
Abstract
To date, no drug is approved for the treatment of Fragile X Syndrome (FXS) although many drugs are used to manage challenging behaviors from a symptomatic perspective in this population. While our understanding of FXS pathophysiology is expanding, efforts to devise targeted FXS-specific treatments have had limited success in placebo-controlled trials. Compounds aimed at rectifying excessive glutamate and deficient gamma-aminobutyric acid (GABA) neurotransmission, as well as other signaling pathways known to be affected by Fragile X Mental Retardation Protein (FMRP) are under various phases of development in FXS. With the failure of several metabotropic glutamate receptor subtype 5 (mGlur5) selective antagonists under clinical investigation, no clear single treatment appears to be greatly effective. These recent challenges call into question various aspects of clinical study design in FXS. More objective outcome measures are under development and validation. Future trials will likely be aimed at correcting multiple pathways known to be disrupted by the loss of FMRP. This review offers a brief summary of the prevalence, phenotypic characteristics, genetic causes and molecular functions of FMRP in the brain (as these have been extensively reviewed elsewhere), discusses the most recent finding in FXS drug development, and summarizes FXS trials utilizing symptomatic treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Despite advances in our understanding of FXS pathophysiology, effective targeted therapies remain elusive. |
A wide array of drug mechanisms is currently being assessed in FXS clinical study, although most are still in early phases. |
Future clinical trials are focusing on optimization of study design including careful consideration of outcome measures as well as participant characteristics including age and molecular, electrophysiological, behavioral, and genetic (epigenetic) phenotypes. |
1 Introduction
Full mutation Fragile X Syndrome (FXS) is caused by the epigenetic silencing of the fragile X mental retardation 1 gene (FMR1) and subsequent loss of fragile X mental retardation protein (FMRP) expression [1]. The silencing of the FMR1 gene is typically caused by methylation of a cytosine guanine guanine (CGG) trinucleotide repeat expansion (greater than 200 repeats is termed full mutation FXS) in the 5′ untranslated region (UTR) of the FMR1 gene on the X chromosome [2, 3]. In rare cases, loss of FMRP expression and FXS can also occur as a point mutation or deletion in the FMR1 gene [4]. Full mutation FXS can result from maternal transmission in which a mother’s permutation allele, (55–200 CGG repeats; typical population has fewer than 45 repeats) undergoes the above-mentioned CGG repeat expansion and is passed on to the next generation. Pre-mutation individuals, also referred to as carriers, were initially thought to be unaffected. However, it is now known that an expansion reaching 150 CGG repeats is associated with an eight-fold increase in FMR1 mRNA and can sometimes be associated with a slight decrease in FMRP levels [5–7]. Along with changes in mRNA and protein, pre-mutation individuals can exhibit clinical features not seen in individuals with the full mutation, such as primary ovarian insufficiency (FXPOI) [8, 9], fragile X-associated tremor ataxia syndrome (FXTAS) [10, 11], and neuropathy [12] among other features.
1.1 Prevalence and Description
FXS is the most prevalent known single gene cause of autism and heritable developmental disability, with a prevalence of 1:4000 males and 1:4000–6000 females [13–17]. Patients with FXS experience an array of physical, neurological, behavioral, and cognitive problems. Physically, FXS is characterized by a long and narrow face, high arched palate, flat feet, hyperextensible joints, low muscle tone, soft skin and macro-orchidism [18]. Neurobehaviorally, individuals with FXS can suffer from sleep issues, aggression, anxiety, attention deficit hyperactivity disorder (ADHD), self-injurious behavior, hypersensitivity to auditory and other stimuli, perseverative language and increased risk of seizures, among other features [16, 19, 20]. In a small study, full scale IQ scores were shown to be inversely correlated with methylation status and were positively correlated with FMRP expression in persons with FXS [21]. Additionally, FXS is often associated with autism spectrum disorder (ASD). Approximately two in every three males diagnosed with FXS will have behavioral characteristics that align with the broader autism phenotype [15, 17, 22, 23]. Females with a full mutation allele on one of their X chromosomes tend to be less severely affected than full mutation males. However, the severity of the syndrome depends on the location and degree to which the full mutation allele is expressed following X-inactivation. The FMR1 genotype of an individual can be further complicated by mosaic characteristics in which some cells express differential repeat numbers (somatic mosaicism) or differential degrees of methylation (methyl mosaicism) compared to other cells that express the full expansion mutation or are fully methylated [24].
1.2 Role of FMRP
FMRP is expressed in a variety of tissues, but is most enriched in the testes and brain. In the brain, FMRP is developmentally expressed in microglia and oligodendrocytes while expression persists in mature astrocytes and neurons [25]. Within neurons, FMRP is found in the soma, spines, and dendrites [26]. FMRP is an mRNA binding protein, with selectivity targeted to approximately 4 % of the mRNAs transcribed in the brain [27]. FMRP contains three RNA binding regions, two hnRNP K-homology KH domains and an RGG (arginine-glycine-glycine) box allowing FMRP to bind to a broad range of mRNAs in a selective manner [28]. FMRP also contains a nuclear localization signal and a nuclear export signal that allow it to transport mRNAs bound in the nucleus to the cytoplasm [29]. Protein interaction domains in FMRP may also be present allowing FMRP to associate with protein complexes, such as polyribosomes, regulating translation particularly at the synapse [30, 31]. Though FMRP has been implicated in stabilization [32] and transportation of mRNA [33], it is believed to mainly function as a translational repressor of its mRNA targets [30, 31, 34]. While the loss of FMRP in FXS causes translation of many FMRP RNA targets to increase, protein expression of other targets is unchanged or even decreased indicating that other compensatory mechanisms must come into play in the absence of FMRP [28].
1.3 FXS-Associated Deficits in Synaptic Plasticity
Research in both humans and animal models has led to advances in the understanding of the neurobiology of FXS, resulting in the development of novel targets and drug optimization in the field. Children with FXS have been reported to have a generalized increase in brain size compared with controls as well as discernable differences in specific brain regions compared to children with autism (larger temporal lobe white matter, cerebellar gray matter, and caudate nucleus; smaller amygdala) [35]. At the microscopic level, loss of FMRP has been shown to alter the structure of dendritic spines which are the sites of synaptic plasticity. These alterations appear to be brain region and activity dependent (reviewed by [36]), often manifesting as increased spine density with longer, spindly, and immature morphology frequently reported in postmortem human and Fmr1 KO mouse brain tissue [37–39]. The loss of FMRP observed in the Fmr1 KO mouse model of FXS also negatively impacts synaptic plasticity with enhanced long-term depression (LTD) [40–43] and brain region-specific alterations in long-term potentiation (LTP) [44–47]. Many signaling cascades involved in synaptic plasticity and learning and memory are known to be altered in the Fmr1 KO mouse including phosphoinositide 3-kinase (PI3K) [48–50], extracellular signal-related kinase (ERK1/2) [51–53], and mammalian target of rapamycin (mTOR) [54–56]. Human tissues have also shown alterations in these signaling molecules [57–60]. The study of molecular, cellular, and behavioral alterations in the FXS animal models has greatly increased our understanding of FMRP function and human disease pathophysiology while providing the potential for targeted treatments in FXS, ASD, and other related developmental disorders.
Many of the recent targeted clinical trials in FXS have attempted to rectify the excitatory/inhibitory imbalance believed to contribute to the pathophysiology of FXS. In the FXS brain, there is likely an excess of excitatory, glutamatergic signaling coupled with deficiencies in inhibitory, GABAergic signaling. Clinical trials are summarized based on proposed drug mechanism followed by symptomatic treatment studies. Table 1 lists drug treatments by trial type, and details the degree of study completion.
2 Modulating Excitatory Neurotransmission
Altered signaling and/or localization of several glutamatergic receptors have been reported in Fmr1 KO mice. Several targeted clinical trials have focused on reducing excitatory neurotransmission by antagonism of group I metabotropic glutamate receptors (mGluRs), particularly mGluR5 (Fenobam, AFQ056, RO4917523). Additional drugs with effects at the N-methyl-d-aspartate (NMDA) receptor (memantine) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) (CX516) have also been studied in humans with FXS.
The mGluR theory of FXS proposes that excessive signaling through mGluRs is significantly contributing to behavioral, electrophysiological, and molecular dysfunction associated with FXS [61]. The work by Bear and colleagues was one of the first discoveries in FXS that describes alterations in components and molecules known to be important for synaptic plasticity in the normal brain. The mGluR theory was based upon the observations that 1) FMRP was shown to repress protein translation at the synapse [62], 2) synaptic protein synthesis can be triggered by activation of mGluRs [26], 3) FMRP loss leads to increased downstream effects of mGluR signaling [40, 63], and 4) many of these downstream effects are dependent on mRNA translation at the synapse [64–67]. The mGluR theory is corroborated by studies showing that strong mGluR5 antagonists, namely MPEP, can improve FXS phenotypes in animal models including abnormalities noted in AMPAR expression, behavioral measures, and dendritic spine morphology [43, 68]. Further corroboration came from a study demonstrating that reduction of mGluR5 levels in Fmr1 KO mice can normalize protein synthesis, dendritic spines, and some behavior [69]. However, a more recent and thorough behavioral assessment of genetically reducing mGluR5 in Fmr1 KO mice showed limited behavioral improvement suggesting that mGlur5 manipulation may not result in as robust of an improvement as originally indicated [70]. It should also be noted that the mGluR5 antagonists used in preclinical Fmr1 KO studies were shown to maintain analgesic effects in mGluR5 KO mice. This finding indicates that these molecules likely have additional molecular targets that weren’t considered in the preclinical Fmr1 KO mouse work as potentially contributing to the favorable treatment effects observed [71].
2.1 Fenobam
Fenobam [N-(3-chlorophenyl)-N′-(4,5-dihydro-1-methyl-4-oxo-1H-imidazole-2-yl)urea] is a non-benzodiazepine anxiolytic drug and negative allosteric modulator of mGluR5. It acts similarly to MPEP, binding in a non-competitive manner and has mGluR5 inverse agonist properties [72]. An open-label, single-dose study was completed to evaluate its safety, pharmacokinetic properties, and explore effects on sensory gating, attention, and inhibition in adult males and females with FXS [73]. Subjects included six males and six females with FXS, aged 18.7–30.7 years. The primary outcome of pre-pulse inhibition (PPI) was tested at baseline and after treatment with a single dose of fenobam. There were no significant adverse events and the medication was well tolerated. Six out of twelve (50 %) subjects met response criterion of at least 20 % improvement over baseline on PPI at 120 ms. Importantly, reports of fenobam treatment in non-FXS individuals taking high doses (four times the daily highest dose in the FXS trial) for a period of 4 weeks reported odd CNS effects including hallucinations, vertigo, paresthesia, and insomnia [74]. This suggests that fenobam is not likely going to be considered for future study as a long-term treatment option in FXS.
2.2 Mavoglurant/AFQ056
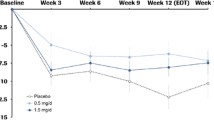
AFQ056 (Mavoglurant) is a noncompetitive mGluR5 antagonist developed by Novartis Pharmaceuticals. Three clinical trials of AFQ056 were completed in subjects with FXS. The first, a 30-subject double-blind crossover study involving 20 days of treatment, failed to find any effect of the drug within the full study population on any primary or secondary outcome measures. However, when the investigators limited their analysis to a small sub-set of seven individuals with complete FMR1 promoter methylation, they found significant improvement across a variety of outcome measures [75]. It is worth noting that these seven individuals showed little if any improvement while in the placebo arm of the study, as measured by the Aberrant Behavior Checklist (ABC), a finding which may have contributed to this post-hoc subgroup effect. Based upon these initial results, two multinational studies were conducted: (1) a Phase IIb FXS double-blind, placebo-controlled, parallel group, 3-month trial in adult males and females aged 18–45 years, (2) a similarly controlled Phase III trial, in adolescents aged 12–17 years. Subjects were assigned to 25 mg twice daily, 50 mg twice daily, 100 mg twice daily, or placebo to evaluate the safety and efficacy of the three doses for treating the behavioral symptoms of FXS. The primary outcome measure was the ABC total score, with the Clinical Global Impression-Improvement (CGI-I) scale and Repetitive Behavior Scale-Revised (RBS-R) as secondary outcome measures. Neither study met significance on the primary endpoint. The sponsor, Novartis Pharmaceuticals, subsequently terminated the open-label extension portion of the study in adolescents and discontinued their development program of AFQ056 for the treatment of FXS (ClinicalTrials.gov Identifiers: NCT01253629, NCT01357239; [76]).
2.3 Basimglurant/RO4917523
Another mGluR5 antagonist, RO4917523 (basimglurant), was studied in a Phase II clinical trial for subjects aged 14–50 years with FXS (ClinicalTrials.gov Identifier: NCT01517698) and in an additional trial in children and adolescents aged 5–13 years evaluating the drug for safety and tolerability (ClinicalTrials.gov Identifiers: NCT01015430, NCT01750957). Due to lack of efficacy, although trial data remain unpublished at this time, the sponsor of the trials, Hoffman–La Roche, subsequently terminated its program for the development of basimglurant as a treatment for FXS [77].
With the disappointing results from both the mavoglurant and basimglurant trials, pharmaceutical companies have largely moved away from pursuing selective mGluR5 antagonists as targeted treatments for FXS. The mGluR5 theory has not been proven or disproven with such short-term trials; however, these results do suggest that pharmacological manipulation of mGlur5 signaling, alone, in humans with FXS is not a short-term approach to attenuating interfering symptoms in the ages studied. Future trials of mGluR5 antagonists could target young subjects whose brains are still developing. By targeting signaling imbalance in the developing brain it might be possible to augment neural circuitry as it develops, potentially improving clinical outcomes as a result. Additionally, the field is likely moving toward combined pharmacotherapies, possibly including mGlur5 modulators, with additional targeted drugs addressing other potential causes of the FXS behavioral phenotype.
2.4 Memantine
Memantine (3,5-dimethyladamantan-1-amine) is a compound that non-competitively antagonizes the N-methyl-d-aspartic acid (NMDA) receptor. There is evidence of NMDA receptor dysfunction in FXS, but the overall direction of the effect is unclear, appearing to depend on brain region and age [40, 78–80]. Memantine is US Food and Drug Administration (FDA) approved for the management of Alzheimer’s disease with a large body of work to support its effect in this disorder. Treatment with memantine is also being explored in humans with other neurological disorders including ASD and FXS. In FXS, an open-label trial was conducted in six subjects who had a diagnosis of both FXS and pervasive developmental disorder (as diagnosed using criteria of the Diagnostic and Statistical Manual of Mental Disorders, fourth edition) [81]. The subjects received an average of 34.7 weeks of memantine treatment. While four subjects showed global symptom improvement as measured by the CGI-I, there were no significant effects in any specific symptom domains and two of the subjects had to discontinue therapy with memantine due to increased irritability with treatment.
2.5 Cx516
AMPAR is a non-NMDA-type ionotropic receptor for glutamate and mediates fast synaptic transmission. Modulation of AMPA signaling, downstream of mGluR signaling, was proposed as an indirect method of restoring GABA/glutamate signaling balance [61]. In FXS, internalization of AMPARs is increased and suspected to contribute to alterations in LTP and LTD since AMPA signaling is required for proper maintenance of synaptic plasticity. CX516 is a positive allosteric modulator of AMPAR and was studied in a four-week, double-blind, placebo-controlled trial. The trial failed to find significant change in memory, the study’s primary measure, or any secondary measures including measures for language skills, behavior, and the ABC and CGI [82].
3 Modulating Inhibitory Neurotransmission
Along with the increase in glutamatergic signaling, the signaling imbalance in FXS is, in part, due to deficits in inhibitory GABAergic function. In the FXS mouse, deficits in GABAergic signaling have been demonstrated in a variety of brain regions including the hippocampus, striatum, amygdala, and somatosensory cortex [83–86]. These deficits are commonly found as decreases in GABA(A) receptor subunit expression. It has been demonstrated in the Fmr1 KO mouse that positive modulation of GABA(A) receptors can improve some behavioral and neurophysiological alterations in mouse and fly models [87, 88].
3.1 Riluzole
Riluzole is an FDA-approved treatment for amyotrophic lateral sclerosis (ALS) and may be helpful for depression and anxiety [89, 90]. It is hypothesized to work by inhibiting glutamate release [91] and potentiating post-synaptic GABA(A) receptor activity [92]. Riluzole was the first GABA(A) agonist studied in FXS clinical trials, in a six-week open-label prospective pilot study (100 mg/day) with a primary outcome of repetitive, compulsive behavior. It was conducted in six adults with FXS. Treatment with riluzole was associated with clinical response in one of six subjects (17 %). Peripheral extracellular signal-related kinase (ERK) activation, which is known to be altered in fragile X knockout mouse models [93], was significantly corrected in all subjects despite the lack of clinical improvement [94].
3.2 Acamprosate
Acamprosate is a compound FDA approved for the management of alcohol dependence and is currently being tested for efficacy in FXS. While acamprosate was not shown to interact with several tested GABA or glutamate receptor subtypes in an Oocyte model [95] it has the potential to effect the excitatory/inhibitory balance by effecting both GABA(A) receptor function [96] and mGluR signaling [97], likely in a pleiotropic manner [98]. The first study of acamprosate in FXS was a small three-subject open-label trial in which all subjects showed improvement measured by the CGI-I [99]. Acamprosate was then studied in 12 youths with FXS for 10 weeks in an open-label trial. Acamprosate significantly improved performance on a number of outcome measures including various subscales of the ABC, CGI-Severity, Social Responsiveness Scale (SRS), Attention Deficit Hyperactivity Disorder Rating Scale (ADHD-RS), and subdomains of the Vineland Adaptive Behavior Scale (VABS). On the CGI-I, the primary outcome measure, 9 of the 12 participants were either “very much improved” or “much improved” [100]. In the same study, acamprosate was also shown to decrease plasma amyloid precursor protein (APP) and increase brain-derived neurotrophic factor (BDNF) with treatment suggesting positive pleotropic effects of this drug in FXS individuals. On the basis of these results, acamprosate is currently the subject of a Phase II placebo-controlled study in FXS (clinicaltrials.gov, NCT01911455).
3.3 Ganaxolone
Ganaxolone (3a-hydroxy-3B-methyl analog of allopregnanolone) is a neuroactive steroid and positive allosteric modulator at GABA(A) receptors. In rodent models of seizure disorders, ganaxolone has been shown to have anticonvulsant effects at tolerable doses and sedative effects at higher doses [101]. Ganaxolone has been found to improve symptoms including audiogenic seizures in the Fmr1 knockout mouse [102, 103] and is well tolerated in human adults, children, and infants [104, 105]. A Phase II trial of ganaxolone in subjects aged 6–17 years with full mutation FXS is currently underway (ClinicalTrials.gov Identifier: NCT01725152). The study aims to determine the safety, tolerability, and efficacy of ganaxolone for the treatment of anxiety and attention deficits in subjects with FXS by using a randomized, double-blind, placebo-controlled, 6-week crossover design with a 2-week washout period between treatment arms.
3.4 Metadoxine ER
Metadoxine (pyridoxol l-2-pyrrolidone-5-carboxylate) is a non-stimulant, ion-pair salt of pyridoxine (vitamin B6) and 2-pyrrolidone-5-carboxylate (PCA), and has been used around the world for the treatment of acute alcohol intoxication for over 30 years. In animal studies, metadoxine ER (MDX; MG01Cl), an extended-release formulation under development by Alcobra Pharmaceuticals, has been shown to increase striatal dopamine levels [106]. MDX is currently in Phase III development for adults with ADHD (ClinicalTrials.gov Identifier: NCT02477748) and Phase II development for pediatric ADHD (ClinicalTrials.gov Identifier: NCT02189772). In the Fmr1 KO mouse, MDX was described as significantly improving working memory, learning, and social interaction on Alcobra’s website, although the study methods and results have not been published in peer-reviewed form. A randomized, double-blind, placebo-controlled Phase II clinical trial of MDX in FXS was recently completed (ClinicalTrials.gov Identifier: NCT02126995; [107]). The study involved a six-week treatment period and enrolled 62 males and females with full mutation FXS aged between 14 and 55 years (mean age of 24 years), with 57 subjects completing the study. Treatment with MDX did not show a significant difference over placebo on the primary endpoint, the inattentive subscale of the Attention Deficit Hyperactivity Disorder Rating Scale (ADHD RS-IV). It did, however, show significant improvement in the Intent-to-Treat (ITT) population on both the Vineland Adaptive Behavior Scale (VABS) Daily Living Skills Domain (p = 0.044) and the computerized cognitive Test of Attentional Performance for Children (KiTAP) Distractibility subscale (p = 0.017). Many initial clinical trials in FXS have had positive results following post-hoc analysis or in secondary measures. However, subsequent trials following these types of analyses have not demonstrated significant improvement. Only additional, well-planned and controlled trials of MDX in FXS will determine if this new drug may be beneficial in FXS.
3.5 Arbaclofen
The active enantiomer of racemic baclofen, STX209 (arbaclofen), is a GABA(B) agonist that was developed by Seaside Therapeutics and studied in FXS and ASD patient populations. STX209 actions at presynaptic GABA(B) receptors were hypothesized to inhibit glutamatergic release from presynaptic terminals thereby reducing the neuronal hyperexcitability associated with FXS. In the Fmr1 KO mouse model, STX209 was shown to reduce susceptibility to audiogenic seizures and normalize excessive dendritic spine density and protein synthesis [108]. A four-week, Phase II trial of STX209 in 63 subjects aged 6–40 years with full mutation FXS was completed in 2010 with the drug being described as well tolerated [109]. The study was a double-blind, placebo-controlled trial with a two-period crossover conducted at 12 sites across the USA. The drug was flexibly titrated between 1 mg twice daily and up to 10 mg three times daily until the optimal tolerated dose was determined for each subject. STX209 did not show a significant difference over placebo on the primary endpoint, the Irritability Subscale of the Aberrant Behavior Checklist (ABC-I). Post hoc analyses did show a significant improvement in parent-reported problem behaviors on the Visual Analog Scale (VAS) and on the Social Avoidance subscale of the ABC (ABC-SA). In a more socially impaired subset of participants (based on the ABC-LSW at baseline), significant improvement was observed in multiple parent and clinician rated scales leading to two phase III clinical trials in FXS individuals [110]. No significant improvements were detected in the adolescent/adult Phase III trial. The Phase III trial in children found no significant effect on any primary outcome measures, but did find an effect on the ABC-Fragile X Irritability subscale, a secondary outcome measure. TheSTX209 FXS development program was terminated by Seaside Therapeutics potentially due to lack of financial resources.
4 Other Targeted Treatment Mechanisms
4.1 Lithium
Lithium was used off-label to treat aggression and mood instability in FXS based on anecdotal evidence before it was the subject of a clinical trial. The 15-subject, open-label trial, with two months of treatment, showed improvement on a variety of secondary measures such as ABC total scores, the CGI and the VAS. No effect was seen on the primary outcome measure, the Irritability Subscale of the ABC. While there were behavioral improvements, the study also had a large number of adverse events including aggression, bedwetting, and polydipsia [111]. The open-label nature of this study makes it difficult to draw major conclusions from the results. Lithium has been shown to ameliorate a variety of phenotypes in the FXS mouse including hyperactivity, social preference, learning, and aberrant dendritic spines [112]. Lithium is proposed to act in FXS through inhibition of glycogen synthase kinase-3 (GSK-3), which has shown to be altered in the FXS mouse [113, 114]. Lithium has also been shown to rescue synaptic plasticity, protein synthesis, and GSK-3 activity phenotypes in the FXS mouse [115–118]. While these findings are promising, the side-effect profile of lithium likely limits its widespread use in FXS.
4.2 Minocycline
Minocycline is an FDA-approved treatment for acne and is known to have inhibitory effects on matrix metalloproteinase 9 (MMP-9) activity. MMP-9 has been shown to be elevated in the hippocampus of Fmr1 KO mice [119]. Minocycline treatment of Fmr1 KO mice was also shown to reduce hyperactivity and improve the immature dendritic spine phenotype [120]. In humans with FXS, an initial open-label 20-subject trial enrolling 13- to 35-year-olds noted significant improvement with minocycline treatment in CGI, ABC-C and the ABC irritability, hyperactivity, and inappropriate speech subscales after eight weeks of minocycline therapy [121]. These results led to further investigation of minocycline in a randomized, double-blind, placebo-controlled trial in 55 children and adolescents aged 3.5–16 years with FXS. The 3-month trial found a significant difference in CGI-I but no significant improvement was found on any behavior domain specific measures [122]. There were no significant effects of minocycline on VAS scores upon initial analysis, but ad hoc analysis did find a significant effect in anxiety and mood-related symptoms [122]. Minocylcine was also able to attenuate altered event-related potentials (ERPs) in a passive, auditory oddball paradigm in 12 subjects taken from the same trial [123]. This could be indicative of reducing the hypersensitivity to auditory simulation observed in FXS. A larger sample is needed to confirm both the behavioral and electrophysiological findings.
4.3 Lovastatin
The extracellular signal-related kinase (ERK1/2) intracellular signaling pathway is often implicated in FXS pathophysiology. Acting downstream of mGluRs, changes in ERK1/2 activity is required for maintenance of normal synaptic plasticity and regulation activity-dependent protein synthesis [124]. ERK activity has been shown to be increased under baseline conditions in the FXS mouse model and in human brain tissue [42, 55, 58]. Reducing ERK1/2 activation by inhibition of its activating kinase MEK with SL327 effectively rescued the audiogenic seizure phenotype in the FXS mouse [58]. Inhibition of MEK by U0126 was also able to reduce the increased protein synthesis seen in the FXS mouse hippocampus [125].
Lovastatin is a compound that has been FDA approved for the long-term management of familial hypercholesterolemia [126] (with demonstrated effects on intracellular signaling). In cultured rat brain neuroblasts, lovastatin was shown to inhibit Ras signaling, an upstream effect that resulted in reduction in ERK1/2 activation [127]. This supported previous work that was completed in fibroblasts [128]. In the FXS mouse, lovastatin was confirmed to inhibit Ras, reduce increased basal ERK1/2 activation, lower protein synthesis to wild type levels, and ameliorate FXS audiogenic seizure susceptibility [129].
Based upon the known safety profile of lovastatin and the promising preclinical results, lovastatin’s efficacy in FXS was assessed in a 16-patient, open-label trial in children and adolescents. Treatment response was assessed using the ABC-C, CGI-I, and VABSII. Significant improvement was observed after 4 and 12 weeks of treatment, with the VABS scores improving from week 4 to week 12. There was modest improvement on the CGI-I, but the open-label nature of the trial precludes any strong inferences of efficacy at this stage of development [130]. Furthermore, particular importance should be placed on lipid monitoring in future lovastatin trials since individuals with FXS are reported to have lower levels of low- and high-density lipoprotein and total cholesterol [131].
4.4 Insulin Growth Factor 1
NNZ-2566 is a synthetic analog of a naturally occurring peptide derived from insulin-like growth factor 1 (IGF-1). NNZ-2566 has been shown to have neuroprotective qualities; improving recovery, reducing apoptotic cell death, and reducing neuroinflamation in a rat model of traumatic brain injury [132–134]. NNZ-2566 was the subject of a recent Phase II clinical trial in Rett syndrome. Though the study has not yet been published, according to a press report the drug was well tolerated and met prespecified criteria for improvement [135]. Treatment with IGF-1, which NNZ-2566 mimics, has also shown efficacy in Phelan-McDermid syndrome (PMDS) in a mouse model, cultured human neurons, and a Phase I clinical trial in children [136–138]. PMDS is a developmental disorder caused by heterozygous deletion of chromosome 22q13.3, including SHANK3, a gene who’s mutation is associated with ASDs [139, 140]. In the mouse model of FXS, NNZ-2566 has been reported to rescue learning and memory deficits, normalize dendritic morphology, and restore normal extracellular signal-related kinase (ERK) signaling [141]. Recently, a double-blind, placebo-controlled early phase trial of NNZ-2566 has completed recruitment of 12- to 45-year-olds with FXS. This study was designed to investigate the safety and tolerability of a liquid oral formulation of NNZ-2566 in adolescent and adult males with FXS (ClinicalTrials.gov Identifier: NCT01894958).
4.5 Donepazil
FMR1 has been shown to be highly expressed in cholinergic neurons during the course of normal development [142]. Choline levels were shown to be lower in FXS individuals in a small 1H magnetic resonance spectroscopy study [143]. Aberrant cholinergic function has also been demonstrated in the subiculum of Fmr1 KO mice [144]. Donepezil is a drug that has been FDA approved to treat dementia in individuals with Alzheimer’s disease by acting as an acetylcholinesterase inhibitor and increasing choline levels. In a nine-subject open-label trial in FXS involving 6 weeks of treatment, donepezil was well tolerated and significantly improved performance on the ABC hyperactivity, irritability, and total scores [143]. In a double-blind, placebo-controlled trial with 12 weeks of treatment, no effect of treatment was found [145]. It is worth noting that none of the measures that reached significance in the open-label trial were measured in the placebo-controlled study.
5 Symptom-Based Treatments
With targeted, mechanism-based therapies demonstrating limited efficacy in placebo-controlled settings to date, most drug treatment in FXS is based upon targeting the symptoms presented by an individual with the use of drugs that have been FDA approved for indications outside of FXS. While these therapies do not likely target a FXS-specific neural alteration, multi-drug therapies based upon individual phenotype are currently the most common method of treating FXS.
5.1 ADHD Treatments
FXS patients often have a concomitant ADHD diagnosis with 73 % of an all-male cohort scoring 15 or higher on the Conner’s abbreviated scale indicative of ADHD [146]. Even subjects without an ADHD diagnosis often express difficulty focusing in a classroom setting. There have been three clinical trials studying drugs aimed at treating the ADHD symptoms in FXS. The first in FXS, was a double-blind placebo-controlled study of the stimulant medications methylphenidate and dextroamphetamine. Patients received one week each of methylphenidate, dextroampetamine, and placebo. Methylphenidate treatment improved patient performance on the ADD-H: Comprehensive Teacher Rating Scale while no improvement was observed with dextroamphetamine [147]. The second trial looked at the ability of l-acetylcarnitine (LAC) to reduce the ADHD symptoms observed in boys with FXS. The double-blind, placebo-controlled study found significant improvement in both the CGI parent and teacher response scores as well as the VABS Socialization and ABC domains [148]. The most recent trial focusing on ADHD behavior in FXS studied the antiepileptic drug valproic acid (VPA). In an open-label trial of VPA, the only significant change was in hyperactivity measured by the Conner’s Parent Rating Scale [149]. While compounds like LAC and VPA may afford some benefit in treating the ADHD symptoms observed in FXS, stimulants are more commonly prescribed.
5.2 Melatonin
Insomnia is another concern associated with FXS with 32 % of youths with FXS experiencing some form of sleep trouble, in a parent/caregiver report. Awakening throughout the night and delayed falling asleep were the most reported issues [150]. In a four-week double blind, placebo-controlled trial of melatonin in FXS and ASD (12 subjects, 6 with FXS) treatment was found to significantly increase sleep duration, decrease latency to sleep (time elapsed from bedtime to falling asleep), and prompt earlier sleep onset (the clock time when the child fell asleep). Sleep awakenings were decreased but the change did not reach significance [151].
5.3 Oxytocin
Subjects with FXS experience strong, near debilitating social anxiety, which often presents with severe eye gaze avoidance and hyperarousal [152]. The effect of intranasal oxytocin on social anxiety was the subject of a double-blind, placebo-controlled trial in eight subjects with FXS. Outcome measures included heart rate, heart rate variability, eye gaze frequency, and concentration of salivary cortisol in response to a social stressor. Significant improvement was found on eye gaze and salivary cortisol measures [153]. A larger study is needed to adequately assess the benefits of oxytocin in FXS.
5.4 Aripiprazole
Atypical antipsychotics, such as aripiprazole, are commonly prescribed by clinicians to reduce irritability in FXS individuals. Despite their widespread use, there have been few studies looking at their efficacy in FXS. Among the newer generation antipsychotics, only aripiprazole has undergone clinical testing in FXS. In a 12-week, 10-subject open-label trial, aripiprazole monotherapy significantly improved performance on CGI-S, SRS, and the Children’s Yale-Brown Obsessive Compulsive Scale Modified for Pervasive Developmental Disorders (PDDs) [154].
5.5 Selective Serotonin Reuptake Inhibitors
While there have yet to be any clinical trials assessing the efficacy of selective serotonin reuptake inhibitors (SSRIs) in FXS syndrome, there is evidence suggesting they may have beneficial effects. In a case series of low-dose sertraline in ASD, eight of nine subjects demonstrated improvements in irritability, anxiety, and transition-induced behavioral deterioration [155]. A retrospective chart review in 45 children with FXS found that the 11 subjects taking sertraline showed improved language development [156]. In a survey of subjects with FXS, it was found that fluoxetine led to the greatest behavioral activation of any drug in its class [157]. Despite the lack of clinical trials in FXS, SSRIs are commonly prescribed for the management of anxiety.
6 Conclusions
Drug development in FXS has received a large amount of attention from both academic researchers (basic and clinician scientists) as well as large and small pharmaceutical companies. As a rare disorder, the Food and Drug Administration (FDA) as well as the European Medicines Agency (EMA) set the bar for demonstrating drug efficacy at a positive single Phase III clinical trial as opposed to two Phase III trials in disorders affecting larger populations. The primary endpoint, which is approved by the FDA or EMA and determined before the clinical trial begins, must be significantly improved in the active drug group compared to placebo. Despite this lowered barrier to entry, there has yet to be a single treatment medication approved to treat FXS.
In FXS clinical research, the primary endpoints have typically included the Aberrant Behavior Checklist (or sub-section scores), Clinical Global Impressions Improvement of Severity subscales (CGI-I or CGI-S), the Social Responsiveness Scale (SRS), or other parent-, teacher-, or clinician-rated scales. In developmental disability research, it is common to observe a 20–30 % placebo response on the above-mentioned subjective measures, which is often difficult to overcome even with a placebo lead-in phase during the trial. It is becoming apparent that the common outcome measures listed above are inadequate to track improvements in the FXS phenotype, resulting in a push to develop reliable, FXS-specific outcome measures [158]. The FXS-specific Fragile X Symptom Rating Scale is currently being validated in several clinical trials, including the Phase II trial of NNZ-2566 (clinicaltrials.gov NCT01894958). Another scale being validated for use in FXS is the Pediatric Anxiety Rating Scale revised for FXS [159]. The possibility of differential treatment response in various subpopulations defined by factors such as gender and IQ complicates the problem further.
Less subjective measures such as eye-tracking, pre-pulse inhibition, neuroimaging, evoked related potentials (ERP), EEG, and blood biomarkers are beginning to be assessed in early Phase II drug development as less subjective measures of a drug’s efficacy and potential engagement with the pathophysiology of the disorder. These types of measures have not been specified as ‘primary’ endpoints in large placebo-controlled trials in FXS. However, the inclusion of these types of assessments is important for discerning changes associated with drug treatment considering the high placebo response on standard parent/caregiver report rating scales. Ideally, as more data are gathered with respect to correlations between behavioral endpoints/severity and these less-subjective measures, the biomarker work will gain utility in aiding patient stratification in the future.
One question yet to be resolved in FXS treatment development is whether modulation of synaptic imbalance in the adult brain is sufficient to restore cognitive function in FXS. Signaling imbalances, which present developmentally, can cause the circuitry of the brain to form incorrectly. Though adult pharmacological intervention may be able to restore synaptic function, it is unlikely to rewire malformed circuits, possibly limiting the efficacy of treatment over the relatively short duration of a clinical trial. In the hopes of normalizing circuit formation and improving the long-term course of FXS, researchers are beginning to pharmacologically target signaling imbalances in younger children with FXS. The next wave of treatment trials in FXS will likely see a deviation from previous studies as researchers begin to use the information from past unsuccessful trials in an effort to ultimately optimize study design specifically for FXS participants.
References
Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4(4):335–40. doi:10.1038/ng0893-335.
Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047–58. doi:10.1016/0092-8674(91)90283-5 (pii:0092-8674(91)90283-5).
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66(4):817–22. doi:10.1016/0092-8674(91)90125-I (pii:0092-8674(91)90125-I).
Coffee B, Ikeda M, Budimirovic DB, Hjelm LN, Kaufmann WE, Warren ST. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: a case report and review of the literature. Am J Med Genet A. 2008;146A(10):1358–67. doi:10.1002/ajmg.a.32261.
Li Y, Jin P. RNA-mediated neurodegeneration in fragile X-associated tremor/ataxia syndrome. Brain Res. 2012;1462:112–7. doi:10.1016/j.brainres.2012.02.057.
Wang JM, Koldewyn K, Hashimoto R, Schneider A, Le L, Tassone F, et al. Male carriers of the FMR1 premutation show altered hippocampal-prefrontal function during memory encoding. Front Hum Neurosc. 2012;6:297. doi:10.3389/fnhum.2012.00297.
Berry-Kravis E, Hall DA. Executive dysfunction in young FMR1 premutation carriers: forme fruste of FXTAS or new phenotype? Neurology. 2011;77(7):612–3. doi:10.1212/WNL.0b013e3182299f98.
Sherman SL. Premature ovarian failure among fragile X premutation carriers: parent-of-origin effect? Am J Hum Genet. 2000;67(1):11–3. doi:10.1086/302985.
Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol. 2008;68(4):499–509. doi:10.1111/j.1365-2265.2007.03073.x.
Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72(4):869–78. doi:10.1086/374321.
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127–30.
Hunter JE, Rohr JK, Sherman SL. Co-occurring diagnoses among FMR1 premutation allele carriers. Clin Genet. 2010;77(4):374–81. doi:10.1111/j.1399-0004.2009.01317.x.
Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet. 1996;64(1):196–7. doi:10.1002/(SICI)1096-8628(19960712)64:1<196:AID-AJMG35>3.0.CO;2-G.
Song FJ, Barton P, Sleightholme V, Yao GL, Fry-Smith A. Screening for fragile X syndrome: a literature review and modelling study. Health Technol Assess. 2003;7(16):1–106. doi:10.3310/hta7160 (pii:01-32-01).
Bailey DB Jr, Raspa M, Olmsted M, Holiday DB. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A. 2008;146A(16):2060–9. doi:10.1002/ajmg.a.32439.
Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123(1):378–90 10.1542/peds.2008-0317.
Budimirovic DB, Kaufmann WE. What can we learn about autism from studying fragile X syndrome? Dev Neurosci. 2011;33(5):379–94. doi:10.1159/000330213.
Lozano R, Rosero CA, Hagerman RJ. Fragile X spectrum disorders. Intract Rare Dis Res. 2014;3(4):134–46. doi:10.5582/irdr.2014.01022.
Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16(6):666–72. doi:10.1038/ejhg.2008.61.
Tsiouris JA, Brown WT. Neuropsychiatric symptoms of fragile X syndrome: pathophysiology and pharmacotherapy. CNS Drugs. 2004;18(11):687–703 (pii:18111).
Pretto D, Yrigollen CM, Tang HT, Williamson J, Espinal G, Iwahashi CK, et al. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front Genet. 2014;5:318. doi:10.3389/fgene.2014.00318.
Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, et al. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A(3):225–34. doi:10.1002/ajmg.a.30229.
Budimirovic DB, Bukelis I, Cox C, Gray RM, Tierney E, Kaufmann WE. Autism spectrum disorder in Fragile X syndrome: differential contribution of adaptive socialization and social withdrawal. Am J Med Genet A. 2006;140A(17):1814–26. doi:10.1002/ajmg.a.31405.
Genc B, Muller-Hartmann H, Zeschnigk M, Deissler H, Schmitz B, Majewski F, et al. Methylation mosaicism of 5′-(CGG)(n)-3′ repeats in fragile X, premutation and normal individuals. Nucleic Acids Res. 2000;28(10):2141–52.
Gholizadeh S, Halder SK, Hampson DR. Expression of fragile X mental retardation protein in neurons and glia of the developing and adult mouse brain. Brain Res. 2015;1596:22–30. doi:10.1016/j.brainres.2014.11.023.
Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA. 1997;94(10):5395–400.
Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Ann Rev Pathol. 2012;7:219–45. doi:10.1146/annurev-pathol-011811-132457.
Sethna F, Moon C, Wang H. From FMRP function to potential therapies for fragile X syndrome. Neurochem Res. 2014;39(6):1016–31. doi:10.1007/s11064-013-1229-3.
Eberhart DE, Malter HE, Feng Y, Warren ST. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet. 1996;5(8):1083–91.
Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1(1):109–18.
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134(6):1042–54. doi:10.1016/j.cell.2008.07.031.
Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, et al. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nature neuroscience. 2007;10(5):578–87. doi:10.1038/nn1893.
Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell. 2008;14(6):926–39. doi:10.1016/j.devcel.2008.04.003.
Corbin F, Bouillon M, Fortin A, Morin S, Rousseau F, Khandjian EW. The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum Mol Genet. 1997;6(9):1465–72.
Hazlett HC, Poe MD, Lightbody AA, Styner M, MacFall JR, Reiss AL, et al. Trajectories of early brain volume development in fragile X syndrome and autism. J Am Acad Child Adolesc Psychiatry. 2012;51(9):921–33. doi:10.1016/j.jaac.2012.07.003.
He CX, Portera-Cailliau C. The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience. 2013;251:120–8. doi:10.1016/j.neuroscience.2012.03.049.
Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, et al. Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol. 1985;67(3–4):289–95.
Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41(3):289–94. doi:10.1002/ajmg.1320410306.
Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, et al. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci USA. 1997;94(10):5401–4.
Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99(11):7746–50. doi:10.1073/pnas.122205699.
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76(2):325–37. doi:10.1016/j.neuron.2012.07.022.
Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51(4):441–54. doi:10.1016/j.neuron.2006.07.005.
Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74(1):49–56. doi:10.1016/j.neuron.2012.03.009.
Auerbach BD, Bear MF. Loss of the fragile X mental retardation protein decouples metabotropic glutamate receptor dependent priming of long-term potentiation from protein synthesis. J Neurophysiol. 2010;104(2):1047–51. doi:10.1152/jn.00449.2010.
Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci. 2005;25(32):7385–92. doi:10.1523/JNEUROSCI.1520-05.2005.
Yun SH, Trommer BL. Fragile X mice: reduced long-term potentiation and N-Methyl-D-Aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res. 2011;89(2):176–82. doi:10.1002/jnr.22546.
Martin HG, Lassalle O, Brown JT, Manzoni OJ. Age-dependent long-term potentiation deficits in the prefrontal cortex of the fmr1 knockout mouse model of fragile X syndrome. Cereb Cortex. 2015. doi:10.1093/cercor/bhv031.
Gross C, Chang CW, Kelly SM, Bhattacharya A, McBride SM, Danielson SW, et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 2015;11(5):727–36. doi:10.1016/j.celrep.2015.03.060.
Gross C, Raj N, Molinaro G, Allen AG, Whyte AJ, Gibson JR, et al. Selective role of the catalytic PI3K subunit p110beta in impaired higher order cognition in fragile X syndrome. Cell Rep. 2015;11(5):681–8. doi:10.1016/j.celrep.2015.03.065.
Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, et al. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30(32):10624–38. doi:10.1523/JNEUROSCI.0402-10.2010.
Matic K, Eninger T, Bardoni B, Davidovic L, Macek B. Quantitative phosphoproteomics of murine Fmr1-KO cell lines provides new insights into FMRP-dependent signal transduction mechanisms. J Proteome Res. 2014;13(10):4388–97. doi:10.1021/pr5006372.
Curia G, Gualtieri F, Bartolomeo R, Vezzali R, Biagini G. Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front Cell Neurosci. 2013;7:46. doi:10.3389/fncel.2013.00046.
Kim SH, Markham JA, Weiler IJ, Greenough WT. Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci USA. 2008;105(11):4429–34. doi:10.1073/pnas.0800257105.
Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30(2):694–702. doi:10.1523/JNEUROSCI.3696-09.2010.
Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27(51):13958–67. doi:10.1523/JNEUROSCI.4383-07.2007.
Amiri A, Sanchez-Ortiz E, Cho W, Birnbaum SG, Xu J, McKay RM, et al. Analysis of FMR1 deletion in a subpopulation of post-mitotic neurons in mouse cortex and hippocampus. Autism Res. 2014;7(1):60–71. doi:10.1002/aur.1342.
McMillan EL, Kamps AL, Lake SS, Svendsen CN, Bhattacharyya A. Gene expression changes in the MAPK pathway in both Fragile X and Down syndrome human neural progenitor cells. Am J Stem Cells. 2012;1(2):154–62.
Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, et al. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J Neurochem. 2012;121(4):672–9. doi:10.1111/j.1471-4159.2012.07722.x.
Weng N, Weiler IJ, Sumis A, Berry-Kravis E, Greenough WT. Early-phase ERK activation as a biomarker for metabolic status in fragile X syndrome. Am J Med Genet Part B Neuropsychiatr Genet. 2008;147B(7):1253–7. doi:10.1002/ajmg.b.30765.
Kumari D, Bhattacharya A, Nadel J, Moulton K, Zeak NM, Glicksman A, et al. Identification of fragile X syndrome specific molecular markers in human fibroblasts: a useful model to test the efficacy of therapeutic drugs. Hum Mutat. 2014;35(12):1485–94. doi:10.1002/humu.22699.
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–7. doi:10.1016/j.tins.2004.04.009.
Brown V, Jin P, Ceman S, Darnell JC, O’Donnell WT, Tenenbaum SA, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–87.
Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25(35):8048–55. doi:10.1523/JNEUROSCI.1777-05.2005.
Karachot L, Shirai Y, Vigot R, Yamamori T, Ito M. Induction of long-term depression in cerebellar Purkinje cells requires a rapidly turned over protein. J Neurophysiol. 2001;86(1):280–9.
Zho WM, You JL, Huang CC, Hsu KS. The group I metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine induces a novel form of depotentiation in the CA1 region of the hippocampus. J Neurosci. 2002;22(20):8838–49.
Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–7.
Raymond CR, Thompson VL, Tate WP, Abraham WC. Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci. 2000;20(3):969–76.
Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49(7):1053–66. doi:10.1016/j.neuropharm.2005.06.004.
Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56(6):955–62. doi:10.1016/j.neuron.2007.12.001.
Thomas AM, Bui N, Graham D, Perkins JR, Yuva-Paylor LA, Paylor R. Genetic reduction of group 1 metabotropic glutamate receptors alters select behaviors in a mouse model for fragile X syndrome. Behav Brain Res. 2011;223(2):310–21. doi:10.1016/j.bbr.2011.04.049.
Montana MC, Cavallone LF, Stubbert KK, Stefanescu AD, Kharasch ED, Gereau RW 4th. The metabotropic glutamate receptor subtype 5 antagonist fenobam is analgesic and has improved in vivo selectivity compared with the prototypical antagonist 2-methyl-6-(phenylethynyl)-pyridine. J Pharmacol Exp Ther. 2009;330(3):834–43. doi:10.1124/jpet.109.154138.
Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, et al. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315(2):711–21. doi:10.1124/jpet.105.089839.
Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46(4):266–71.
Friedmann CTH, Davis LJ, Ciccone PE, Rubin RT. Phase-ii double-blind controlled-study of a new anxiolytic, fenobam (Mcn-3377) vs placebo. Curr Ther Res Clin E. 1980;27(2):144–51.
Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3(64):64ra1. doi:10.1126/scitranslmed.3001708.
Clapp K. Novartis discontinues development of mavoglurant (AFQ056) for Fragile X syndrome. http://www.fraxa.org/novartis-discontinues-development-mavoglurant-afq056-fragile-x-syndrome/. Accessed 5 Jan 2016.
Santarelli L. Roche letter. http://www.fraxa.org/pdf/RocheLetter.pdf. Accessed 5 Jan 2016.
Hu H, Qin Y, Bochorishvili G, Zhu Y, van Aelst L, Zhu JJ. Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J Neurosci. 2008;28(31):7847–62. doi:10.1523/JNEUROSCI.1496-08.2008.
Pilpel Y, Kolleker A, Berberich S, Ginger M, Frick A, Mientjes E, et al. Synaptic ionotropic glutamate receptors and plasticity are developmentally altered in the CA1 field of Fmr1 knockout mice. J Physiol. 2009;587(Pt 4):787–804. doi:10.1113/jphysiol.2008.160929.
Eadie BD, Cushman J, Kannangara TS, Fanselow MS, Christie BR. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus. 2012;22(2):241–54. doi:10.1002/hipo.20890.
Erickson CA, Mullett JE, McDougle CJ. Open-label memantine in fragile X syndrome. J Autism Dev Disord. 2009;39(12):1629–35. doi:10.1007/s10803-009-0807-3.
Berry-Kravis E, Krause SE, Block SS, Guter S, Wuu J, Leurgans S, et al. Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: a controlled trial. J Child Adolesc Psychopharmacol. 2006;16(5):525–40. doi:10.1089/cap.2006.16.525.
Olmos-Serrano JL, Paluszkiewicz SM, Martin BS, Kaufmann WE, Corbin JG, Huntsman MM. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J Neurosci. 2010;30(29):9929–38. doi:10.1523/JNEUROSCI.1714-10.2010.
El Idrissi A, Yan X, L’Amoreaux W, Brown WT, Dobkin C. Neuroendocrine alterations in the fragile X mouse. Results Probl Cell Differ. 2012;54:201–21. doi:10.1007/978-3-642-21649-7_11.
D’Hulst C, Heulens I, Brouwer JR, Willemsen R, De Geest N, Reeve SP, et al. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009;1253:176–83. doi:10.1016/j.brainres.2008.11.075.
D’Hulst C, De Geest N, Reeve SP, Van Dam D, De Deyn PP, Hassan BA, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121(1):238–45. doi:10.1016/j.brainres.2006.08.115.
Heulens I, D’Hulst C, Van Dam D, De Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229(1):244–9. doi:10.1016/j.bbr.2012.01.031.
Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, et al. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4(4):256–63. doi:10.1038/nchembio.78.
Zarate CA Jr, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry. 2004;161(1):171–4. doi:10.1176/appi.ajp.161.1.171.
Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17(6):761–7. doi:10.1089/cap.2007.0021.
Martin D, Thompson MA, Nadler JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250(3):473–6.
Jahn K, Schlesinger F, Jin LJ, Dengler R, Bufler J, Krampfl K. Molecular mechanisms of interaction between the neuroprotective substance riluzole and GABA(A)-receptors. Naunyn Schmiedeberg’s Arch Pharmacol. 2008;378(1):53–63. doi:10.1007/s00210-008-0290-y.
Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, et al. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA. 2004;101(50):17504–9. doi:10.1073/pnas.0407533101.
Erickson CA, Weng N, Weiler IJ, Greenough WT, Stigler KA, Wink LK, et al. Open-label riluzole in fragile X syndrome. Brain Res. 2011;1380:264–70. doi:10.1016/j.brainres.2010.10.108.
Reilly MT, Lobo IA, McCracken LM, Borghese CM, Gong D, Horishita T, et al. Effects of acamprosate on neuronal receptors and ion channels expressed in Xenopus oocytes. Alcohol Clin Exp Res. 2008;32(2):188–96. doi:10.1111/j.1530-0277.2007.00569.x.
Boismare F, Daoust M, Moore N, Saligaut C, Lhuintre JP, Chretien P, et al. A homotaurine derivative reduces the voluntary intake of ethanol by rats: are cerebral GABA receptors involved? Pharmacol Biochem Behav. 1984;21(5):787–9.
Harris BR, Prendergast MA, Gibson DA, Rogers DT, Blanchard JA, Holley RC, et al. Acamprosate inhibits the binding and neurotoxic effects of trans-ACPD, suggesting a novel site of action at metabotropic glutamate receptors. Alcohol Clin Exp Res. 2002;26(12):1779–93. doi:10.1097/01.ALC.0000042011.99580.98.
Mann K, Kiefer F, Spanagel R, Littleton J. Acamprosate: recent findings and future research directions. Alcoholism, clinical and experimental research. 2008;32(7):1105–10. doi:10.1111/j.1530-0277.2008.00690.x.
Erickson CA, Mullett JE, McDougle CJ. Brief report: acamprosate in fragile X syndrome. J Autism Dev Disord. 2010;40(11):1412–6. doi:10.1007/s10803-010-0988-9.
Erickson CA, Wink LK, Ray B, Early MC, Stiegelmeyer E, Mathieu-Frasier L, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl). 2013;228(1):75–84. doi:10.1007/s00213-013-3022-z.
Carter RB, Wood PL, Wieland S, Hawkinson JE, Belelli D, Lambert JJ, et al. Characterization of the anticonvulsant properties of ganaxolone (CCD 1042; 3α-hydroxy-3β-methyl-5α-pregnan-20-one), a selective, high-affinity, steroid modulator of the γ-aminobutyric acidA receptor. J Pharmacol Exp Therap. 1997;280(3):1284–95.
Heulens I, D’Hulst C, Van Dam D, De Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229(1):244–9.
Braat S, D’Hulst C, Heulens I, De Rubeis S, Mientjes E, Nelson DL, et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle. 2015;14(18):2985–95. doi:10.4161/15384101.2014.989114.
Kerrigan JF, Shields WD, Nelson TY, Bluestone DL, Dodson WE, Bourgeois BF, et al. Ganaxolone for treating intractable infantile spasms: a multicenter, open-label, add-on trial. Epilepsy Res. 2000;42(2):133–9.
Monaghan EP, Navalta LA, Shum L, Ashbrook DW, Lee DA. Initial human experience with ganaxolone, a neuroactive steroid with antiepileptic activity. Epilepsia. 1997;38(9):1026–31.
Fornai F, Grazia Alessandri M, Bonuccelli U, Scalori V, Corsini GU. Effect of metadoxine on striatal dopamine levels in C57 black mice. J Pharm Pharmacol. 1993;45(5):476–8.
Alcobra announces results from phase 2 clinical trial of mdx for fragile x syndrome. http://www.alcobra-pharma.com/releasedetail.cfm?ReleaseID=919218. Accessed 5 Jan 2016.
Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med. 2012;4(152):152ra28. doi:10.1126/scitranslmed.3004218.
Berry-Kravis EM, Hessl D, Rathmell B, Zarevics P, Cherubini M, Walton-Bowen K et al. Effects of STX209 (Arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4(152):152ra27. doi:10.1126/scitranslmed.3004214.
Berry-Kravis E HR, Visootsak J, Budimirovic D, Kaufmann W, Bear M, Walton-Bowen K, Carpenter R, Wang P. Arbaclofen in fragile X syndrome: results of phase 3 trials. Ann Neurol. 2014;76(S174).
Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29(4):293–302. doi:10.1097/DBP.0b013e31817dc447.
Liu ZH, Chuang DM, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol. 2011;14(5):618–30. doi:10.1017/S1461145710000520.
Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7(11):1421–34.
Portis S, Giunta B, Obregon D, Tan J. The role of glycogen synthase kinase-3 signaling in neurodevelopment and fragile X syndrome. Int J Physiol Pathophysiol Pharmacol. 2012;4(3):140–8.
Liu ZH, Huang T, Smith CB. Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol Dis. 2012;45(3):1145–52. doi:10.1016/j.nbd.2011.12.037.
Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–19. doi:10.1016/j.brainres.2010.11.032.
Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol. 2010;79(4):632–46. doi:10.1016/j.bcp.2009.09.023.
Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry. 2014;75(3):198–206. doi:10.1016/j.biopsych.2013.08.003.
Bilousova TV, Rusakov DA, Ethell DW, Ethell IM. Matrix metalloproteinase-7 disrupts dendritic spines in hippocampal neurons through NMDA receptor activation. J Neurochem. 2006;97(1):44–56. doi:10.1111/j.1471-4159.2006.03701.x.
Bilousova T, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, et al. Minocycline promotes dendritic spine maturation and improves behavioral performance in the fragile X mouse model. J Med Genet. 2008;. doi:10.1136/jmg.2008.061796.
Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell IM, et al. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10:91. doi:10.1186/1471-2377-10-91.
Leigh MJ, Nguyen DV, Mu Y, Winarni TI, Schneider A, Chechi T, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behavior Pediatr JDBP. 2013;34(3):147–55. doi:10.1097/DBP.0b013e318287cd17.
Schneider A, Leigh MJ, Adams P, Nanakul R, Chechi T, Olichney J, et al. Electrocortical changes associated with minocycline treatment in fragile X syndrome. J Psychopharmacol. 2013;27(10):956–63. doi:10.1177/0269881113494105.
Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24(20):4859–64. doi:10.1523/JNEUROSCI.5407-03.2004.
Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30(46):15616–27. doi:10.1523/JNEUROSCI.3888-10.2010.
Descamps OS, Tenoutasse S, Stephenne X, Gies I, Beauloye V, Lebrethon MC, et al. Management of familial hypercholesterolemia in children and young adults: consensus paper developed by a panel of lipidologists, cardiologists, paediatricians, nutritionists, gastroenterologists, general practitioners and a patient organization. Atherosclerosis. 2011;218(2):272–80. doi:10.1016/j.atherosclerosis.2011.06.016.
Cerezo-Guisado MI, Garcia-Roman N, Garcia-Marin LJ, Alvarez-Barrientos A, Bragado MJ, Lorenzo MJ. Lovastatin inhibits the extracellular-signal-regulated kinase pathway in immortalized rat brain neuroblasts. Biochem J. 2007;401(1):175–83. doi:10.1042/BJ20060731.
Xu XQ, McGuire TF, Blaskovich MA, Sebti SM, Romero G. Lovastatin inhibits the stimulation of mitogen-activated protein kinase by insulin in HIRcB fibroblasts. Arch Biochem Biophys. 1996;326(2):233–7. doi:10.1006/abbi.1996.0070.
Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77(2):243–50. doi:10.1016/j.neuron.2012.01.034.
Caku A, Pellerin D, Bouvier P, Riou E, Corbin F. Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am J Med Genet A. 2014;164A(11):2834–42. doi:10.1002/ajmg.a.36750.
Berry-Kravis E, Levin R, Shah H, Mathur S, Darnell JC, Ouyang B. Cholesterol levels in fragile X syndrome. Am J Med Genet A. 2015;167A(2):379–84. doi:10.1002/ajmg.a.36850.
Cartagena CM, Phillips KL, Williams GL, Konopko M, Tortella FC, Dave JR, et al. Mechanism of action for NNZ-2566 anti-inflammatory effects following PBBI involves upregulation of immunomodulator ATF3. Neuromol Med. 2013;15(3):504–14. doi:10.1007/s12017-013-8236-z.
Wei HH, Lu XC, Shear DA, Waghray A, Yao C, Tortella FC, et al. NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating ballistic-like brain injury in rats. J Neuroinflammation. 2009;6:19. doi:10.1186/1742-2094-6-19.
Lu XC, Chen RW, Yao C, Wei H, Yang X, Liao Z, et al. NNZ-2566, a glypromate analog, improves functional recovery and attenuates apoptosis and inflammation in a rat model of penetrating ballistic-type brain injury. J Neurotrauma. 2009;26(1):141–54. doi:10.1089/neu.2008.0629.
Pharmaceuticals N. NNZ-2566 in Rett Syndrome. http://www.neurenpharma.com/irm/content/nnz-2566-in-rett-syndrome.aspx?RID=330. Accessed 5 Jan 2016.
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–71. doi:10.1038/nature12618.
Kolevzon A, Bush L, Wang AT, Halpern D, Frank Y, Grodberg D, et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol Autism. 2014;5(1):54. doi:10.1186/2040-2392-5-54.
Bozdagi O, Tavassoli T, Buxbaum JD. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol Autism. 2013;4(1):9. doi:10.1186/2040-2392-4-9.
Phelan K, McDermid HE. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol Syndromol. 2012;2(3–5):186–201. doi:10.1159/000334260. (pii:000334260).
Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D, Dwivedi A, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet EJHG. 2013;21(3):310–6. doi:10.1038/ejhg.2012.175.
Deacon RM, Glass L, Snape M, Hurley MJ, Altimiras FJ, Biekofsky RR, et al. NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome. Neuromolecular medicine. 2015;17(1):71–82. doi:10.1007/s12017-015-8341-2.
Abitbol M, Menini C, Delezoide AL, Rhyner T, Vekemans M, Mallet J. Nucleus basalis magnocellularis and hippocampus are the major sites of FMR-1 expression in the human fetal brain. Nat Genet. 1993;4(2):147–53. doi:10.1038/ng0693-147.
Kesler SR, Lightbody AA, Reiss AL. Cholinergic dysfunction in fragile X syndrome and potential intervention: a preliminary 1H MRS study. Am J Med Genet A. 2009;149A(3):403–7. doi:10.1002/ajmg.a.32697.
D’Antuono M, Merlo D, Avoli M. Involvement of cholinergic and gabaergic systems in the fragile X knockout mice. Neuroscience. 2003;119(1):9–13.
Sahu JK, Gulati S, Sapra S, Arya R, Chauhan S, Chowdhury MR, et al. Effectiveness and safety of donepezil in boys with fragile x syndrome: a double-blind, randomized, controlled pilot study. J Child Neurol. 2013;28(5):570–5. doi:10.1177/0883073812449381.
Baumgardner TL, Reiss AL, Freund LS, Abrams MT. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics. 1995;95(5):744–52.
Hagerman RJ, Murphy MA, Wittenberger MD. A controlled trial of stimulant medication in children with the fragile X syndrome. Am J Med Genet. 1988;30(1–2):377–92.
Torrioli MGVS, Mariotti P, Bianchi E, Calvani M, De Gaetano A, Chiurazzi P, Neri G. Double-blind, placebo-controlled study of l-acetylcarnitine for the treatment of hyperactive behavior in fragile X syndrome. Am J Med Genet. 1999;87(4):4.
Torrioli M, Vernacotola S, Setini C, Bevilacqua F, Martinelli D, Snape M, et al. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am J Med Genet A. 2010;152A(6):1420–7. doi:10.1002/ajmg.a.33484.
Kronk R, Bishop EE, Raspa M, Bickel JO, Mandel DA, Bailey DB Jr. Prevalence, nature, and correlates of sleep problems among children with fragile X syndrome based on a large scale parent survey. Sleep. 2010;33(5):679–87.
Wirojanan J, Jacquemont S, Diaz R, Bacalman S, Anders TF, Hagerman RJ, et al. The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J Clin Sleep Med JCSM. 2009;5(2):145–50.
Reiss AL, Hall SS. Fragile X syndrome: assessment and treatment implications. Child Adolesc Psychiatr Clin N Am. 2007;16(3):663–75. doi:10.1016/j.chc.2007.03.001.
Hall SS, Lightbody AA, McCarthy BE, Parker KJ, Reiss AL. Effects of intranasal oxytocin on social anxiety in males with fragile X syndrome. Psychoneuroendocrinology. 2012;37(4):509–18. doi:10.1016/j.psyneuen.2011.07.020.
Erickson CA, Stigler KA, Posey DJ, McDougle CJ. Aripiprazole in autism spectrum disorders and fragile X syndrome. Neurotherapeutics. 2010;7(3):258–63. doi:10.1016/j.nurt.2010.04.001.
Steingard RJ, Zimnitzky B, DeMaso DR, Bauman ML, Bucci JP. Sertraline treatment of transition-associated anxiety and agitation in children with autistic disorder. J Child Adolesc Psychopharmacol. 1997;7(1):9–15.
Indah Winarni T, Chonchaiya W, Adams E, Au J, Mu Y, Rivera SM, et al. Sertraline may improve language developmental trajectory in young children with fragile x syndrome: a retrospective chart review. Autism Res Treatment. 2012;2012:104317. doi:10.1155/2012/104317.
Hagerman RJ, Fulton MJ, Leaman A, Riddle J, Hagerman K, Sobesky W. A Survey of fluoxetine therapy in fragile-X syndrome. Dev Brain Dysfunct. 1994;7(2–3):155–64.
Berry-Kravis E, Hessl D, Abbeduto L, Reiss AL, Beckel-Mitchener A, Urv TK, et al. Outcome measures for clinical trials in fragile X syndrome. J Dev Behav Pediatr JDBP. 2013;34(7):508–22. doi:10.1097/DBP.0b013e31829d1f20.
Russo-Ponsaran NM, Yesensky J, Hessl D, Berry-Kravis E. Feasibility, reproducibility, and clinical validity of the pediatric anxiety rating scale-revised for fragile X syndrome. Am J Intellect Develop Disabil. 2014;119(1):1–16. doi:10.1352/1944-7558-119.1.1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding sources were used to assist with the preparation of this review.
Conflicts of interest
Dr. Schaefer receives support from Confluence Pharmaceuticals and FRAXA Research Foundation. Dr. Erickson receives support from Alcobra Pharma, American Academy of Child and Adolescent Psychiatry, Autism Speaks, Cincinnati Children’s Hospital Medical Center, FRAXA Research Foundation, Hoffmann-La Roche Inc., Neuren Pharmaceuticals Limited, Simons Foundation Autism Research Initiative, Stemina Biomarker Discovery, Inc., SynapDx, The John Merck Fund, the National Fragile X Foundation, and the US Department of Defense. Dr. Erickson is currently a consultant for Confluence Pharmaceuticals and Neurotrope and has stock or equity in Confleunce Pharmaceuticals. In addition, he was a consultant for Novartis Pharmaceutical Corporation, the Roche Group, and Alcobra Pharmaceuticals in the past. All other authors (Matthew Davenport, Katherine Friedman and Sarah Fitzpatrick) declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Davenport, M.H., Schaefer, T.L., Friedmann, K.J. et al. Pharmacotherapy for Fragile X Syndrome: Progress to Date. Drugs 76, 431–445 (2016). https://doi.org/10.1007/s40265-016-0542-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0542-y