
Abstract
The mitogen-activated protein kinase kinases (MAPKK) MEK1 and MEK2 are integral members of the MAPK/ERK signaling pathway and are of interest in the development of anti-cancer therapeutics. The MAPK/ERK pathway is dysregulated in more than 30 % of cancers, predominately by mutations in RAS and BRAF proteins, and MEK serves as a potential downstream target for both of these. The biology of MEK inhibition is complex, as the molecule is differentially regulated by upstream RAS or RAF. This has impacted on the past development of MEK inhibitors as treatments for cancer and may be exploited in more rational, molecularly selected drug development plans in the future. The role of MEK in cancer and the mechanism of action of MEK inhibitors is reviewed. Furthermore, MEK inhibitors that are available in standard practice, as well as those most advanced in clinical development, are discussed. Finally, next steps in the development of MEK inhibitors are considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
MEK1 and MEK2 are integral elements of the mitogen-activated protein kinase (MAPK)/ERK pathway and are important molecules for targeted therapy drug development in cancer. |
The activity of MEK inhibitors is differentially regulated by upstream activation of RAS as opposed to BRAF, and the activity of MEK inhibitors will be structure dependent. |
Trametinib was the first MEK1/2 inhibitor approved for cancer therapy; however, several other MEK inhibitors have demonstrated interesting activity as single agents as well as in combination with other cancer therapies. |
1 Introduction
With the development of clinical sequencing platforms for cancer genotyping, the role for oncogene-targeted therapies has grown rapidly. Mutations in multiple growth pathways have been identified and there is a growing list of small molecules that have been successfully developed as targeted therapies for cancer. Here we focus on the role of MEK in cancer biology and review agents in clinical development that target MEK. We further discuss the benefits and downsides of combination strategies involving the inhibition of MEK.
2 Role of MEK in Cancer
The mitogen-activated protein kinase (MAPK) pathway plays a critical role in multiple cellular functions and is constitutively activated in a wide range of human cancers. Canonical MAPK signaling results from activation of receptor tyrosine kinases (RTKs) on the cell surface such as the epidermal growth factor receptor, HER kinases, MET and the fibroblast-growth factor receptor [1]. Upon activation of an RTK by a mitogen (growth factors, cytokines, etc.), RTKs generally undergo dimerization and transphosphorylation, leading to their activation. Thereafter, small adaptor proteins, such as SHC, son of sevenless (SOS), and GRB, associate with activated RTKs recruiting guanine nucleotide exchange factors (GEF). At the cell membrane, these GEFs activate RAS GTP-ases (H-, K-, N-RAS) by promoting the exchange of GDP for GTP by RAS [2, 3]. When GTP is bound, RAS-GTP triggers activation signals down multiple growth pathways including MAPK as well as phosphoinositide 3-kinase (PI3K)/AKT and RalGEF-Ral [4, 5]. RAS activity is regulated under physiologic conditions due to an inherent low-level GTP-ase activity of the RAS protein that facilitates a normal exchange of GTP to GDP, eventually extinguishing signaling by RAS GTP.
After RAS activation, RAF kinases (A- B-, C-RAF [RAF-1]) are recruited to the cell membrane in association with RAS [6, 7], are differentially phosphorylated [8], and undergo dissociation from RAF kinase inhibitory protein [9]. RAFs are activated via phosphorylation of homo- or heterodimer RAF complexes [6], and then act as serine/threonine MAPK kinase kinases (MAPKKK), directly activating MAPKK (MEK1/MEK2) [10]. Among RAF isoforms, B-RAF has been associated with a much higher potency for MEK phosphorylation relative to other isoforms. Other MAPKKK proteins also exist and can modulate MEK activity via phosphorylation. Notable examples of this include MAP3K8 (COT/Tpl2) and PAK1, which have both been implicated in cancer processes [11].
After MEK activation, MEK acts as an MAPKK via dual-specificity tyrosine and serine/threonine kinase activity on MAPK (ERK1/ERK2) [12]. ERKs are the only known phosphorylation targets of MEKs. The consequences of ERK activation by different MEK isoforms (e.g. MEK1 vs. MEK2) are under investigation; however, potential differences are not yet clearly relevant in the development of MEK inhibitors for cancer therapy. Phosphorylated MAPK/ERK proteins function as serine/threonine kinases, activating many cytoplasmic and nuclear mediators such as kinases, phosphatases, and transcription factors. The activity of signal transduction via RAF-MEK-ERK, and eventual downstream ERK output, is dependent on scaffold proteins notably including the IQGAP family. Of the three IQGAP members, IQGAP1 is understood the best; more than 90 partner molecules have been found that associate with IQGAP1, including many oncogenic proteins [13]. In the context of scaffold support, activated ERK then facilitates multiple cellular processes such as cell proliferation, motility, differentiation, and survival. Examples of downstream ERK targets include cell cycle proteins such as cyclin D, transcription factors such as MYC and FOS, as well as the regulatory protein families SPRY and DUSP, which mediate negative feedback on the MAPK pathway [14, 15].
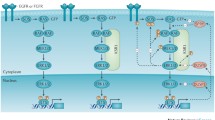
While canonical signaling through the MAPK pathway appears uni-linear at first glance, feedback regulation of the pathway and cross-talk between MAPK proteins and other pathways, such as PI3K, c-Jun N-terminal kinase (JNK), nuclear factor (NF)-κB, p38, and others, complicate attempts to develop inhibitors of this pathway as anti-cancer agents [16, 17]. Feedback regulation of the MAPK pathway has recently gained interest due to the development of inhibitors of oncogenic MAPK component proteins such as V600E BRAF. After suppression of oncogenic BRAF, a rebound of ERK activity eventually results in resistance to upstream BRAF inhibition. This has been attributed to relief of BRAF inhibitor-induced physiologic ERK-dependent feedback via SPRY and DUSP activity, reactivation of RTK by ligand binding, increased Ras-GTP, and generation of physiologic RAF dimers that are insensitive to selective V600E BRAF inhibitors [18]. Similar feedback, as well as cross-talk with other pathways, may influence the ability of MEK inhibitors to effectively inhibit their targets as well (Fig. 1).

Inhibition of MEK with a small-molecule inhibitor leads to induction of a feedback loop, via SPRY and DUSP proteins, and reactivation of the MAPK pathway. RAS mutant (a) but not BRAF mutant (b) tumors exhibit a relative abundance of wild-type RAF proteins. Thus, in RAS mutant tumors, MEK inhibitor-induced feedback mediates activation of wild-type RAF proteins (predominately CRAF) through the formation of CRAF–MEK complexes. This results in feedback phosphorylation of MEK and an eventual increase in downstream MAPK pathway activation. BRAF mutant tumors exhibit a relative lack of wild-type RAF proteins, attenuating feedback-induced ERK reactivation. BRAF mt mutant BRAF, RAS mt mutant RAS, RTK receptor tyrosine kinase
The importance of the MAPK pathway is emphasized by the recurrent mutations found in RTKs that activate this pathway as well as the specific signaling components that make it up. The MAPK pathway is activated in at least 30 % of human cancers through activating mutations in RAS or BRAF [19]. Of these, aberrant RAS activity plays a major role, with RAS mutations being commonly associated with carcinomas of the pancreas (63–90 %), colon (36–50 %), lung (19–30 %), and melanoma (15–20 %) [20, 21]. Mutations in RAS are frequently found in codons 12, 13, and 61, with variability by RAS isoform and cancer histology [22]. KRAS mutations occur at codon 12 at a frequency of approximately 80 %, while many fewer mutations occur in codon 61. This contrasts with NRAS, where codon 61 is mutated in 60 % of cases, while less than 35 % are mutated at codon 12. Mutation frequency in HRAS is less polarized, with a nearly 50 % split between codons 12 and 61, though the incidence of HRAS mutation in human cancers is much less frequent than K- and NRAS [21]. The association of some of these mutations with specific cancer types has been determined to be due to particular genetic insults. For example, ionizing radiation skews toward RAS Q61 mutations via the formation of pyrimidine dimers, as seen in cutaneous melanoma [23], whereas chemical carcinogenesis, by tobacco smoking, leads to the development of the DNA adducts and bias toward RAS G12 mutations, as commonly seen in non-small-cell lung cancer (NSCLC) [24].
BRAF is the most frequently mutated protein kinase in human malignancies [25]. Mutations of BRAF have been characterized as approximately 50 % of driver mutations in cutaneous melanoma [25, 26], and several other tumor types such as 10–15 % of colorectal carcinomas [25], 3 % of NSCLC [27], 3 % of breast cancers [28], 20–50 % of serous ovarian cancers [29–31], and 29–69 % of papillary thyroid cancers [32, 33]. The presence of mutations in BRAF has also been correlated with an unfavorable prognosis [34, 35]. Why BRAF, as opposed to ARAF or CRAF, is recurrently mutated is not clear. However, structural analysis of BRAF suggests that a single mutation in V600 can introduce a stable mutated catalytic site of the BRAF protein, whereas multiple mutations would be predicted to be required for a stable mutated form of ARAF or CRAF [36, 37].
Oncogenic mutations in MEK are much less frequent than RAS or BRAF mutations; ERK mutations have not been described. In NSCLC, mutations in MEK1 (Q56P, K57N, D67N) have been described as constituting less than 1 % of mutations [38]. In melanoma, mutations in MEK1 and MEK2 have been described in up to 8 % of patient samples [39], and appear to co-exist with mutations in RAS and BRAF in some cases [40]. In the setting of anti-cancer treatment directed toward BRAF, MEK mutations as secondary resistance mechanisms have also been observed. After treatment with a selective BRAF inhibitor in V600E BRAF cell lines and patient samples, mutations in MEK1 C121S, F129L, and P124L as well as MEK2 C125S have conferred resistance to inhibitors of both BRAF and MEK [41–44]. Similarly, after a combination strategy of BRAF and MEK inhibitors, a MEK2 Q60P mutation has been observed in a post-treatment patient sample [45].
3 Mechanism of Action of MEK Inhibitors
MEK, the MAPK kinase, is a dual-specific tyrosine and serine/threonine kinase, with ERK being the only known phosphorylation target. Regulation of MEK is via phosphorylation by RAF on serine residues in the catalytic domain at codons 217 and 221 [46]. Stoichiometric analysis of phosphorylation on MEK by RAF confirms that RAF facilitates phosphorylation at both sites with rapid kinetics, ruling out intrinsic autophosphorylation of MEK protein. All RAFs are able to phosphorylate MEKs; however, the degree of MEK activation that results is variable: BRAF is the most potent, followed by CRAF and ARAF [10].
RAF-mediated activation of MEK is differentially regulated in KRAS and BRAF mutant molecular environments, as illustrated by the differential anti-proliferative effects of various MEK inhibitors and especially in the variable activity of the small-molecule MEK inhibitors that have been brought into clinic to date [47–50]. This difference may be partly understood by dissecting the feedback induced via inhibition of the MAPK pathway at different levels (Fig. 1). In the setting of MEK inhibition of KRAS mutant cells, release of feedback on upstream RTK by SPRY and DUSP leads to physiologic signaling through MAPK (due to abundant wild-type RAF), resulting in an increase of phosphorylated MEK [50, 51]. This is in contrast to V600 BRAF mutant cells where the low level of wild-type RAF does not lead to increased MEK phosphorylation, thus allowing persistent suppression of MEK [49, 51]. In KRAS mutant cells, BRAF-CRAF heterodimers have been described as the predominant RAF complexes as depletion of B- or ARAF, but not CRAF, has been linked to MEK phosphorylation after MEK inhibitor treatment, leading to downstream ERK phosphorylation and reactivation of the MAPK pathway with an impact on the anti-neoplastic effect [46, 52].
In this context, the mechanism of MEK inhibition by different small molecules has the potential to determine impact the anti-neoplastic effect (Fig. 2). Two mechanisms for MEK inhibition then become obvious, one being inhibition of RAF phosphorylation activity of MEK or an allosteric association of an inhibitor with MEK impeding the conformational shift induced by phosphorylation on serine 217 and 221. Some data to support such models exist in which MEK inhibitors have been described to directly inhibit RAF phosphorylation of MEK, [49, 53] though the exact mechanism has been unclear.

In KRAS mutant tumors, the MEK inhibitor PD0325901 leads to formation of CRAF–MEK complexes, resulting in MEK phosphorylation and reactivation of the MAPK pathway. Trametinib induces formation of CRAF–MEK complexes, but inhibits MEK phosphorylation by CRAF (by inducing a conformational change in MEK). CH5126766 does not induce relevant CRAF/MEK interaction (by increasing dissociation of MEK from CRAF), thereby preventing phosphorylation of MEK. As a result, both trametinib and CH5126766 lead to more sustained ERK phosphorylation relative to older-generation MEK inhibitors such as PD0325901
In a series of elegant modeling and functional experiments, Hatzivassiliou et al. [46] have described a mechanism for differential potency of different MEK inhibitors. Using crystal structures and serine mutagenesis of MEK, they demonstrated that formation of a strong hydrogen bond by higher potency MEK inhibitors to serine 212 of MEK disrupts the hydrogen-bond network between the serine 212 side chain, the activation loop helix, and the glutamate 114 of the α-C helix of the MEK protein. This prevents access of RAF to serine 217 of MEK, decreases phosphorylation, and stabilizes the RAF-MEK complex. RAF-MEK complexes are specific to wild-type RAF as V600 BRAF signals downstream as a monomer [54]. As such, this RAF-MEK stabilization has a more powerful anti-neoplastic effect in KRAS mutant models. This is consistent with previous reports, which have also described an interaction of MEK inhibitors with serine 212 [55, 56]. In V600E BRAF models, a correlation was observed between high MEK inhibitor-binding affinity and MEK inhibition due to the high basal MEK activity in V600 BRAF cancers [46]. This high binding affinity was described as secondary to interactions between a high-affinity binding MEK inhibitor and the α-C helix and catalytic loop of MEK. These data are also consistent with previous experiments demonstrating the differential activation of MEK by V600 BRAF versus wild-type RAF in (B- and CRAF) in KRAS mutant models [57, 58]. These experiments suggest that the chemical structure of different MEK inhibitors could be rationally tailored in their clinical benefit as anti-neoplastic activity could be predicted prior to dosing of human subjects based on BRAF or KRAS mutation.
The activity of MEK inhibitors in the setting of mutant RAS has been further elucidated by Lito et al. [59] in the specific setting of KRAS mutant tumors. By administering the MEK inhibitors PD0325901, selumetinib, and refametinib in conjunction with a library of doxycycline-inducible short hairpin RNAs against components of RAS effector pathways, they identified CRAF as the primary mediator of MEK activation in KRAS mutant tumors. Further, after blockade of ERK signaling, as by MEK inhibitor treatment, CRAF was observed to robustly reactivate and mediate rebound in ERK phosphorylation in KRAS as compared with V600E BRAF mutant cells. Using an engineered FLAG-MEK1, they found that treatment with allosteric MEK inhibitors in the setting of RAS mutant cell lines leads to formation of RAF-MEK complexes (predominately CRAF-MEK), whereas this is not the case in BRAF mutant cells. This RAF-MEK complex is then less sensitive to MEK inhibition in terms of reduction of phosphorylated ERK as compared with the free MEK observed in the BRAF mutation setting. Thus, MEK inhibitors activate CRAF by relieving ERK-dependent negative feedback and cause the formation of CRAF-MEK complexes, leading to attenuation of MEK inhibition in KRAS mutant tumors.
Thereafter, Lito et al. [59] show that the efficacy of MEK inhibition in the RAS mutation setting can be modulated by two approaches that are exemplified by newer MEK inhibitors: trametinib and CH5126766, a novel MEK inhibitor. Using surface plasmon resonance to define the dissociation constant of MEK1 with B- or CRAF, they show that trametinib reduces the formation of RAF-MEK complexes. This is despite a proposed similar mechanism of action of trametinib to the older MEK inhibitor PD0325901 in inhibiting the catalytic activity of MEK. Interruption of RAF-MEK complexes then leads to reduction in phosphorylated ERK rebound and prevention of MAPK reactivation. In contrast, CH5126766 does not inhibit the catalytic site of MEK and promotes RAF-MEK complexes, yet also inhibits ERK rebound. Using a structure guided approach, Lito et al. [59] demonstrated that CH5126766 interacts with serine 222 and asparagine 221 on MEK. This prevents MEK phosphorylation by CRAF, leading to MEK inhibitor-bound, dephosphorylated MEK binding to RAF and a subsequent increase of RAF-MEK complexes. Notably, this model of CH5126766 activity is, to some extent, at odds with the model proposed by Hatzivassiliou et al. [46]. The different findings are potentially explained by the fact that Lito et al. [59] completely solved the ternary structure of CH5126766-bound MEK1, as opposed to a limited structure used previously, showing that CH5126766 forms interactions at multiple positions beyond serine 212 of MEK and displaces the activation segment of MEK1. Nevertheless, both of these models will likely be useful in the future development of more potent MEK inhibitors.
4 Approved and Investigational MEK Inhibitors of Significance in Clinical Development
The first generation of MEK1/2 inhibitors, such as CI-1040, demonstrated poor exposure in human subjects, limiting their utility [60]. Second-generation inhibitors, such as PD325901, overcame this problem, but had a poor safety profile, with dose-limiting toxicities such as neurological and eye toxicities [61]. Subsequent generations of MEK inhibitors are now being investigated and appear to be more potent and more tolerable. The agents that are furthest along in clinical development are compared in Table 1 and reviewed here.
4.1 Trametinib Dimethyl Sulfoxide (GSK1120212B, JTP-74057)
4.1.1 Preclinical Data
Trametinib dimethyl sulfoxide (trametinib) is an allosteric, non-ATP competitive inhibitor of both MEK isoforms (half maximal inhibitory concentration [IC50] of MEK1: 0.7, MEK2: 0.9 ηM) [49]. Trametinib inhibits phosphorylation by RAF on serine 217 of MEK1 and suppresses phosphorylated ERK. The MEK kinase-specific suppressive activity of trametinib has been broadly validated across a panel of kinases [62]. Trametinib has also shown differential activity against BRAF and NRAS mutants, as compared with wild-type, cell lines and xenografts [62, 63].
4.1.2 Pharmacokinetics and Drug Metabolism
Dose escalation of trametinib resulted in a maximum tolerated dose of 3 mg continuous daily dosing and a recommended phase II dose (RP2D) of 2 mg daily [63]. Fasting pharmacodynamic monitoring demonstrated dose proportionality up to 6 mg daily, with an effective half-life of approximately 4.5 days. Steady state drug levels were reached by day 15 of dosing. At 2 mg daily, on day 15, the mean area under the curve (AUC) over 24 h was 376 ng·h/mL and maximum concentration (C max) was 23 ng/mL. The trough concentrations ranged from a mean of 10.0 to 18.9 ng/mL. Trametinib exhibits non-cytochrome P450 (CYP)-mediated metabolism, primarily via deacetylation and glucuronidation.
4.1.3 Clinical Experience
In a phase I study of 206 patients with advanced solid tumors, a response rate (RR) for trametinib was determined as 10 % [63]. Within a melanoma-only cohort of the study, 97 patients were treated. These included V600E/K BRAF (total of 36 patients, 30 BRAF inhibitor naive), wild-type BRAF (39 patients), BRAF status unknown (six patients), and uveal (16 patients) [64]. An RR of 33 % was determined for BRAF inhibitor-naive patients with median progression-free survival (PFS) of 5.7 months. In BRAF wild-type patients, the RR was 10 %; no responses were observed in patients with uveal melanoma.
The phase III METRIC study subsequently evaluated trametinib as compared with dacarbazine (DTIC) or paclitaxel in patients with melanoma harboring a V600E/K BRAF mutation [65]. A total of 322 patients were randomized in a two-to-one ratio favoring trametinib. Cross-over to trametinib was allowed at progression of disease. A median PFS of 4.8 months was determined in the intention-to-treat population of the trametinib-treated arm versus 1.5 months (p < 0.001) for the DTIC- or paclitaxel-treated arm. An early suggestion of improvement in survival with trametinib as compared with chemotherapy was also noted, with an immature hazard ratio (HR) for death of 0.54 (95 % confidence interval [CI] 0.32–0.92, p = 0.01) despite cross-over of nearly half of the study population.
Combination therapy with inhibitors of BRAF and MEK via dabrafenib and trametinib has also been explored. In a phase I/II study, doses of 150 mg twice daily of dabrafenib and 2 mg daily of trametinib were found to be the RP2D. Only patients with V600E/K BRAF-mutant melanoma were enrolled, and the phase II portion of the study evaluated doses of 150 and 2 mg (dabrafenib/trametinib) as well as 150 and 1 mg compared with dabrafenib 150 mg twice daily. An improvement in both RR (76 vs. 54 %, p = 0.03.) and PFS (9.4 vs. 5.8 months, p < 0.001) was observed when dabrafenib and trametinib (150/2) were compared with dabrafenib monotherapy [66]. Additionally, fewer adverse events for the combination were observed when compared with dabrafenib alone, including the development of squamous cell carcinoma (7 vs. 19 %). Of note, pyrexia (71 vs. 26 %) was increased with the combination. Based on this, the drug combination was approved by the US FDA in 2014.
Data from the phase III clinical trial of dabrafenib and trametinib combination therapy versus dabrafenib and placebo are eagerly awaited. In January 2014, a communication from the drug manufacturer GlaxoSmithKline revealed that the primary endpoint of PFS had been met. The median PFS was described as 9.3 months in the dabrafenib and trametinib arm as compared with 8.8 months in the dabrafenib arm (HR 0.75, p = 0.035). The combination therapy was described to have an RR of 67 versus 51 % for dabrafenib. Toxicity was similar to the previously described phase II results. Data on overall survival (OS) were not yet mature [67].
4.1.4 Safety Profile
Common toxicities for trametinib were similar across studies, with the dominant events in skin (rash), gastrointestinal (diarrhea), general (edema), and constitutional (fatigue) systems [63–65]. Dose-limiting toxicities were similar but were also observed to include central serous retinopathy. At doses of 2 mg, skin toxicity was observed at a rate of 48–91 % across studies and diarrhea with an incidence of 28–58 %. Other notable toxicities at 2 mg included cardiac toxicity (ejection fraction reduction or ventricular dysfunction in 3–21 % of patients), ocular toxicity (blurred vision, reversible chorioretinopathy or retinal vein occlusion in 6–21 % of patients), hepatitis (7–15 % of patients), and pneumonitis (<5 %).
4.2 Selumetinib (AZD6244, ARRY-142886)
4.2.1 Preclinical Data
Selumetinib is a selective, ATP non-competitive inhibitor of MEK1 and MEK2 at an IC50 of approximately 10–14 ηM [50, 68]. Selumetinib is metabolized to N-desmethyl AZD6244, a compound with greater MEK-inhibiting capacity, three times greater downstream ERK suppression and three times greater reduction of cell viability relative to the parent compound. The selectivity of selumetinib for MEK has been documented against a panel of kinases up to concentrations of 10 µM [68]. Selumetinib has anti-tumor activity in BRAF and NRAS mutant preclinical models including cell lines and xenografts from melanoma, pancreatic, lung, colon, breast, and hepatocellular carcinoma [50, 68, 69].
4.2.2 Pharmacokinetics and Drug Metabolism
The initial formulation of selumetinib was an oral suspension requiring reconstitution from free base in an aqueous solution of sulphobutylether β-cyclodextrin (Captisol®) [70]. Despite pharmacological animal studies suggesting good oral bioavailability, absorption was dose limited and drug administration was inconvenient. The drug was therefore reformulated as the hydrogen sulfate salt (AZD6244 Hyd-Sulfate). Subsequent studies suggest dose-proportional bioavailability [71]. Steady-state pharmacokinetic evaluation suggests a C max of approximately 1 h, a half-life of 12 h, and time-independent kinetics based on comparison of day 1 and day 15 drug levels. Excretion of selumetinib is predominantly via the feces, with the majority of metabolites undergoing glucuronidation. Selumetinib is metabolized by CYP, with predominant activity by CYP1A2, 2C19, and 3A4 [72].
4.2.3 Clinical Experience
Phase I experience with selumetinib confirmed RP2D of 100 mg twice daily on a 28-day administration schedule [71]. Selumetinib was then evaluated in a wide range of unselected but MAPK-activated enriched tumors including biliary, colorectal, pancreatic, NSCLC, and melanoma. In patients with melanoma, a similar RR (5.8 %) and PFS (2.5 months) were observed for selumetinib as compared with temozolomide [73]. In patients with BRAF mutant tumors, the RR was 11.1 %, which is not different from that for temozolomide. In patients with pancreatic cancer, OS in the second line after gemcitabine with selumetinib or capcitabine was found to be similar (5.4 months) [74]. In patients with NSCLC who had progressed through at least two prior lines of treatment, selumetinib appeared similar to pemetrexed in terms of median PFS (3 months) [75]. Similarly, in colorectal cancer, PFS of selumetinib was similar to capecitabine (2.8 months) [76]. In patients with biliary tract cancers who had progressed on one line of prior chemotherapy, a single-arm phase II study suggested a RR of 12 % and PFS of 3.7 months [77]. In previously untreated patients with advanced hepatocellular carcinoma, median time to progression was 8 weeks; three transient objective tumor responses were observed [78]. In an open-label phase II study of patients with low-grade serous carcinoma of the ovary, a disease commonly characterized by mutations in BRAF, an RR of 15 % was observed. This was considered useful when compared with the historical standard of care with chemotherapy where responses are very rare [79].
The clinical activity of selumetinib was more impressive when patients were selected based on mutational status of KRAS or BRAF. In melanoma, trials including patients with both BRAF V600E and BRAF wild-type tumors in particular have been pursued. In V600E BRAF patients, a phase II combination of DTIC with and without selumetinib reported a significant improvement in the PFS of patients treated with selumetinib as opposed to placebo: 5.6 months versus 3.0 months (p = 0.021), respectively. No improvement in OS was seen, and toxicity consistent with both agents was observed [80]. In a phase II study of selumetinib in BRAF V600E melanoma stratified by phosphorylated AKT (high vs. low), no responses were seen in the AKT high cohort, whereas a 20 % RR was observed in the AKT low cohort [81]. In BRAF wild-type melanoma, the combination of docetaxel with selumetinib had a similar PFS to docetaxel alone (4 months). The RR for the combination was higher, but toxicity was also increased [82]. In uveal melanoma, a chemotherapy-resistant melanoma subtype where mutations in the G-coupled protein receptors GNAQ and GNA11 are found in 85 % of tumors eventually leading to MAPK activation [83], tumor regression and RR were found to be 50 and 15 %, respectively [84]. In KRAS mutant NSCLC, the combination of docetaxel and selumetinib showed a significantly improved RR (37 vs. 0 %, p < 0.0001) and PFS (5.3 vs. 2.1 months, p = 0.014). OS was not improved, and toxicity was greater with the combination treatment [85].
Finally, selumetinib appears to influence the biology of thyroid cancer. In iodine-refractory papillary thyroid cancer, a median PFS of 32 weeks was observed. In patients with tumors harboringV600E BRAF mutation, PFS was longer (33 vs. 11 weeks) [86]. Further, a phase II study was performed in which selumetinib was administered with the intent of reversing refractory status to radioiodine in patients with metastatic thyroid cancer [87]. In four of nine patients with BRAF mutant tumors and five of five patients with NRAS mutant tumors, treatment with selumetinib led to increased uptake of iodine-124. In 8 of 12 patients, the dosimetry threshold for radioiodine therapy was reached and, in these eight patients, five patients experienced partial response (PR) and three patients experienced stable disease (SD). All of these patients demonstrated decreased levels of serum thyroglobulin levels, with a mean reduction of 89 %.
4.2.4 Safety Profile
The most common adverse events reported in human trials of selumetinib include fatigue (71 %), nausea (54 %), vomiting (27 %), diarrhea (48.2), edema (43 %), and dyspnea (29 %) [71]. Serious adverse events associated with selumetinib have notably included hypoxia (<1 %), pyrexia (<1 %), and visual disturbances (including serous retinal detachment, 10 %).
4.3 Cobimetinib (GDC-0973, XL518)
4.3.1 Preclinical Data
Cobimetinib is a potent and highly selective allosteric inhibitor of MEK1/2, with a MEK1 IC50 of 4.2 ηM [88]. Efficacy in vivo has been demonstrated in BRAF and KRAS mutant cell lines [88, 89]. Cobimetinib showed inhibition of tumor growth in a dose-dependent fashion up to 10 mg/kg in Colo205 (V600E BRAF) tumor xenografts. Pharmacodynamic parameters estimated the 50 % knock-down concentration (KC50) as 0.39 μM. Total concentration required for tumor stasis was 0.05 μM [90]. An estimated IC50 after single doses of cobimetinib of phosphorylated ERK in xenograft mice were 0.78 and 0.52 μmol/L in tumor models WM-266-4 and A375, respectively. Multiple dosing increased the IC50 [91].
4.3.2 Pharmacokinetics and Drug Metabolism
Cobimetinib has exhibited variable absorption in cancer patients. Absolute bioavailability of cobimetinib is described as 46.2 %, with mean clearance of 11.7 L/h. The half-life of cobimetinib is described as demonstrating dose-proportional and time-independent pharmacokinetics, with a half-life of 2.0 days [92]. A high-fat diet delays absorption with prolonged time to C max (T max) but does not affect exposure or AUC. Overall, oral administration of cobimetinib is not affected by food or gastric pH.
4.3.3 Clinical Experience
In the phase I monotherapy study of cobimetinib, multiple schedules were evaluated with maximum tolerated doses of cobimetinib established as 100 mg 14 days on/14 days off and 60 mg 21 days on/7 days off [93]. No efficacy data of cobimetinib as a single agent in the treatment of human cancers have been released to date. However, cobimetinib has been evaluated in multiple schedules in combination with the V600 BRAF inhibitor vemurafenib [94]. A multi-factorial dose-escalation phase I study was pursued in which vemurafenib at both 720 or 960 mg twice daily was evaluated with cobimetinib at 60, 80, or 100 mg in schedules including 14 days on/14 days off, 21 days on/7 days off, or continuously. Two dose levels were expanded, including vemurafenib 720 mg and 960 mg twice daily plus cobimetinib 60 mg daily 21 days on/7 days off. When assessing benefit in patients previously treated with vemurafenib, a 15 % RR and 43 % SD rate were observed in the expansion arms. The median PFS for patients who had previously progressed on vemurafenib was 2.8 months. In those patients not previously treated with BRAF inhibitors, an RR of 85 and 10 % complete RR was observed. In BRAF inhibitor-untreated patients, the median PFS was not reached at a median follow-up of 10 months.
4.3.4 Safety Profile
Common adverse events associated with single-agent cobimetinib include diarrhea, rash, pruritus, nausea, vomiting, blurred and impaired vision, fatigue, and abdominal pain [93]. In combination with vemurafenib, the most frequent adverse events observed included diarrhea (48 %), non-acneiform rash (35 %), nausea (32 %), photosensitivity/sunburn (29 %), fatigue (28 %), and liver test abnormality (25 %) [94].
4.4 Refametinib (BAY 86-9766, RDEA119)
4.4.1 Preclinical Data
Refametinib is a potent, non-ATP-competitive, highly selective allosteric inhibitor of MEK1 and MEK2 with an IC50 of 19 and 47 ηmol/L, respectively. Refametinib has exhibited activity against V600E BRAF mutant cell lines, with 50 % growth inhibition concentration (GI50) values ranging from 40 to 84 ηmol/L, and tumor reduction in A375 melanoma, Colo205 colorectal, and OCIP19 pancreatic carcinoma xenografts [95, 96].
4.4.2 Pharmacokinetics and Drug Metabolism
Refametinib demonstrates a median T max of 2 h with an elimination half-life (T ½) of 12 h after single dosing. Dose-proportional C max and steady-state AUC were observed between a 2–100 mg dosing range on a daily schedule [97].
4.4.3 Clinical Experience
The RP2D of refametinib is 100 mg daily [97]. Activity was observed in the phase I study in BRAF and KRAS mutant tumors. Refametinib was also dose escalated in combination with gemcitabine 1,000 mg/m2 daily, where an RP2D of 30 mg twice per day was determined [98].
4.4.4 Safety Profile
Common treatment-related adverse events include dermatologic toxicity such as primarily acneiform dermatitis (33 %), maculopapular rash (20 %), as well as nausea (29 %), vomiting (26 %), diarrhea (32 %), edema (28 %), and fatigue (26 %) [97].
4.5 Binimetinib (MEK162, ARRY-438162)
4.5.1 Preclinical
Binimetinib inhibits MEK1 and MEK2 in a potent and selective non-ATP competitive manner (IC50 = 12 ηM). Binimetinib has been documented to inhibit proliferation and reduce tumor volume in KRAS, NRAS, and BRAF mutant as well as non-MAPK activities in vitro and in vivo cell line and tumor models [99].
4.5.2 Pharmacokinetics and Metabolism
The pharmacokinetics of binimetinib are linear, and the drug exhibits moderate oral bioavailability in all species tested. It is highly protein bound in plasma and somewhat more distributed in plasma than blood. Binimetinib has multiple routes of metabolism; however, glucuronidation (predominately through UGT1A1) appears to be most common, with excretion nearly evenly split between urine and feces. Binimetinib has limited effects on relevant CYP enzyme family members. The human T ½ is approximately 8 h [100].
4.5.3 Clinical Experience
Binimetinib is dosed as 45 or 60 mg daily or twice daily [100, 101]. In a phase II study of binimetinib in melanoma, a 20 % RR was observed for both patients with advanced NRAS mutant and those with BRAF mutant melanoma [101]. The activity of binimetinib is also being evaluated in biliary tract tumors, where a PR and a complete response have been observed. Notably, these patients were found not to harbor RAS- or RAF-activating mutations [102]. Results from a combination phase I/II study of binimetinib with the selective BRAF inhibitor LGX818 have also described a dosing regimen of LGX818 at 450 mg daily with binimetinib 45 mg twice daily as a tolerable regimen. Early indications of efficacy for this combination in melanoma have described an RR of 71 % was seen in patients with non-previously treated BRAF mutant disease as well as 22 % in those who were previously treated with a BRAF inhibitor [103].
4.5.4 Safety
Frequently reported adverse events associated with binimetinib include rash (79 %), diarrhea (32 %), edema (32 %), nausea (43 %), fatigue (29 %), and vomiting (36 %) [102].
4.6 Pimasertib (AS703026, MSC1936369B)
4.6.1 Preclinical
Pimasertib is a selective and potent oral ATP uncompetitive inhibitor of MEK1 and MEK2 that has demonstrated anti-tumor activity in multiple preclinical models, with special focus on RAS-driven tumors (myeloma, pancreas, lung, colon cancers) [104, 105]. Biomarker analysis of 12-O-Tetradecanoylphorbol-I3-acetate-stimulated (MAPK-activated) leukocytes indicated a consistent inhibition of ERK phosphorylation by pimasertib [105]. Further, pimasertib has shown a potential for combination therapy. Studies with PI3K pathway inhibitors or various chemotherapies have shown synergistic activity on tumor growth inhibition [106].
4.6.2 Pharmacokinetics and Metabolism
Human pharmacokinetic data have not been publicly disclosed regarding pimasertib. One study evaluating drug exposure in murine brain and glioblastoma models has suggested a T max in the brain of 1 h associated with a 90 % inhibition of phosphorylated ERK [107].
4.6.3 Clinical Activity
Pimasertib has been evaluated in several RAS and RAF mutant tumors. In a phase I study of pimasertib with 5-fluorouracil (5-FU), irinotecan, and leucovorin, dose-limiting toxicity limited the escalation of pimasertib, and this combination was abandoned [108]. Other combination approaches have included dose escalation with the PI3K/mammalian target of rapamycin (mTOR) inhibitor SAR245409 and gemcitabine. In combination with SAR245409, doses of pimasertib 60 mg twice daily with 70 mg of SAR245409 was determined to be the RP2D, with PRs being observed in patients with KRAS mutant colorectal carcinoma and low-grade ovarian cancer [109]. Activity of pimasertib has also been observed in combination with gemcitabine in pancreatic carcinoma. In combination with gemcitabine, pimasertib 60 mg twice daily, continuous dosing was deemed the RP2D; PR was observed in 19 % of patients, and SD for greater than 3 months was seen in 25 % of patients [110].
4.6.4 Safety
Toxicity has yet to be widely reported; however, common toxicities have been similar to MEK inhibitor class effects and include retinal vein occlusion, ocular events, diarrhea, asthenia, edema, vomiting, and nausea.
4.7 RO4987655
4.7.1 Preclinical
RO4987655 is a potent, highly selective non-ATP-competitive inhibitor of MEK1 and MEK2. Anti-tumor activity in preclinical models of melanoma, pancreatic carcinoma, and colorectal cancer has been observed with RO4987655 as a single agent as well as in combination with chemotherapies such as paclitaxel, gemcitabine, and cisplatin [111].
4.7.2 Pharmacokinetics and Metabolism
RO4987655 demonstrates dose-proportional pharmacokinetics by C max and AUC as well as rapid bioavailability, with a median T max of approximately 1 h (range 0.5–2) [112]. The human mean terminal T ½ was demonstrated as 12 h with an effective T ½ of 9 h. RO4987655 demonstrates biphasic elimination with multiple routes of elimination; however, renal elimination is minimal. The major metabolite of RO4987655 is a ring-open structure, although oxidative metabolites are also observed.
4.7.3 Clinical Activity
RO4987655 has been evaluated in several expansion arms of the phase I dose-escalation study including melanoma, NSCLC, and colorectal carcinoma [113]. Among patients with melanoma, four of 18 (24 %) BRAF mutant and four of 20 (20 %) BRAF WT had a response, with PR as best response in these cohorts. Among patients with KRAS mutant NSCLC, two of 18 patients had response (all PR), while none of the 18 patients with KRAS mutant colorectal carcinoma had a response.
4.7.4 Safety
Toxicities observed with RO4987655 have been similar to those described with other MEK inhibitors [113]. Common toxicities have included those that were skin related (rash and acneiform dermatitis), gastrointestinal disorders (nausea, vomiting, diarrhea), ocular (serous retinal detachment and blurred vision), and general (peripheral edema and asthenia). The majority of grade 3–4 adverse events included asymptomatic increase of creatine phosphokinase (23 %), rash (16 %), diarrhea (8 %), folliculitis (7 %), super-infected dermatitis (1 %), and serous retinal detachment (7 %).
5 Next Steps for MEK Inhibitors
With the approval of trametinib, MEK inhibition has entered the clinical area as a standard of care option. Multiple studies are ongoing with MEK inhibitors as single agents (Table 2). Beyond V600 BRAF, other melanoma subtypes may also be sensitive enough to MEK monotherapy to justify approval of MEK inhibitors for clinical practice. In other molecular subsets, such as RAS mutant histologies, this question is more open.
A robust preclinical literature suggests that combining MEK inhibition may be additive or synergistic with other treatments. While monotherapy with MEK inhibition does have interesting activity in several diseases, combination strategies with other treatment approaches such as chemotherapy, targeted therapy, and immunotherapy also appear promising. Table 3 describes ongoing combination trials with MEK inhibitors. To date, the development of these approaches has been limited by toxicity, and this highlights an important difference between MEK inhibition and V600 BRAF inhibition. Mutation of BRAF at V600 generates a new enzyme target for drug development. This tumor-specific mutation facilitates a wider ‘therapeutic window’, allowing administration of a much higher dosage of BRAF inhibition prior to the development of dose-limiting toxicity [114]. This is in contrast to MEK inhibition, which targets the physiologic MAPKK proteins on which nearly all cells depend to some degree. As such, the therapeutic window for MEK inhibition is much smaller than for V600 BRAF and limits dose escalation.
This difference in mutant versus physiologic protein targets manifests particularly in the setting of combination therapy. For reasons that have been well described previously [19, 54], combination approaches of V600 BRAF and MEK inhibition are synergistic and reduce the toxicity of BRAF inhibition alone [66]. However, synergy is not necessarily expected with other combinations (e.g. MEK inhibitor plus PI3K, AKT inhibitor or chemotherapy, etc.). This has borne out in phase I studies where it has not been possible to optimally dose escalate MEK inhibitors with other drugs and the observed efficacy has been limited [115, 116].
The utility of MEK inhibition will also have to be closely considered in the setting of a new class of immunotherapeutic agents that are of increasing interest in cancer treatment. These immune-checkpoint blockade approaches, such as anti-CTLA-4, anti-PD1 and anti-PD-L1 antibodies, rely on an activated T-cell response as the anti-tumor agent. Laboratory experiments have suggested that inhibition of MEK may have immune-dampening effects via blockade of the MAPK pathway during T-cell activation. Whether the combination of MEK inhibitors and immunotherapy is an appropriate approach for optimal anti-tumor effect will have to be closely investigated [117]. Thus, much is yet to be learned regarding the combining of MEK inhibitors with chemotherapy and immunotherapy [118].
With these caveats, MEK inhibition is clearly an important area of drug development that has advanced tremendously over the past decade. While the development of MEK inhibitors as single agents may be appropriate in some settings, it seems likely that combination approaches with other agents will be the most highly efficacious strategies in many cancers. Furthermore, it may be that the dosing schedule of MEK inhibitors is an important consideration in their clinical utility. Preclinical models have suggested that intermittent dosing approaches may be more appropriate to avoid genetic and molecular feedback resistance through the MAPK pathway [119]. Clearly, further research into these areas will be required to optimize the utility of MEK inhibition in the treatment of patients with cancer.
References
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40.
Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, et al. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112(5):685–95.
Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, et al. A Ras-induced conformational switch in the Ras activator son of sevenless. Proc Natl Acad Sci. 2006;103(45):16692–7.
Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. The RalGEF-Ral effector signaling network: the road less traveled for anti-Ras drug discovery. Genes Cancer. 2011;2(3):275–87.
Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773(8):1177–95.
Luo Z, Tzivion G, Belshaw PJ, Vavvas D, Marshall M, Avruch J. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature. 1996;383(6596):181–5.
Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273(37):24052–6.
Fabian JR, Daar IO, Morrison DK. Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol. 1993;13(11):7170–9.
Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401(6749):173–7.
Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13(7):1610–9.
Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–72.
Kocieniewski P, Lipniacki T. MEK1 and MEK2 differentially control the duration and amplitude of the ERK cascade response. Phys Biol. 2013;10(3):035006. doi:10.1088/1478-3975/10/3/035006.
White CD, Brown MD, Sacks DB. IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009;583(12):1817–24.
Leboeuf R, Baumgartner JE, Benezra M, Malaguarnera R, Solit D, Pratilas CA, et al. BRAFV600E mutation is associated with preferential sensitivity to mitogen-activated protein kinase kinase inhibition in thyroid cancer cell lines. J Clin Endocrinol Metab. 2008;93(6):2194–201.
Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16(1):45–54.
Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40(12):2707–19.
De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–27.
Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22(5):668–82.
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5.
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–9.
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67.
Quinlan MP, Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol. 2009;5(1):105–16.
Tormanen VT, Pfeifer GP. Mapping of UV photoproducts within ras proto-oncogenes in UV-irradiated cells: correlation with mutations in human skin cancer. Oncogene. 1992;7(9):1729–36.
Le Calvez F, Mukeria A, Hunt JD, Kelm O, Hung RJ, Taniere P, et al. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct patterns in never, former, and current smokers. Cancer Res. 2005;65(12):5076–83.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54.
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–47.
Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29(15):2046–51.
Santarpia L, Qi Y, Stemke-Hale K, Wang B, Young EJ, Booser DJ, et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res Treat. 2012;134(1):333–43.
Singer G, Oldt R 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95(6):484–6.
Singer G, Kurman RJ, Chang HW, Cho SK, Shih Ie M. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol. 2002;160(4):1223–8.
Sieben NL, Macropoulos P, Roemen GM, Kolkman-Uljee SM, Jan Fleuren G, Houmadi R, et al. In ovarian neoplasms, BRAF, but not KRAS, mutations are restricted to low-grade serous tumours. J Pathol. 2004;202(3):336–40.
Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63(7):1454–7.
Lupi C, Giannini R, Ugolini C, Proietti A, Berti P, Minuto M, et al. Association of BRAF V600E mutation with poor clinicopathological outcomes in 500 consecutive cases of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92(11):4085–90.
Caronia LM, Phay JE, Shah MH. Role of BRAF in thyroid oncogenesis. Clin Cancer Res. 2011;17(24):7511–7.
Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29(10):1261–70.
Fransen K, Klintenas M, Osterstrom A, Dimberg J, Monstein HJ, Soderkvist P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25(4):527–33.
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67.
Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68(14):5524–8.
Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44(2):133–9.
Shi H, Moriceau G, Kong X, Koya RC, Nazarian R, Pupo GM, et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov. 2012;2(5):414–24.
Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B, et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011;71(16):5535–45.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci. 2009;106(48):20411–6.
Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–96.
Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109.
Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–8.
Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501(7466):232–6.
Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–62.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6.
Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000.
Friday BB, Yu C, Dy GK, Smith PD, Wang L, Thibodeau SN, et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res. 2008;68(15):6145–53.
Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci. 2009;106(11):4519–24.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22.
Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270(46):27489–94.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30.
Heald RA, Jackson P, Savy P, Jones M, Gancia E, Burton B, et al. Discovery of novel allosteric mitogen-activated protein kinase kinase (MEK) 1,2 inhibitors possessing bidentate Ser212 interactions. J Med Chem. 2012;55(10):4594–604.
Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11(12):1192–7.
Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20(6):963–9.
Roring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012;31(11):2629–47.
Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25(5):697–710.
Lorusso PM, Adjei AA, Varterasian M, Gadgeel S, Reid J, Mitchell DY, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23(23):5281–93.
LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16(6):1924–37.
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39(1):23–31.
Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):773–81.
Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
GlaxoSmithKline, plc. GSK announces headline results for Phase III study of the combination of Tafinlar® (dabrafenib) and Mekinist® (trametinib) in metastatic melanoma. 24 Jan 2014 [online]. Available from URL: http://us.gsk.com/html/media-news/pressreleases/2014/gsk-announces-headline-results-for-phase-iii-study-of-the-combin.html.
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–83.
Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007;6(1):138–46.
Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26(13):2139–46.
Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16(5):1613–23.
Leijen S, Soetekouw PM, Jeffry Evans TR, Nicolson M, Schellens JH, Learoyd M, et al. A phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor Selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68(6):1619–28.
Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18(2):555–67.
Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib [AZD6244 (ARRY-142886)] versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–23.
Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5(10):1630–6.
Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011;29(5):1021–8.
Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29(17):2357–63.
O’Neil BH, Goff LW, Kauh JS, Strosberg JR, Bekaii-Saab TS, Lee RM, et al. Phase II study of the mitogen-activated protein kinase 1/2 inhibitor selumetinib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2011;29(17):2350–6.
Farley J, Brady WE, Vathipadiekal V, Lankes HA, Coleman R, Morgan MA, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14(2):134–40.
Robert C, Dummer R, Gutzmer R, Lorigan P, Kim KB, Nyakas M, et al. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. Lancet Oncol. 2013;14(8):733–40.
Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–64.
Gupta A, Love S, Schuh A, Shanyinde M, Larkin JM, Plummer R, et al. DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Ann Oncol. 2014;25(5):968–74.
Patel M, Smyth E, Chapman PB, Wolchok JD, Schwartz GK, Abramson DH, et al. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin Cancer Res. 2011;17(8):2087–100.
Carvajal R, Sosman J, Quevedo F, Milhem M, Joshua A, Kudchadkar R et al. Phase II study of selumetinib (sel) versus temozolomide (TMZ) in gnaq/Gna11 (Gq/11) mutant (mut) uveal melanoma (UM). J Clin Oncol. 2013;31 Suppl. [Abstract CRA9003].
Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47.
Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18(7):2056–65.
Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623–32.
Belvin M, Berry L, Chan J, den Otter D, Friedman L, Hoeflich K, et al. 132 Intermittent dosing of the MEK inhibitor, GDC-0973, and the PI3K inhibitor, GDC-0941, results in prolonged accumulation of Bim and causes strong tumor growth inhibition in vivo. Eur J Cancer Suppl. 2010;8(7):48.
Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649–64.
Choo EF, Belvin M, Boggs J, Deng Y, Hoeflich KP, Ly J, et al. Preclinical disposition of GDC-0973 and prospective and retrospective analysis of human dose and efficacy predictions. Drug Metab Dispos. 2012;40(5):919–27.
Wong H, Vernillet L, Peterson A, Ware JA, Lee L, Martini JF, et al. Bridging the gap between preclinical and clinical studies using pharmacokinetic-pharmacodynamic modeling: an analysis of GDC-0973, a MEK inhibitor. Clin Cancer Res. 2012;18(11):3090–9.
Musib L, Choo E, Deng Y, Eppler S, Rooney I, Chan IT, et al. Absolute bioavailability and effect of formulation change, food, or elevated pH with rabeprazole on cobimetinib absorption in healthy subjects. Mol Pharm. 2013;10(11):4046–54.
Rosen L, LoRusso P, Ma W, Goldman J, Weise A, Dimitrios Colevas A et al. A first-in-human phase 1 study to evaluate the MEK1/2 inhibitor GDC-0973 administered daily in patients with advanced solid tumors. Cancer Res. 2011;71(8 Suppl. 1) [abstract 4706].
Daud A, Ribas A, Gonzalez R, Pavlick A, Hamid O, Puzanov I, et al. Phase 1B (BRIM7) results combining vemurafenib (VEM) and cobimetinib, a MEK inhibitor, in patients with advanced BRAFV600-mutated melanoma. Society for melanoma research [abstract 201]: Philadelphia; 2013.
Iverson C, Larson G, Lai C, Yeh LT, Dadson C, Weingarten P, et al. RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009;69(17):6839–47.
Chang Q, Chapman MS, Miner JN, Hedley DW. Antitumour activity of a potent MEK inhibitor RDEA119/BAY 869766 combined with rapamycin in human orthotopic primary pancreatic cancer xenografts. BMC Cancer. 2010;10:515.
Weekes CD, Von Hoff DD, Adjei AA, Leffingwell DP, Eckhardt SG, Gore L, et al. Multicenter phase I trial of the mitogen-activated protein kinase 1/2 inhibitor BAY 86-9766 in patients with advanced cancer. Clin Cancer Res. 2013;19(5):1232–43.
Van Laethem J, Heinemann V, Martens U, Jassem J, Michl P, Peeters M, et al. A phase I/II study of the MEK inhibitor BAY 86-9766 (BAY) in combination with gemcitabine (GEM) in patients with nonresectable, locally advanced or metastatic pancreatic cancer (PC): Phase I dose-escalation results. J Clin Oncol. 2012; 30 Suppl. [abstract 4050].
Winski S, Anderson D, Bouhana K, Impastato R, Woessner R, Zuzack J, et al. 162 MEK162 (ARRY-162), a novel MEK 1/2 inhibitor, inhibits tumor growth regardless of KRas/Raf pathway mutations. EJC Suppl. 2010;8(7):56.
Bendell J, Papadopoulos K, Jones S, Barrett E, Guthrie K, Kass C, et al. Abstract B243: A phase I dose-escalation study of MEK inhibitor MEK162 (ARRY-438162) in patients with advanced solid tumors. Mol Cancer Ther. 2011;10(11 Suppl. 1) [abstract B243].
Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–56.
Finn R, Javle M, Tan B, Weekes C, Bendell J, Patnaik A, et al. A phase I study of MEK inhibitor MEK162 (ARRY-438162) in patients with biliary tract cancer. J Clin Oncol. 2012; 30 Suppl. 4 [abstract 220].
Kefford R, Miller W, Tan D, Sullivan R, Long G, Dienstmann R, et al. Preliminary results from a phase Ib/II, open-label, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK1/2 inhibitor MEK162 in BRAF V600-dependent advanced solid tumors. J Clin Oncol. 2013; 31 Suppl. [abstract 9029].
Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S, et al. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol. 2010;149(4):537–49.
Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res. 2011;71(2):445–53.
Martinelli E, Troiani T, D’Aiuto E, Morgillo F, Vitagliano D, Capasso A, et al. Antitumor activity of pimasertib, a selective MEK 1/2 inhibitor, in combination with PI3K/mTOR inhibitors or with multi-targeted kinase inhibitors in pimasertib-resistant human lung and colorectal cancer cells. Int J Cancer. 2013;133(9):2089–101.
Shaw JV, Zhang H, Carden R, Qiu D, Tian H, Ma J, Clark A, et al. Abstract LB-456: Evaluation of brain pharmacokinetics as a potential differentiation factor for the MEK inhibitors, MSC2015103 and pimasertib. Cancer Res. 2012;72(8 Suppl. 1).
Macarulla T, Tabernero J, Cervantes A, Roselló S, Van Cutsem E, Tejpar S, et al. Phase I/II study of FOLFIRI plus the MEK1/2 inhibitor pimasertib (MSC1936369b) as second-line treatment for kras mutated metastatic colorectal cancer. Ann Oncol. 2012; 23 Suppl. 4:PD-0024.
Heist R, Gandhi L, Shapiro G, Rizvi N, Burris H, Bendell J, et al. Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: Results of a phase Ib dose-escalation trial. J Clin Oncol. 2013;31 Suppl. [abstract 2530].
Verslype C, Hammel P, Hidalgo M, Macarulla T, Garcia-Carbonero R, André T et al. Pimasertib plus gemcitabine in metastatic pancreatic adenocarcinoma: Results of a safety run-in part of a phase II trial. J Clin Oncol. 2013; 31 Suppl. [abstract 4041].
Aida S, Yoshimura Y, Ishii N. CH4987655 (RO4987655), a selective orally bioavailable MEK inhibitor, shows highly potent and broad antitumor activity as a monotherapy and in combination with other antitumor agents. [Abstract no. 2011], American Association of Cancer Research: Denver; 2009.
Lee L, Niu H, Rueger R, Igawa Y, Deutsch J, Ishii N, et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of CH4987655 in healthy volunteers: target suppression using a biomarker. Clin Cancer Res. 2009;15(23):7368–74.
Zimmer L, Barlesi F, Martinez-Garcia M, Dieras V, Schellens JH, Spano JP, et al. Phase I expansion and pharmacodynamic study of the oral MEK inhibitor RO4987655 (CH4987655) in selected patients with advanced cancer with RAS-RAF mutations. Clin Cancer Res. 2014;20(16):4251–61.
Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci. 2010;107(33):14903–8.
Bendell J, LoRusso P, Cho D, Musib L, Yan Y, Chang I, et al. Clinical results of a phase Ib dose-escalation study of the Mek inhibitor cobimetinib (GDC-0973) and the Akt inhibitor ipatasertib (GDC-0068) in patients (pts) with solid tumors [abstract no. CT328], American Association of Cancer Research: San Diego; 2014.
Speranza G, Kinders R, Khin S, Weil M, Do K, Horneffer Y, et al. Pharmacodynamic biomarker-driven trial of MK-2206, an AKT inhibitor, with AZD6244 (selumetinib), a MEK inhibitor, in patients with advanced colorectal carcinoma (CRC). J Clin Oncol. 2012; 30 Suppl. [abstract 3529].
Ott PA, Henry T, Baranda SJ, Frleta D, Manches O, Bogunovic D, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. 2013;62(4):811–22.
Luke JJ, Ott PA. Kinase inhibitors and immune check-point blockade for the treatment of metastatic melanoma and advanced cancer: synergistic or antagonistic? Expert Opin Pharmacother. 2013;14(18):2457–62.
Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res. 2012;72(1):210–9.
Disclosures
Jason J. Luke has acted as a paid consultant to Bayer and Genentech and has received clinical trial support to institution from EMD Serono, GlaxoSmithKline, and Novartis for MEK inhibitor-based studies. Patrick A. Ott has no relevant disclosures. Geoffrey I. Shapiro has received clinical trial support to institution from Pfizer, GlaxoSmithKline, and Genentech for MEK inhibitor-based studies. No specific funding was received for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Luke, J.J., Ott, P.A. & Shapiro, G.I. The Biology and Clinical Development of MEK Inhibitors for Cancer. Drugs 74, 2111–2128 (2014). https://doi.org/10.1007/s40265-014-0315-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0315-4