of ≥2 %). θ = incidence of 0
In a retrospective analysis (data available as an abstract), the rate of hospitalisations for all serious adverse events in the RECOURSE trial was 28 % in the trifluridine/tipiracil group and 33 % in the placebo groups [38].
Results from a post-marketing surveillance study suggested that the safety profile of trifluridine/tipiracil in Japanese patients with mCRC (n = 3420) in clinical practice is consistent with that observed in clinical trials, and there were no unexpected safety signals [39].
5.1 Haematological Adverse Events
The most common grade 3 or 4 TEAEs in RECOURSE were haematological in nature (Fig. 1) [23]. The most frequent adverse events leading to trifluridine/tipiracil treatment interruptions or delays and/or dose reductions in this trial were a decreased neutrophil count (20.5 %), neutropenia (19.9 %) and anaemia (5.4 %) [10]. Neutropenia was generally controlled with reductions in the dose or delays in cycle commencement, and the use of granulocyte colony-stimulating factors (required in 9.4 % of trifluridine/tipiracil recipients) [10, 40]. In patients in the trifluridine/tipiracil group who began at least 2 cycles of treatment, 53 % had a delay of 4 days or more in beginning their next cycle due to toxicity; approximately half of this subgroup had delays of 8 days or more [23]. Only 14 % the trifluridine/tipiracil group required dose reductions [23].
In the post-marketing surveillance study, serious neutropenia and febrile neutropenia events generally occurred around day 15 of the first cycle and non-serious neutropenia generally occurred from days 19–25 in the first cycle. Thus, patients with serious neutropenia around day 15 should be carefully monitored to prevent febrile neutropenia during the first treatment cycle [39]. One trifluridine/tipiracil recipient (0.2 %) in RECOURSE died from septic shock due to neutropenic infection [23]. The US prescribing information contains warnings about the risk of severe and life-threatening myelosuppression [7]. Complete blood counts should be conducted prior to and on day 15 of each treatment cycle [7].
6 Dosage and Administration of Trifluridine/Tipiracil
The recommended starting trifluridine/tipiracil dosage is 35 mg/m2 twice daily administered on days 1–5 and days 8–12 of each 28-day treatment cycle (calculated according to body surface area) [7–9]. The dosage should be rounded to the nearest 5 mg increment and should not exceed 80 mg/dose (based on the trifluridine component) [7–9]. Trifluridine/tipiracil should be taken within 1 h of morning and evening meals [7–9]. Treatment should be continued until disease progression or unacceptable toxicity occurs [7, 8]. Dose modifications or interruptions may be required based on individual safety and tolerability considerations, with a maximum of three dose reductions to a minimum dosage of 20 mg/m2 twice daily (the trifluridine/tipiracil dosage should not be increased after it has been decreased) [7, 8]. Trifluridine/tipiracil is associated with a risk of severe myelosuppression and embryo-fetal toxicity [7–9].
Local manufacturer’s prescribing information should be consulted for detailed information, including starting dose calculation (according to body surface area), recommended dosage modifications (according to blood counts), warnings, precautions, contraindications and use in special populations.
7 Current Status of Trifluridine/Tipiracil in Metastatic Colorectal Cancer
For patients with unresectable mCRC, fluoropyrimidines have been the backbone of all chemotherapy schedules, both as initial therapy and as an important option in subsequent therapy, alone and in combination with other drugs (including biological targeted agents). Treatment is administered until the disease progresses, recurs, or the toxicity of therapy is deemed intolerable and the patient therefore requires palliative treatment [3, 4].
Trifluridine/tipiracil is the second oral agent to be approved in the palliative setting for the treatment of mCRC [7–9]. Although trifluridine is a fluoropyrimidine, and as such has many similarities to 5-FU, differences in the mechanism of action (e.g. more readily incorporated into DNA than 5-FU) mean that trifluridine has a distinct cytotoxic effect (Sect. 2) [13, 16]. In addition, the presence of tipiracil prevents the otherwise rapid degradation of the trifluridine, allowing for the maintenance of adequate plasma levels of trifluridine (Sect. 3). The combined properties of trifluridine/tipiracil, therefore, provide a novel treatment opportunity for patients that are refractory to traditional fluoropyrimidines. The importance of these findings is reflected in the latest ESMO Consensus Guidelines for the treatment of mCRC, which recommends the use of trifluridine/tipiracil as a third-line therapy in fit, pretreated patients and in RAS wild-type patients with EGFR antibodies [4]. The National Institute for Health and Care Excellence in the UK recommends trifluridine/tipiracil as an option in patients who have had previous treatment with, or are not considered candidates for, currently available therapies [41].
Trifluridine/tipiracil was initially approved in Japan [9] on the basis of results of the phase II trial (Sect. 4.1), and elsewhere based on the results from the pivotal phase III RECOURSE trial (Sect. 4.2) [7, 8]. In RECOURSE, the addition of trifluridine/tipiracil to BSC resulted in a modest, but clinically meaningful and statistically significant improvement in the median OS compared with placebo plus BSC in patients with mCRC who were refractory to or intolerant of all approved standard therapies (Sect. 4.2). Furthermore, the OS benefit seen with trifluridine/tipiracil was maintained across various prespecified subgroups, including patients refractory to fluoropyrimidines and those with KRAS mutations. Trifluridine/tipiracil was also associated with a significantly longer PFS and a higher disease control rate than placebo. In addition to prolonging survival, the other aim of palliative care is to maintain the health-related quality of life of the patient [3, 4]. However, no health-related quality of life data or other patient-reported outcomes (e.g. use of analgesics, pain control and other specific disease-related symptoms) are currently available from patients treated with trifluridine/tipiracil; such data would be of interest.
Trifluridine/tipiracil has a manageable tolerability profile, with the most commonly reported adverse events of any grade being haematological or GI in nature (Sect. 5). Severe myelosuppression was the most serious safety concern identified during trifluridine/tipiracil treatment and was managed with dose or cycle modification or granulocyte-colony stimulating factor administration (Sect. 5). In general, the most common adverse events emerged during the first treatment cycle [37].
Overall, the benefit-risk of trifluridine/tipiracil treatment was considered positive [10]. Further analyses to fully determine the extent of the correlation between trifluridine/tipiracil exposure and the efficacy and tolerability (neutropenia) of the drug (Sect. 2) are required.
Regorafenib is the only other agent approved for the treatment of refractory mCRC in a palliative setting [3–5] and the latest ESMO guidelines confirm its status as a third-line therapy [4]. Regorafenib provides an improvement in survival of a similar magnitude to trifluridine/tipiracil, although toxicity concerns exist with regorafenib in frail patients and close and frequent monitoring for toxicity is recommended [4].
The tolerability and safety profiles of trifluridine/tipiracil and regorafenib differ in terms of adverse events. However, the most common grade ≥3 adverse events with either drug are considered manageable [haematological events with trifluridine/tipiracil and skin reactions (hand-foot, rash or desquamation), fatigue, diarrhoea and hypertension with regorafenib] [5, 7–9, 23, 42].
As trifluridine/tipiracil and regorafenib differ in their mechanisms of action [5, 8–10], it is hypothesised that sequential use of both agents may be beneficial. As yet, there is no consensus regarding which agent would be the better first choice [3, 4]. To date only limited data from subgroup analyses from RECOURSE (Sect. 4.2) and a retrospective analysis are available [43] demonstrating that the clinical benefit seen with trifluridine/tipiracil is maintained regardless of prior regorafenib treatment. The optimal sequential use of trifluridine/tipiracil and regorafenib warrants further evaluation.
In conclusion, oral trifluridine/tipiracil significantly improves survival compared with placebo and is associated with a manageable tolerability profile. The acceptable benefit-risk profile of trifluridine/tipiracil, combined with a paucity of treatment options and the seriousness of the condition, make trifluridine/tipiracil a useful additional treatment option for the management of mCRC in patients who are refractory to, or are not considered candidates for, currently available therapies.
Data selection sources:
Relevant medical literature (including published and unpublished data) on trifluridine/tipiracil (Lonsurf) was identified by searching databases including MEDLINE (from 1946), PubMed (from 1946) and EMBASE (from 1996) [searches last updated 9 August 2016], bibliographies from published literature, clinical trial registries/databases and websites. Additional information was also requested from the company developing the drug.
Search terms: Lonsurf, TAS-102, trifluridine, trifluorothymidine, TFT, tipiracil, mCRC, metastatic, metastasis, metastases, advanced, colorect, colon, rectal, bowel.
Study selection: Studies in patients with metastatic colorectal cancer with disease progression after treatment with standard therapies who received on trifluridine/tipiracil (Lonsurf). When available, large, well designed, comparative trials with appropriate statistical methodology were preferred. Relevant pharmacodynamic and pharmacokinetic data are also included.
References
Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2016. doi:10.1136/gutjnl-2015-310912.
Wilkes GM. Metastatic colorectal cancer: management challenges and opportunities. Oncology. 2011;25(7):32–44.
National Comprehensive Cancer Network. NCCN Guidelines Version 2.2016 Colon Cancer. 2016. https://www.nccn.org/. Accessed 9 Aug 2016.
Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–422.
Bayer. STIVARGA® (regorafenib) tablets: US prescribing infomation. 2015. http://labeling.bayerhealthcare.com/html/products/pi/Stivarga_PI.pdf. Accessed 9 Aug 2016.
Emura T, Suzuki N, Fujioka A, et al. Potentiation of the antitumor activity of alpha, alpha, alpha-trifluorothymidine by the co-administration of an inhibitor of thymidine phosphorylase at a suitable molar ratio in vivo. Int J Oncol. 2005;27(2):449–55.
Taiho Oncology Inc. LONSURF (trifluridine and tipiracil) tablets for oral use: US prescribing infomation 2015. https://www.taihooncology.com/us/prescribing-information.pdf. Accessed 9 Aug 2016.
European Medicines Agency. Lonsurf® (trifluridine/tipiracil): summary of product characteristics. 2016. http://www.ema.europa.eu/. Accessed 13 June 2016.
Taiho Oncology Inc. Lonsurf combination tablet: Japanese prescribing information. 2015. http://di.taiho.co.jp/taiho/hp/fileDownloadMediaContent.do?_contentGroupId=2919&_pspCode=80&_pspSubCode=02 Accessed 9 Aug 2016.
European Medicines Agency. European public assessment report (EPAR) for Lonsurf. 2016. http://www.ema.europa.eu. Accessed 9 Aug 2016.
Sun W, Rosen LS, Rasco DW, et al. An open-label, randomized, parallel-group study of the pharmacokinetics of trifluridine as a component of TAS-102 versus FTD alone [abstract no. 751]. J Clin Oncol. 2016;34(Suppl 4).
Matsuoka K, Iimori M, Niimi S, et al. Trifluridine induces p53-dependent sustained G2 phase arrest with its massive misincorporation into DNA and Few DNA Strand Breaks. Mol Cancer Ther. 2015;14(4):1004–13.
Tanaka N, Sakamoto K, Okabe H, et al. Repeated oral dosing of TAS-102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep. 2014;32(6):2319–26.
Emura T, Murakami Y, Nakagawa F, et al. A novel antimetabolite, TAS-102 retains its effect on FU-related resistant cancer cells. Int J Mol Med. 2004;13(4):545–9.
Yamashita F, Komoto I, Oka H, et al. Exposure-dependent incorporation of trifluridine into DNA of tumors and white blood cells in tumor-bearing mouse. Cancer Chemother Pharmacol. 2015;76(2):325–33.
Sakamoto K, Yokogawa T, Ueno H, et al. Crucial roles of thymidine kinase 1 and deoxyUTPase in incorporating the antineoplastic nucleosides trifluridine and 2′-deoxy-5-fluorouridine into DNA. Int J Oncol. 2015;46(6):2327–34.
Suzuki N, Emura T, Fukushima M. Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int J Oncol. 2011;39(1):263–70.
Suzuki S, Iwaizumi M, Hamaya Y, et al. Trifluridine to enhance cytotoxicity by DNA mismatch repair deficiency and subsequent MBD4 frameshift mutation in colorectal cancer [abstract no. 608]. J Clin Oncol. 2016;34(Suppl 4).
Emura T, Nakagawa F, Fujioka A, et al. An optimal dosing schedule for a novel combination antimetabolite, TAS-102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med. 2004;13(2):249–55.
Doi T, Ohtsu A, Yoshino T, et al. Phase I study of TAS-102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012;107(3):429–34.
Ohtsu A, Yoshino T, Falcone A, et al. Onset of neutropenia as an indicator of treatment response in the phase III RECOURSE trial of TAS-102 vs placebo in patients with metastatic colorectal cancer [abstract no. 3556]. J Clin Oncol. 2016;34(Suppl 4).
Kasi PM, Kotani D, Cecchini M, et al. Chemotherapy induced neutropenia at 1-month mark is a predictor of overall survival in patients receiving TAS-102 for refractory metastatic colorectal cancer: a cohort study. BMC Cancer. 2016;16:467.
Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372(20):1909–19.
Bendell JC, Patel MR, Yoshida K, et al. Phase 1 study of cardiac safety of TAS-102 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(6):1275–83.
Cleary JM, Mayer RJ, Van Cutsem E, et al. Population pharmacokinetic (PK) analysis of TAS-102 in patients (pts) with metastatic colorectal cancer (mCRC): Results from 3 phase 1 trials and the phase 3 RECOURSE trial [abstract no. 2579]. J Clin Oncol. 2015;33(15 Suppl 1).
Yoshino T, Kojima T, Bando H, et al. The effect of food on the pharmacokinetics of TAS-102 and its efficacy and safety in patients with advanced solid tumors. Cancer Sci. 2016;2016(107):659–65.
Lee JJ, Seraj J, Yoshida K, et al. Human mass balance study of TAS-102 using (14)C analyzed by accelerator mass spectrometry. Cancer Chemother Pharmacol. 2016;77(3):515–26.
Yoshino T, Mizunuma N, Yamazaki K, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13(10):993–1001.
Ohtsu A, Yoshino T, Wahba MM, et al. Phase III RECOURSE trial of TAS-102 versus placebo with best supportive care in patients with metastatic colorectal cancer: Geographic subgroups [abstract no. 646]. J Clin Oncol. 2016;34(Suppl 4).
Mayer RJ, Ohtsu A, Yoshino T, et al. TAS-102 versus placebo plus best supportive care in patients with metastatic colorectal cancer refractory to standard therapies: Final survival results of the phase III RECOURSE trial [abstract no. 634]. J Clin Oncol. 2016;34(Suppl 4).
Hochster H, Hager S, Pipas JM, et al. KRAS and BRAF gene subgroup analysis in the Phase 3 RECOURSE trial of TAS-102 versus placebo in patients with metastatic colorectal cancer [abstract no. O-010]. Ann Oncol. 2015;26(Suppl 4):iv111.
Van Cutsem E, Benedetti FM, Mizuguchi H, et al. TAS-102 vs placebo (PBO) in patients (pts) >65 years (y) with metastatic colorectal cancer (mCRC): an age-based analysis of the RECOURSE trial [abstract no. 638]. J Clin Oncol. 2016;34(Suppl 4).
Yoshino T, Shinozaki E, Yamazaki K, et al. Final survival results and onset of neutropenia as an indicator of therapeutic effect in phase 2 of TAS-102 vs placebo with metastatic colorectal cancer (J003-10040030) [abstract no. PD-014]. Ann Oncol. 2016;27(Suppl 2):ii107.
Hong DS, Abbruzzese JL, Bogaard K, et al. Phase I study to determine the safety and pharmacokinetics of oral administration of TAS-102 in patients with solid tumors. Cancer. 2006;107(6):1383–90.
Overman MJ, Varadhachary G, Kopetz S, et al. Phase 1 study of TAS-102 administered once daily on a 5-day-per-week schedule in patients with solid tumors. Invest New Drugs. 2008;26(5):445–54.
Bendell JC, Rosen LS, Mayer RJ, et al. Phase 1 study of oral TAS-102 in patients with refractory metastatic colorectal cancer. Cancer Chemother Pharmacol. 2015;76(5):925–32.
Shinozaki E, Laurent S, Gravalos C, et al. Timing of adverse events (AEs) in the Phase 3 RECOURSE trial of TAS-102 versus placebo in patients (pts) with metastatic colorectal cancer (mCRC) [abstract no. 2151]. Eur J Cancer. 2015;51(Suppl 3):S383–4.
Falcone A, Garcia-Carbonero R, Tabernero J, et al. Low rates of hospitalizations with TAS-102 in the European (EU) subregion of the Phase 3 RECOURSE trial in patients (pts) with metastatic colorectal cancer (mCRC) [abstract no. 2150]. Eur J Cancer. 2015;51(Suppl 3):S383.
Yoshino T, Uetake H, Furuta T. TAS-102 safety in metastatic colorectal cancer: results from the first post-marketing surveillance study. Clin Colorectal Cancer. 2016. doi:10.1016/j.clcc.2016.04.004.
Mayer R, Van Cutsem E, Yoshino T, et al. Supportive treatment for hematologic toxicities in the phase 3 RECOURSE trial of TAS-102 vs placebo with best supportive care in patients with metastatic colorectal cancer [abstract no. e15021]. J Clin Oncol. 2016;34(Suppl 4).
National Institute for Health and Care Excellence. Trifluridine-tipiracil hydrochloride for previously treated metastatic colorectal cancer. 2016. http://www.nice.org.uk. Accessed 25 Aug 2016.
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–12.
Kotani D, Shitara K, Kawazoe A, et al. Safety and efficacy of trifluridine/tipiracil monotherapy in clinical practice for patients with metastatic colorectal cancer: experience at a single institution. Clin Colorectal Cancer. 2015. doi:10.1016/j.clcc.2015.11.005.
Acknowledgments
During the peer review process, the manufacturer of trifluridine/tipiracil was offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Celeste Burness and Sean Duggan are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: A.B. Benson III, Robert H. Lurie Comprehensive Cancer Center, Northwestern University Chicago, IL, USA; A. Grothey, Division of Medical Oncology, Mayo Clinic, Rochester, MN, USA; F. Opdam, Department of Clinical Pharmacology, The Netherlands Cancer Institute, Amsterdam, Netherlands; T. Price, Department Medical Oncology, The Queen Elizabeth Hospital and University of Adelaide, Adelaide, Australia; A. Zaniboni, Medical Oncology Department, Fondazione Poliambulanza, Brescia, Italy.
Rights and permissions
About this article

Cite this article
Burness, C.B., Duggan, S.T. Trifluridine/Tipiracil: A Review in Metastatic Colorectal Cancer. Drugs 76, 1393–1402 (2016). https://doi.org/10.1007/s40265-016-0633-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0633-9