
Abstract
The Ras/Raf/MEK/ERK and PI3K/Akt/mTOR cascades are often activated by genetic alterations, either by mutations in upstream signaling molecules or by mutations in intrinsic pathway components. Upstream mutations in one signaling pathway or even in downstream components of the same pathway can alter the sensitivity of the cells to certain small molecule inhibitors. These pathways have profound effects on proliferative, apoptotic, and differentiation pathways. Dysregulation of components of these pathways can contribute to: malignant transformation, resistance to other pathway inhibitors, and chemotherapeutic drug resistance. This chapter will first briefly describe these pathways and then evaluate potential uses of Raf, MEK, PI3K, Akt, and mTOR inhibitors that have been investigated in preclinical and clinical investigations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Epidermal Growth Factor Receptor
- KRAS Mutation
- mTOR Inhibitor
- PI3K Inhibitor
- Epidermal Growth Factor Receptor Inhibitor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
13.1 Introduction
Since the discovery of the RAS, RAF, MEK1, PIK3CA, and AKT oncogenes and neurofibromin 1 (NF1), PTEN, TSC1, and TSC2 tumor suppressor genes, the Ras/Raf/MEK/ERK, and Ras/PI3K/PTEN/Akt/mTOR signaling cascades have been extensively investigated with the ultimate goal of determining how these genes become activated/inactivated and whether it is possible to suppress their activity in human cancer and other diseases [1]. Furthermore, these pathways are also implicated in the resistance and sometimes sensitivity to therapy [2]. There have been breakthroughs in the discovery of complex interacting pathway components, and their genetic and epigenetic regulation. Furthermore, elucidation of the mechanisms by which mutations of components of the pathways can lead to aberrant signaling, uncontrolled proliferation, and in some cases confer sensitivity to targeted therapy has greatly advanced the field. This chapter will review some of the current inhibitors, their targets, and how they are being used to treat cancer and overcome therapeutic resistance.
Usually signaling commences upon ligation of a growth factor/cytokine/interleukin/mitogen (ligand) to its cognate receptor at the cell surface. This event can result in the activation of many downstream signaling cascades including the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways. These pathways can further transmit their signals to different subcellular components, namely to the nucleus to control gene expression, to the translational apparatus to enhance the translation of “weak” mRNAs, to the apoptotic machinery to regulate apoptosis, or to other events involved in the regulation of cellular proliferation (e.g., interactions with the p53 pathway to regulate cell cycle progression). Regulation of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways is mediated by a series of kinases, phosphatases, GTP:GDP exchange, and scaffolding proteins. There are also many tumor suppressor proteins which interact with these cascades which frequently serve to fine tune or limit activity (e.g., NF1, PTEN, RKIP, PP2A, TSC1, and TSC2). Mutations occur in many of the genes in these pathways leading to uncontrolled regulation and aberrant signaling.
13.2 The Ras/Raf/MEK/ERK Pathway
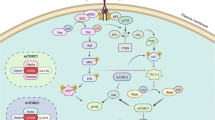
An overview of the Ras/Raf/MEK/ERK pathway and the sites where small molecule inhibitors act is presented in Fig. 13.1. This figure serves to illustrate the flow of information through this pathway from a growth factor to a specific receptor to phosphorylation of appropriate transcription factors as well as affect proteins involved in translation and apoptosis. Following the stimulation of a receptor with a growth factor/cytokine/mitogen, a Src homology 2 domain-containing protein (Shc) adaptor protein becomes associated with the C-terminus of the activated Growth Factor Receptor (GFR), for example, Epidermal Growth Factor Receptor (EGFR), Insulin-like Growth Factor-1 Receptor (IGF-1R), Vascular Endothelial Growth Factor Receptor (VEGFR) and many others [1, 2]. Shc recruits the growth Factor receptor–bound protein 2 (Grb2) protein and the Son of Sevenless (SOS) homolog protein [a Guanine nucleotide Exchange Factor (GEF)], resulting in the loading of the membrane-bound GDP:GTP exchange protein (GTPase). GEFs promote Ras activation by displacing GDP from Ras which leads to GTP binding. Ras activation is suppressed by the GTPase-Activating Proteins (GAPs) that stimulate the GTPase activity of Ras. There are two prominent GAP proteins, p120GAP and NF1. Ras can also be activated by Growth Factor Receptor Tyrosine Kinases (GFRTK), such as Insulin Receptor (IR), via intermediates like Insulin Receptor Substrate (IRS) proteins that bind Grb2 [3]. Ras:GTP then recruits the serine/threonine (S/T) kinase Raf to the membrane where it becomes activated [1, 2].

Overview of the Ras/Raf/MEK/ERK cascade and small molecule inhibitors used for targeting this pathway. Activation of this pathway can occur by mutations in upstream Growth Factor Receptors (GFR) or by stimulation by the appropriate Growth Factors (GF). In addition, mutations can occur in intrinsic members of the pathway (RAS, RAF, MEK1, or the tumor suppressor Neurofibromin (NF1)). Sites where NF1, Protein Phosphatase 2A (PP2A), Raf Kinase Inhibitory Protein (RKIP), Kinase Suppressor of Ras (KSR) interact with this pathway are on the right hand side of the Ras/Raf/MEK/ERK pathway. NF1, PP2A, and RKIP are depicted in black rectangles as they normally serve to dampen the activity of this pathway. Sites where various small molecule inhibitors function are in black octagons on the left hand side of the pathway. Representative inhibitors are listed in boxes next to the octagons
Both RAS and RAF are members of multi-gene families, and there are three RAS members (KRAS, NRAS, and HRAS) and three RAF members (BRAF, RAF1 (a.k.a c-Raf), and ARAF) [1, 2]. Raf-1 and A-Raf are activated, in part, by a Src-family kinase, while B-Raf does not require the Src-family kinase for activation. Raf-1 can be regulated by dephosphorylation by the Protein serine/threonine Phosphatase 2A (PP2A). PP2A has been reported to positively and negatively regulate Raf-1 [4, 5].
Raf is responsible for S/T phosphorylation of Mitogen-activated protein Kinase kinase-1 (MEK1) (a dual specificity kinase (T/Y) [1, 2]. Other proteins such as Kinase Suppressor of Ras (KSR) have recently been shown to phosphorylate MEK1 [6]. KSR has scaffolding properties and interacts with Raf, MEK, and ERK which regulates ERK activation. KSR can form dimers with various Raf proteins which alter the effects of Raf inhibitors. KSR competes with Raf-1 for Raf inhibitor–induced binding to B-Raf which decreases the normal ERK activation observed after Raf-inhibitor treatment [7].
MEK1 phosphorylates Extracellular signal-Regulated Kinases 1/2 (ERK1 and 2) at specific T and Y residues [1, 2]. MEK1 was originally not thought to be mutated frequently in human cancer. However, recent large-scale mutation screening studies and studies aimed at determining mechanisms of resistance to small molecule inhibitors have observed that MEK1 is mutated in certain human cancers and also is mutated in certain inhibitor-resistant cells [8].
Activated ERK1 and ERK2 serine S/T kinases phosphorylate and activate a variety of substrates, including p90 Ribosomal six kinase-1 (p90Rsk1) [2]. ERK also phosphorylates MAPK signal-integrating kinases (Mnk1/2) which can in turn phosphorylate eukaryotic translation Initiation Factor 4E (eIF4E), a key protein involved in the translation of difficult mRNAs [9].
p90Rsk1 can activate the cAMP-Response Element-Binding protein (CREB) transcription factor as well as proteins involved in regulation of protein translation (e.g., Mnk-1, p70 ribosomal S6 Kinase (p70S6K), eukaryotic translation Initiation Factor 4B, (eIF4B), and ribosomal protein S6 (rpS6) [10].
The number of ERK1/2 substrate/targets is easily in the hundreds. These substrates/targets include different types of molecules including other kinases, transcription factors, or proteins involved in protein translation or apoptosis. Suppression of MEK and ERK can have profound effects on cell growth, inflammation, and aging. Activated ERK can also phosphorylate “upstream” Raf-1 and MEK1 which alter their activity. Depending upon the site phosphorylated on Raf-1, ERK phosphorylation can either enhance [11] or inhibit [12] Raf-1 activity. In contrast, some studies have indicated that when MEK1 are phosphorylated by ERK, their activity decreases [13]. Recent studies indicate that ERK does not negatively feedback-inhibit B-Raf [14].
These phosphorylation events induced by ERK serve to alter the stability and/or activities of the proteins. These examples of feedback loops become important in consideration of whether to just target MEK or to target both Raf and MEK in various cancers. It is important that the reader realize that certain phosphorylation events can either inhibit or repress the activity of the affected protein. This often depends on the particular residue on the protein phosphorylated which can confer a different configuration to the protein or target the protein to a different subcellular localization that may result in proteasomal degradation or association with certain scaffolding proteins.
13.3 The Ras/PI3K/PTEN/Akt/mTOR Pathway
An introductory overview of the Ras/PI3K/PTEN/Akt/mTOR pathway is presented in Fig. 13.2. Also outlined in this diagram are common sites of intervention with signal transduction inhibitors. Many of these inhibitors have been evaluated in various clinical trials, and some are currently being used to treat patients with specific cancers. Extensive reviews of many inhibitors targeting these pathways have been recently published [1, 15, 16].

Overview of the PI3 K/Akt/mTOR cascade and small molecule inhibitors used for targeting this pathway. Activation of this pathway can occur by mutations in upstream growth factor receptors (GFR) or by stimulation by the appropriate GF. In addition, mutations can occur in intrinsic members of the pathway (RAS, PIK3CA, AKT, or the tumor suppressors (NF1, PTEN, TSC1, TSC2). Sites where NF1, PTEN, TSC1, TSC2 are depicted in black rectangles as they normally serve to dampen the activity of this pathway. Sites where various small molecule inhibitors function are in black octagons. Representative inhibitors are listed in boxes next to the octagons
Phosphatidylinositol 3-Kinase (PI3K) is a heterodimeric protein with an 85 kDa regulatory subunit and a 110 kDa catalytic subunit (PIK3CA) [1, 2, 66–69]. PIK3CA is frequently mutated in certain cancers such as breast, ovarian, colorectal, endometrial, and lung [2, 17].
PI3K serves to phosphorylate a series of membrane phospholipids including Phosphatidylinositol 4-Phosphate (PtdIns(4)P) and Phosphatidylinositol 4,5-bisPhosphate (PtdIns(4,5)P2), catalyzing the transfer of ATP-derived phosphate to the D-3 position of the inositol ring of membrane phosphoinositides, thereby forming the second messenger lipids Phosphatidylinositol 3,4-bisPhosphate (PtdIns(3,4)P2) and Phosphatidylinositol 3,4,5-trisPhosphate (PtdIns(3,4,5)P3) [2, 15]. Most often, PI3K is activated via the binding of a ligand to its cognate receptor, whereby p85 associates with phosphorylated tyrosine residues on the receptor via a Src Homology 2 (SH2) domain. After association with the receptor, the p110 catalytic subunit then transfers phosphate groups to the aforementioned membrane phospholipids [15]. It is these lipids, specifically PtdIns [3–5] P3, that attract a series of kinases to the plasma membrane, thereby initiating the signaling cascade.
Downstream of PI3K is the primary effector molecule of the PI3K signaling cascade, Akt/Protein Kinase B (PKB) which is a 57 kDa S/T kinase that phosphorylates many targets on RxRxxS/T (R = Arginine) consensus motifs [18]. Akt was discovered originally as the cellular homolog of the transforming retrovirus AKT8 and as a kinase with properties similar to protein kinases A and C [19]. Akt contains an amino-terminal Pleckstrin Homology (PH) domain that serves to target the protein to the membrane for activation [15]. Within its central region, Akt has a large kinase domain and is flanked on the carboxyl-terminus by hydrophobic and proline-rich regions [15]. Akt-1 is activated via phosphorylation of two residues: T308 and S473. Akt-2 and Akt-3 are highly related molecules and have similar modes of activation. Akt-1 and Akt-2 are ubiquitously expressed, while Akt-3 exhibits a more restricted tissue distribution and is found abundantly in nervous tissue [20].
The Phosphatidylinositol-Dependent Kinases (PDKs) are responsible for the activation of Akt. PDK1 is the kinase responsible for phosphorylation of Akt-1 at T308 [18]. Akt-1 is also phosphorylated at S473 by the mammalian Target Of Rapamycin (mTOR) complex referred to as (Rapamycin-insensitive companion of mTOR/mLST8 complex) mTORC2 [15]. Therefore, phosphorylation of Akt is complicated as it is phosphorylated by a complex that lies downstream of activated Akt itself [15]. Thus, as with the Ras/Raf/MEK/ERK pathway, there are feedback loops that serve to regulate the activity of the Ras/PI3K/PTEN/Akt/mTOR pathway. Once activated, Akt leaves the cell membrane to phosphorylate intracellular substrates.
After activation, Akt is able to translocate to the nucleus [15] where it affects the activity of a number of transcriptional regulators. Some examples of molecules which regulate gene transcription that are phosphorylated by Akt include CREB [21], E2F [22], Nuclear Factor kappa from B cells (NF-κB) via Inhibitor kappa B protein Kinase (Iκ-K) [23], and the forkhead transcription factors [24]. These are all either direct or indirect substrates of Akt and each can regulate cellular proliferation, survival, and other important biologic processes. Besides transcription factors, Akt targets a number of other molecules to affect the survival state of the cell including the proapoptotic molecule Bcl-2-Associated Death promoter (BAD) [25] and Glycogen Synthase Kinase-3β (GSK-3β) [26].
Negative regulation of the PI3K pathway is primarily accomplished through the action of the Phosphatase and TENsin homolog deleted on chromosome 10 (PTEN) tumor suppressor protein. PTEN encodes a lipid and protein phosphatase whose primary lipid substrate is PtdIns(3,4,5)P3 [27]. The purported protein substrate(s) of PTEN are more varied, including Focal Adhesion Kinase (FAK), the Shc exchange protein, the transcriptional regulators E-Twenty Six-2 (ETS-2) [28] and Sp1 and the Platelet-Derived Growth Factor Receptor (PDGF-R) [29].
Next, we discuss some of the key targets of Akt that can also contribute to abnormal cellular growth by the regulation of protein translation. Akt-mediated regulation of mTOR activity is a complex multi-step phenomenon. Akt inhibits Tuberous Sclerosis 2 (TSC2 or tuberin) function through direct phosphorylation [30]. TSC2 is a GTPase-Activating Protein (GAP) that functions in association with the putative Tuberous Sclerosis 1 (TSC1 or hamartin) to inactivate the small G protein Ras homolog enriched in brain (Rheb) [31]. TSC2 phosphorylation by Akt represses GAP activity of the TSC1/TSC2 complex, allowing Rheb to accumulate in a GTP-bound state. Rheb-GTP then activates, through a mechanism not yet fully elucidated, the protein kinase activity of mTOR which complexes with Raptor (Regulatory-associated protein of mTOR) adaptor protein, DEP domain-containing mTOR-interacting protein (DEPTOR) and mLST8, a member of the Lethal-with-Sec-Thirteen gene family, first identified in yeast, FK506-Binding Protein 38 (FKBP38) and Proline-Rich Akt Substrate 40 kDa protein (PRAS40). mTORC1 is sensitive to rapamycin and, importantly, inhibits Akt via a negative feedback loop which involves, at least in part, p70S6K [31]. This is due to the negative effects that p70S6K has on IRS1. DEPTOR may be a tumor suppressor gene as decreased expression of DEPTOR results in increased mTORC1 activity [32].
The mechanism by which Rheb-GTP activates mTORC1 has not been fully elucidated yet; however, it requires Rheb farnesylation and can be blocked by Farnesyl Transferase (FT) inhibitors. It has been proposed that Rheb-GTP would relieve the inhibitory function of FKBP38 on mTOR, thus leading to mTORC1 activation [33].
As stated previously, TSC1 and TSC2 have important roles in the regulation of mTORC1. Two additional molecules important in this regulation are Liver Kinase B (LKB1 also known as STK11). LKB1 is an upstream activator of 5′AMP-activated Protein Kinase (AMPK) which activates TSC2 that negatively regulates mTORC1 [34]. LKB1 mediates the effects of the diabetes drug metformin [35]. Metformin has also been shown to be effective in suppressing the developments of certain cancers [36–38].
Akt also phosphorylates PRAS40, an inhibitor of mTORC1, and by doing so, it prevents the ability of PRAS40 to suppress mTORC1 signaling (recently reviewed in [15]). Thus, this could be yet another mechanism by which Akt activates mTORC1. Moreover, PRAS40 is a substrate of mTORC1 itself, and it has been demonstrated that mTORC1-mediated phosphorylation of PRAS40 prevents the inhibition of additional mTORC1 signaling [31].
Ras/Raf/MEK/ERK signaling positively impinges on mTORC1. Both p90Rsk−1 and ERK 1/2 phosphorylate TSC2, thus suppressing its inhibitory function [31]. Moreover, mTORC1 inhibition resulted in ERK 1/2 activation, through p70S6K/PI3K/Ras/Raf/MEK [39].
The relationship between Akt and mTOR is further complicated by the existence of the mTOR/Rictor complex (mTORC2), which, in some cell types, displays rapamycin-insensitive activity. mTORC2 is comprised of Rapamycin-insensitive companion of mTOR (Rictor), mTOR, DEPTOR, mLST8, Stress-activated protein kinase Interacting protein 1 (SIN1), and Protein observed with Rictor (Protor). mTORC2 phosphorylates Akt on S473 in vitro which facilitates T308 phosphorylation [15]. Thus, mTORC2 can function as the elusive PDK-2 which phosphorylates Akt-1 on S473 in response to growth factor stimulation [40]. Akt and mTOR are linked to each other via positive and negative regulatory circuits, which restrain their simultaneous hyperactivation through mechanisms involving p70S6K and PI3K [15, 31]. Assuming that equilibrium exists between these two complexes, when the mTORC1 complex is formed, it could antagonize the formation of the mTORC2 complex and reduce Akt activity. Thus, at least in principle, inhibition of the mTORC1 complex could result in Akt hyperactivation. This is one problem associated with therapeutic approaches using rapamycin or modified rapamycins (rapalogs) that block some actions of mTOR but not all.
mTOR is a 289 kDa S/T kinase. mTOR was the first identified member of the Phosphatidylinositol 3-Kinase-related Kinase (PIKK) family [15]. mTOR has been referred to as the gatekeeper of autophagy [41]. mTOR regulates translation in response to nutrients and growth factors by phosphorylating components of the protein synthesis machinery, including p70S6K and eukaryotic Initiation Factor (eIF)-4E Binding Protein-1 (4EBP-1), the latter resulting in the release of the eukaryotic Initiation Factor-4E (eIF-4E) allowing eIF-4E to participate in the assembly of a translational initiation complex [2]. p70S6K phosphorylates the 40S rpS6, leading to translation of “weak” mRNAs. Integration of a variety of signals (mitogens, growth factors, hormones) by mTOR assures cell cycle entry only if nutrients and energy are sufficient for cell duplication [15].
Unphosphorylated 4E-BP1 interacts with the cap-binding protein eIF4E and prevents the formation of the 4F translational initiation complex (eIF4F) by competing for the binding of eukaryotic Initiation Factor 4G (eIF4G) to eIF4E. 4E-BP1 phosphorylation by mTORC1 results in the release of the eIF4E, which then associates with eIF4G to stimulate translation initiation [15].
eIF4E is a key component for translation of 5′ capped mRNAs, which include transcripts mainly encoding for proliferation and survival promoting proteins, such as c-Myc, cyclin D1, Cyclin-Dependent Kinase-2 (CDK-2), Signal activator and Transducer of Transcription-3 (STAT-3), ornithine decarboxylase, surviving, B-cell lymphoma 2 (Bcl-2), Bcl-xL, Myeloid cell leukemia-1 (Mcl-1) [2].
13.4 Overview of Pathway Inhibitors
Sites of intervention with signal transduction inhibitors in the Ras/Raf/MEK/ERK are presented in Fig. 13.1. Some of the inhibitors are currently being used to treat patients with specific cancers, and others have been or are being evaluated in numerous clinical trials with many different types of cancer patients. Effective inhibitors, specific for many of the key components of the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/mTOR pathways, have been developed [1, 15]. In many cases, these inhibitors have been examined in clinical trials. Furthermore, inhibitors that target the mutant protein more than the Wild-Type (WT) protein of various genes (e.g., BRAF and PIK3CA) either have been or are being characterized.
13.4.1 Raf Inhibitors
Raf inhibitors have been developed, and some are being used for therapy while others are being evaluated in clinical trials. Raf inhibitors have, in general, exhibited greater response rates in clinical trials than MEK inhibitors which may be related to the broader therapeutic index of Raf inhibitors that suppress ERK activity in a mutant-allele-specific fashion as opposed to MEK inhibitors which suppress MEK activity in tumor and normal cells [42].
13.4.1.1 Sorafenib
Sorafenib (Bayer) was initially thought to specifically inhibit Raf but has been subsequently shown to have multiple targets (e.g., VEGF-R, Flt-3, PDGF-R) [43]. However, that does not preclude its usefulness in cancer therapy. Sorafenib is approved for the treatment for certain cancers (e.g., Renal Cell Carcinoma (RCC) and patients with unresectable HCC and was further evaluated in the Sorafenib Hepatocellular carcinoma Assessment Randomized Protocol (SHARP) trial, which demonstrated that the drug was effective in prolonging median survival and time to progression in patients with advanced HCC [44, 45]. Sorafenib is generally well tolerated in HCC patients with a manageable adverse events profile [44]. While sorafenib is not considered effective for the treatment for most melanomas with BRAF V600E mutations, it may be effective in the treatment for a minority of melanomas with G469E and D594G mutations which express constitutive ERK1/2 but low levels of MEK. These melanomas are sensitive to sorafenib, potentially because they signal through Raf-1. Raf-1 also exerts anti-apoptotic effects at the mitochondrion in association with Bcl-2 family members [46].
13.4.1.2 Vemurafenib
Vemurafenib (Zelboraf, PLX-4032, Plexxikon/Roche) is a B-Raf inhibitor that has and is being evaluated in many clinical trials [47–49]. Vemurafenib has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with unresectable or metastatic melanoma carrying the BRAF(V600E) mutation. For vemurafenib to be clinically effective, it needs to suppress downstream ERK activation essentially completely [47].
13.4.1.3 Dabrafenib
Dabrafenib (GSK2118436) is an ATP-competitive inhibitor of mutant B-Raf, WT B-Raf, and WT Raf-1 developed by GlaxoSmithKlein (GSK) [50]. Dabrafenib is in clinical trials [51, 52].
13.4.1.4 CCT239065
CCT239065 is a mutant B-Raf inhibitor developed at the Institute of Cancer Research in London, UK. It inhibits mutant BRAF V600E signaling and proliferation more than those cells containing WT BRAF [53]. Its effects are more selective for cells containing mutant BRAF than WT BRAF. CCT239065 is well tolerated in mice and had good oral bioavailability. It suppressed tumors containing BRAF-mutant gene but not WT BRAF tumors in mice tumor xenograft studies.
13.4.1.5 GDC-0879
GDC-0879 is a BRAF-mutant allele-selective inhibitor developed by Genentech [54]. The efficacy GDC-0879 is related to the BRAF V600E mutational status in the cancer cells and inhibition of downstream MEK and ERK activity.
13.4.1.6 AZ628
AZ628 is a selective Raf inhibitor developed by Astra Zeneca. It has been shown that when BRAF-mutant melanoma cells, which are normally very sensitive to AZ628, are grown for prolonged periods of time, they become resistant to AZ628 by upregulating the expression of Raf-1 [55].
13.4.1.7 XL281
XL281 is an oral active wild-type and mutant RAF kinase-selective inhibitor developed by Exelixis and Bristol-Myers Squibb. It has been examined in clinical trials primarily with patients having BRAF mutations (ColoRectal cancers (CRC), melanoma, Papillary Thyroid Cancers (PTC), and NSCLC) [56].
13.4.1.8 PLX5568
PLX5568 is a selective Raf kinase inhibitor developed by Plexxicon. It is being examined for the treatment for Polycystic Kidney Disease (PKD). In the kidney, Raf-1 is localized to the tubular cells where it is linked with many physiologically important functions. PLX5568 suppressed cyst enlargement in a rat model of PKD but did not improve kidney function as fibrosis was not suppressed [57].
13.4.1.9 Raf-265
Raf-265 is an ATP-competitive pan-Raf inhibitor developed by Novartis. Treatment for bronchus carcinoid NCI-H727 and CM-insulinoma cells with Raf-265 enhanced sensitivity to TRAIL-induced apoptosis. These cells were normally resistant to PI3K/mTOR inhibitors when combined with TRAIL. Raf-265 was shown to decrease Bcl-2 levels which correlated with their sensitivity to TRAIL-mediated apoptosis. This approach may be effective in the therapy of neuroendocrine tumors [58].
13.4.1.10 Regorafenib
Regorafenib (BAY 73-4506) is an oral multi-kinase inhibitor of angiogenic, stromal, and oncogenic RTKs developed by Bayer. Regorafenib inhibits RTKs such as VEGF-R2, VEGF-R1/3, PDGF-Rβ, fibroblast growth factor receptor-1 as well as mutant Kit, RET, and B-Raf. The effects of regorafenib on tumor growth have been evaluated in human xenograft models in mice, and tumor shrinkages were observed in breast MDA-MB-231 and renal 786-O carcinoma models [59].
13.4.2 MEK Inhibitors
Most MEK inhibitors differ from most other kinase inhibitors as they do not compete with ATP binding (non-ATP competitive), which confers a high specificity [60–62]. Most MEK inhibitors are specific and do not inhibit many different protein kinases [62] although as will be discussed below, certain MEK inhibitors are more specific than others.
Molecular modeling studies indicate that many MEK bind to an allosteric binding site on MEK1/MEK2. The binding sites on MEK1/MEK2 are relatively unique to these kinases and may explain the high specificity of MEK inhibitors. This binding may lock MEK1/2 in an inactivate conformation that enables binding of ATP and substrate, but prevents the molecular interactions required for catalysis and access to the ERK activation loop [61].
A distinct advantage of inhibiting MEK is that it can be targeted without knowledge of the precise genetic mutation that results in its aberrant activation. This is not true with targeting Raf as certain Raf inhibitors will activate Raf and also certain B-Raf-specific inhibitors will not be effective in the presence of RAS mutations.
An advantage of targeting MEK is that the Ras/Raf/MEK/ERK pathway is a convergence point where a number of upstream signaling pathways can be blocked with the inhibition of MEK. For example, MEK inhibitors, such as Selumetinib (AZD6244), are also being investigated for the treatment for pancreatic cancers, breast cancers, and other cancers such as hematopoietic malignancies, including multiple myeloma [1, 63].
13.4.2.1 Selumetinib
Selumetinib inhibits MEK1 in vitro with an IC50 value of 14.1 ± 0.79 nM [64–67]. It is specific for MEK1 as it did not appear to inhibit any of the approximately 40 other kinases in the panel tested. Selumetinib is not competitive with ATP. Selumetinib inhibited downstream ERK1/ERK2 activation in in vitro cell line assays with stimulated and unstimulated cells and also inhibited activation in tumor transplant models. Selumetinib did not prevent the activation of the related ERK5 that occurs with some older MEK1 inhibitors, which are not being pursued in clinical trials. Inhibition of ERK1/2 suppresses their ability to phosphorylate and modulate the activity of Raf-1, B-Raf, and MEK1 but not MEK2 as MEK2 lacks the ERK1/ERK2 phosphorylation site. In essence, by inhibiting ERK1/2 the negative loop of Raf-1, B-Raf, and MEK phosphorylation is suppressed, and hence, there will be an accumulation of activated Raf-1, B-Raf, and MEK [67]. This biochemical feedback loop may provide a rationale for combining Raf and MEK inhibitors in certain therapeutic situations. Selumetinib has also been shown to suppress cetuximab-resistant CRCs which had KRAS mutations both in vitro and in vivo models [68].
13.4.2.2 PD-0325901
The PD-0325901 MEK inhibitor is an orally active, potent, specific, non-ATP-competitive inhibitor of MEK. PD-0325901 demonstrated improved pharmacological and pharmaceutical properties compared with PD-184352, including a greater potency for inhibition of MEK and higher bioavailability and increased metabolic stability. PD-0325901 has a Ki value of 1 nM against MEK1 and MEK2 in in vitro kinase assays. PD-0325901 inhibits the growth of cell lines that proliferate in response to elevated signaling of the Raf/MEK/ERK pathways [62]. PD-0325901 has undergone phase I clinical trials [62, 69–71]. Although the initial trial results were not encouraging, it was determined that some tumors which proliferate in response to the Raf/MEK/ERK pathways may be sensitive to PD0325901 [72]. Although the clinical trials with PD-0325901 were initially suspended, there are now some clinical trials with PD-0325901 in combination with other pathway inhibitors.
13.4.2.3 Refametinib
Refametinib (RDEA119) is a more recently described MEK inhibitor developed by Ardea Biosciences [73]. It is a highly selective MEK inhibitor that displays a >100-fold selectivity in kinase inhibition in a panel of 205 kinases. In contrast, in the same kinase specificity analysis, other recently developed MEK inhibitors (e.g., PD0325901) also inhibited the Src and RON kinases.
13.4.2.4 Trametinib
Trametinib (GSK1120212) is an allosteric MEK inhibitor developed by GSK. It has been shown to be effective when combined with dabrafenib in certain dabrafenib-resistant BRAF V600 melanoma lines that also had mutations at NRAS or MEK1 [52]. The combination of trametinib and the PI3K/mTOR dual inhibitor GSK2126458 also enhanced cell growth inhibition in these B-Raf inhibitor-resistant BRAF-mutant melanoma lines.
13.4.2.5 GDC-0973
GDC-0973 (XL518) is a potent and selective MEK inhibitor developed by Genentech [74]. The effects of combining GDC-0973 and the PI3K inhibitor GDC-0941 on the proliferation of BRAF and KRAS mutant cancer cells indicated the combination efficacy both in vitro and in vivo.
13.4.2.6 AS703026
AS703026 (MSC1936369B) is a MEK inhibitor developed by EMD Serono. AS703026 suppressed cetuximab-resistant CRCs which had KRAS mutations both in vitro and in vivo models [68]. AS703026 inhibited growth and survival of Multiple Myeloma (MM) cells and cytokine-induced differentiation more potently than selumetinib, and importantly, AS703026 was cytotoxic, where as most MEK inhibitors are cytostatic [75]. AS703026 sensitized MM cells to a variety of conventional (dexamethasone, melphalan) and novel (lenalidomide, perifosine, bortezomib, rapamycin) drugs used to treat MM.
13.4.2.7 RO4987655
RO4987655 (CH4987655) is an allosteric, orally available MEK inhibitor developed by Roche/Chiron. It has been tested in humans and determined to inhibit active ERK levels. At the levels of RO4987655 administered, it was determined to be safe in healthy volunteers [76].
13.4.2.8 TAK-733
TAK-733 is a potent and selective, allosteric MEK inhibitor developed by Takeda San Diego [77]. TAK-733 is being investigated in clinical trials.
13.4.2.9 MEK162
MEK162 (ARRY-162) is a MEK inhibitor developed by Novartis. It is in clinical trials.
13.4.2.10 SL327
SL337 is a MEK inhibitor that has been used in many neurological and drug addiction studies [78].
13.4.2.11 Other MEK Inhibitors
Other MEK inhibitors are being developed. RG422 is one such inhibitor.
13.4.3 Combining Raf and MEK Inhibitors
The possibility of treating certain patients with Raf and MEK inhibitors is a concept which is gaining more acceptance as it may be a therapeutic possibility to overcome resistance [42]. Raf inhibitors induce Raf activity in cells with WT RAF if Ras is active [79]. The addition of a MEK inhibitor would suppress the activation of MEK and ERK in the normal cells of the cancer patient. Thus, B-Raf would be suppressed by the B-Raf-selective inhibitor in the cancer patient, while the consequences of Raf activation in the normal cells would be suppressed by the MEK inhibitor. These concepts are being examined in clinical trials.
13.4.4 Combining MEK and Bcl-2 Inhibitors
The effects of combining MEK and Bcl-2/Bcl-XL inhibitors have been examined in preclinical studies with AML cell lines and patient samples [80]. The Bcl-2 inhibitor, ABT-737, was observed to induce ERK activation and Mcl-1 expression. However, when the ABT-737 inhibitor was combined with the MEK inhibitor PD0325901, a synergistic response was observed in terms of the induction of cell death both on AML cell lines and on primary tumor cells with the properties of leukemia stem cells. Furthermore, these studies were also extended into tumor transplant models with the MOLT-13 cell line, and synergy between ABT-737 and PD0325901 were also observed in vivo.
13.4.5 ERK Inhibitors
There are at least two ERK molecules regulated by the Raf/MEK/ERK cascade, ERK1 and ERK2. Little is known about the differential in vivo targets of ERK1 and ERK2. The development of specific ERK1 and ERK2 inhibitors is ongoing and may be useful in the treatment for certain diseases such as those leukemias where elevated ERK activation is associated with a poor prognosis (e.g., AML, ALL) [81]. ERK inhibitors have been described [82].
13.4.5.1 AEZS-131
AEZS-131 has been reported on the Internet to be a highly selective ERK 1/2 inhibitor developed by Aeterna Zentaris and has been examined on human breast cancer cells.
13.4.5.2 Pyrimidylpyrrole ERK Inhibitors
A novel series of pyrimidylpyrrole ERK inhibitors has been developed at Vertex Pharmaceuticals [82]. A lead compound, ERKi, has been evaluated for its ability to overcome MEK inhibitor resistance [83]. These studies performed in breast and CRCs demonstrated that dual inhibition of MEK and ERK by small molecule inhibitors was synergistic. Furthermore, inhibition of both MEK and ERK acted to suppress the emergence of resistance and overcome the acquired resistance to MEK inhibitors in these breast and CRC cell line models.
13.4.5.3 SCH772984
SCH772984 is reported to be an ERK inhibitor.
13.4.6 PI3K/Akt/mTOR Inhibitors
Numerous PI3K, Akt, mTOR, and dual PI3K/mTOR inhibitors have been developed and evaluated. The PI3K and mTOR inhibitors have been used in basic science studies for years and have provided much information about the role of the PI3K/Akt/mTOR pathway in many biologic and diseases processes. We will focus on the newer inhibitors of this pathway and how they are now being used in clinical trials.
13.4.6.1 PX-866
The modified wortmannin PX-866 has been evaluated as a PI3K inhibitor [84]. It is being evaluated in phase II clinical trials for patients with advanced metastatic prostate cancer by Oncothyreon.
13.4.6.2 GDC-0941
GDC-0941 is a PI3K inhibitor developed by Genentech. GDC-0941 inhibited the metastatic characteristics of thyroid carcinomas by targeting both PI3K and Hypoxia-Inducible Factor 1α (HIF-1α) pathways [85]. GDC-0941 synergized with the MEK inhibitor UO126 in inhibiting the growth of NSCLC [86]. It is being evaluated in a clinical trial for advanced cancers or metastatic breast cancers which are resistant to aromatase inhibitor therapy.
13.4.6.3 IC87114
IC87114 is a selective p110δ PI3K inhibitor. It decreased cell proliferation and survival in AML cells and increased sensitivity to etoposide [87–90].
13.4.6.4 CAL-101
CAL-101(GS-1101) is a derivative of IC87114 [91–93]. CAL-101 is an oral p110δ PI3K inhibitor developed by Calistoga Pharmaceuticals and Gilead Sciences. CAL-101 is currently undergoing clinical evaluation in patients with various hematopoietic malignancies including relapsed or refractory indolent B-cell NHL, mantle cell lymphoma or CLL. An additional clinical trial will examine the effects of combining CAL-101 with chemotherapeutic drugs and the αCD20 Monoclonal Ab (MoAb).
13.4.6.5 XL-147 (SAR245408)
XL-147 (SAR245408) is a PI3K inhibitor developed by Exelixis/Sanofi-Aventis [94]. It is in clinical trials, either as a single agent or in combination with erlotinib, hormonal therapy, chemotherapy, or MoAb therapy for various cancers including lymphoma, breast, endometrial, glioblastoma, astrocytoma, or other solid cancers.
13.4.6.6 Novartis PI3K Inhibitors
NVP-BKM120 is an orally available pan-class I PI3-kinase inhibitor developed by Novartis [95]. It is in many clinical trials, either as a single agent or in combination with other drugs or signal transduction inhibitors [96]. NVP-BKM120 is clinical trial with patients having advanced cancers such as CRC, NSCLC, breast, prostate, endometrial, squamous cell carcinoma of the head and neck, GIST, RCC, melanoma, and advanced leukemias.
NVP-BYL719 (BYL719) is a PI3Kα-selective inhibitor developed by Novartis. It is in clinical trials for patients with advanced solid tumors, some containing mutations at PIK3CA. It is also being examined in a clinical trial in combination with the MEK-162 inhibitor for patients with advanced CRC, esophageal, pancreatic, NSCLC, or other advanced solid tumors containing RAS or BRAF mutations.
13.4.7 Dual PI3K/mTOR Inhibitors
The catalytic sites of PI3K and mTOR share a high degree of sequence homology. This feature has allowed the synthesis of ATP-competitive compounds that target the catalytic site of both PI3K and mTOR. Several dual PI3K/mTOR inhibitors have also been developed. In preclinical settings, dual PI3K/mTOR inhibitors displayed a much stronger cytotoxicity against leukemic cells than either PI3K inhibitors or allosteric mTOR inhibitors, such as rapamycin and its derivatives (rapalogs). In contrast to rapamycin/rapalogs, dual PI3K/mTOR inhibitors targeted both mTOR complex 1 and mTOR complex 2 and inhibited the rapamycin-resistant phosphorylation of eIF4B-1 and inhibited protein translation of many gene products associated with oncogenesis (enhanced proliferation) in leukemic cells. The dual inhibitors strongly reduced the proliferation rate and induced an important apoptotic response [16].
The kinase selectivity profile of the dual PI3K/mTOR modulators is consistent with the high sequence homology and identity in the ATP-catalytic cleft of these kinases. Dual PI3K/mTOR inhibitors have demonstrated significant, concentration-dependent cell proliferation inhibition and induction of apoptosis in a broad panel of tumor cell lines, including those harboring PI3K p110α (PIK3CA) activating mutations [97].
Moreover, the in vitro activity of these ATP-competitive PI3K/mTOR modulators has translated well in in vivo models of human cancer xenografted in mice. They were well tolerated and achieved disease stasis or even tumor regression when administered orally [98]. In spite of their high lipophilicity and limited water solubility, the pharmacological, biologic, and preclinical safety profiles of these dual PI3K/mTOR inhibitors supported their clinical development.
There may be some benefits to treating patients with an inhibitor that can target both PI3K and mTOR as opposed to treating patients with two inhibitors, that is, one targeting PI3K and another specifically mTOR. An obvious benefit could be lowered toxicities. Treatment with a single drug could have fewer side effects than treatment with two separate drugs. The effects of detrimental Akt activation by mTOR inhibition might be avoided upon treatment with a dual kinase inhibitor. Furthermore, the negative side effects of mTOR inhibition on the activation of the Raf/MEK/ERK pathway might be eliminated with the PI3K inhibitor activity in the dual inhibitor. There remains, however, considerable uncertainty about potential toxicity of compounds that inhibit both PI3K and mTOR enzymes whose activities are fundamental to a broad range of physiological processes. Although it should be pointed out that there are some clinical trials in progress to determine whether it is beneficial to treat cancer patients with a PI3K/mTOR dual inhibitor and an mTORC1 blocker such as NVP-BEZ235 and RAD001, preclinical studies have documented the benefits of combining RAD001 with NVP-BEZ235 [99].
13.4.7.1 PI-103
PI-103 was the first reported ATP-competitive kinase inhibitor of mTOR which also blocked the enzymatic activity of PI3K p110 isoforms. It was developed at UCSF in 2006. PI-103 exhibits good selectivity over the rest of the human kinome in terms of non-selective inhibition of other kinases [100, 101]. PI-103 is a pan-class I PI3K inhibitor with IC50 values in the 2 nm (p110α PI3K) to 15 nm range (p110γ PI3K) PI-103 inhibits both mTORC1 (IC50 = 0.02 μm) and mTORC2 (IC50 = 0.083 μm).
13.4.7.2 Novartis Dual PI3K/mTOR Inhibitors
NVP-BEZ235 is a dual PI3 K/mTOR inhibitor developed by Novartis. Importantly and in contrast to rapamycin, NVP-BEZ235 inhibited the rapamycin-resistant phosphorylation of 4E-BP1, causing a marked inhibition of protein translation in AML cells. This resulted in the reduced levels of the expression of c-Myc, cyclin D1, and Bcl-xL known to be regulated at the translation initiation level [102]. NVP-BEZ235 suppressed proliferation and induced an important apoptotic response in AML cells without affecting healthy CD34+ cell survival. Importantly, it suppressed the clonogenic activity of leukemic, but not healthy, CD34+ cells [103]. NVP-BEZ235 targeted the Side Population (SP) of both T-ALL cell lines and patient lymphoblasts, which might correspond to Leukemia-Initiating Cells (LIC), and synergized with several chemotherapeutic agents (cyclophosphamide, cytarabine, dexamethasone) currently used for treating T-ALL patients [104]. Also, NVP-BEZ235 reduced chemoresistance to vincristine induced in Jurkat cells by co-culturing with MS-5 stromal cells, which mimic the bone marrow microenvironment [105]. In this study, NVP-BEZ235 was cytotoxic to T-ALL patient lymphoblasts displaying pathway activation, where the drug dephosphorylated 4E-BP1, in contrast to the results with obtained rapamycin. Taken together, these findings indicated that longitudinal inhibition at two nodes of the PI3K/Akt/mTOR network with NVP-BEZ235, either alone or in combination with chemotherapeutic drugs, may be an effective therapy for of those T-ALLs that have aberrant upregulation of this signaling pathway.
NVP-BEZ235 has been evaluated also in a mouse model consisting of BA/F3 cells overexpressing either WT BCR-ABL or its imatinib-resistant BCR-ABL mutants (E255K and T315I) [106]. NVP-BEZ235 inhibited proliferation of both cytokine-independent WT BCR-ABL and mutant BCR-ABL (E255K and T315I) overexpressing cells, whereas parental cytokine-dependent Ba/F3 cells were much less sensitive. The drug also induced apoptosis and inhibited both mTORC1 and mTORC2 signaling. Remarkably, the drug displayed cytotoxic activity in vivo against leukemic cells expressing the E255K and T315I BCRABL mutant forms. However, in this experimental model, NVP-BEZ235 induced an overactivation of MEK/ERK signaling, most likely due to the well-known compensatory feedback mechanism that involves p70S6K [39]. NVP-BEZ235 has been intensively investigated and is in clinical trials for patients with advanced cancers [107]. In some trials, NVP-BEZ235 is being evaluated in combination with either paclitaxel or trastuzumab (herceptin). NVP-BTG226 is a recently developed PI3K/mTOR inhibitor [15].
13.4.7.3 Pfizer Dual PI3K/mTOR Inhibitors
PKI-587, also known as PF-05212384, inhibited class I PI3Ks, PI3Kα mutants, and mTOR. PKI-587-suppressed proliferation of approximately 50 diverse human tumor cell lines at IC50 values less than 100 nmol/L. PKI-587-induced apoptosis in cell lines with elevated PI3K/Akt/mTOR signaling. PKI-587 inhibited the tumor growth in various models including breast (MDA-MB-361, BT474), colon (HCT116), lung (H1975), and glioma (U87MG). The efficacy of PKI-587 was enhanced when administered in combination with the MEK inhibitor, PD0325901, the topoisomerase I inhibitor, irinotecan, or the HER2 inhibitor, neratinib [108].
PF-04691502 is an ATP-competitive PI3K/Akt inhibitor which suppresses the activation of Akt. PF-04691502 suppressed the transformation of avian cells in response to either WT or mutant PIK3CA. PF-04691502 inhibited tumor growth in various xenograft models including U87 (PTEN null), SKOV3 (PIK3CA mutation) and gefitinib (EGFR inhibitor) and erlotinib-resistant NSCLC [109]. Both PKI-587 and PF-04691502 are in clinical trials to treat endometrial cancers.
PKI-402 is a selective, reversible, ATP-competitive, PI3K and mTOR inhibitor. It suppress PI3Ks, PI3Kα mutant, and mTOR equally. PKI-402 inhibited the growth of many human tumor cell lines including breast, glioma, pancreatic, and NSCLC [110].
13.4.7.4 XL765
XL765 (SAR25409) is a dual PI3K/mTOR inhibitor developed by Exelixis/Sanofi-Aventis. XL765 has been investigated in brain and pancreatic cancer models either as a single agent or in combination with temozolomide [111] or the autophagy inhibitor chloroquine [112]. XL765 downregulated the phosphorylation of Akt induced by PI3K/mTORC2 and reduced brain tumor growth [111]. Combining XL765 with chloroquine suppressed autophagy and induced apoptotic cell death in pancreatic tumor models [112]. Clinical trials are being performed with XL765 in combination with temozolomide to treat patients with glioblastoma or in combination with erlotinib to treat NSCLC patients.
13.4.7.5 Genentech Dual PI3K/mTOR Inhibitors
GNE-477 is a dual PI3K/mTOR inhibitor developed by Genentech [113]. GDC-0980 is similar to GNE-477 and has been shown to have high activity in cancer models driven by PI3K pathway activation [114]. GDC-0980 is in a clinical trial for patients with advanced cancers or metastatic breast cancers which are resistant to aromatase inhibitor therapy.
13.4.7.6 GSK Dual PI3K/mTOR Inhibitors
GSK2126458 is a dual PI3K/mTOR inhibitor developed by GSK [52]. It is in at least two clinical trials with advanced cancer patients. In one trial, it is being combined with the MEK inhibitor GSK1120212. GSK1059615 is a dual PI3K/mTOR inhibitor developed by GSK. It was in a clinical trial with patients with solid tumors, metastatic breast cancer, endometrial cancers, and lymphomas which was terminated.
13.4.7.7 WJD008
WJD008 (Chinese Academy of Sciences, Shanghai) is a dual PI3K/mTOR [115]. WJD008 inhibited the increased activity of the PI3K pathway normally induced by PIK3CA H1047R and suppressed proliferation and colony formation of transformed RK3E cells containing PIK3CA H1047R.
13.4.8 PDK Inhibitors
Some compounds have been reported to be PDK inhibitors, including the osteoarthritis drug celecoxib [116], the modified celecoxib, OSU-03012 [76, 117], and 2-O-BN-InsP(5) [118]. Celecoxib (Celebrex, Pfizer) obviously has other targets than PDK, such as Cyclooxygenase-2 (Cox-2). Celecoxib is used to treat CRC patients to reduce the number of polyps in the colon. OSU-03012 is reported not to inhibit Cox-2 [117]. 2-O-BN-InsP(5) is based on the structure of inositol 1,3,4,5,6-pentakisphosphate, it may inhibit both PDK and mTOR [118].
13.4.9 Akt Inhibitors
Many attempts to develop Akt inhibitors have been performed over the years. In many of the earlier attempts, the various Akt inhibitors either lacked specificity or had deleterious side effects. Part of the deleterious side effects is probably related to the numerous critical functions that Akt plays in normal physiology. Namely, some Akt inhibitors will alter the downstream effects of insulin on Glut-4 translocation and glucose transport.
13.4.9.1 Triciribine
Triciribine (API-2) is an Akt inhibitor that has been used in many studies: at least 92 are listed on PubMed. Triciribine suppressed the phosphorylation of all three Akt isoforms in vitro and the growth of tumor cells overexpressing Akt in mouse xenograft models [119]. The mechanism(s) by which triciribine inhibits Akt activity are not clear. The drug has been evaluated in a phase I clinical trial in patients with advanced hematologic malignancies, including refractory/relapsed AML. In this trial, triciribine was administered on a weekly schedule. The drug was well tolerated, with preliminary evidence of pharmacodynamic activity as measured by decreased levels of activated Akt in primary blast cells [120]. Triciribine has also been examined in clinical trial with Akt+ metastatic cancers.
13.4.9.2 MK-2206
MK-2206 (Merck) is an allosteric Akt inhibitor which inhibits both T308 and S473 phosphorylation. It also inhibits the downstream effects of insulin on Glut-4 translocation and glucose transport [121]. MK-2206 decreased T-Acute Lymphocytic Leukemia (T-ALL) cell viability by the cells in the G0/G1 phase of the cell cycle and inducing apoptosis. MK-2206 also induced autophagy in the T-ALL cells. MK-2206 induced a concentration-dependent dephosphorylation of Akt and its downstream targets, GSK-3α/β and FOXO3A. MK-2206 also was cytotoxic to primary T-ALL cells and induced apoptosis in a T-ALL patient cell subset (CD34+/CD4−/CD7−) which is enriched in LICs. [122]. MK-2206 is in at least 43 clinical trials either as a single agent or in combination with other small molecule inhibitors or chemotherapeutic drugs with diverse types of cancer patients.
13.4.9.3 GSK Akt Inhibitors
GSK690693 is a pan-Akt inhibitor developed by GSK. GSK690693 is an ATP-competitive inhibitor effective at the low nanomolar range. Daily administration of GSK690693 resulted in significant anti-tumor activity in mice bearing various human tumor models including SKOV-3 ovarian, LNCaP prostate and BT474 and HCC-1954 breast carcinoma. The authors also noted that GSK690693 resulted in acute and transient increases in blood glucose level [123]. The effects of GSK690693 were also examined 112 cell lines representing different hematologic neoplasia. Over 50 % of the cell lines were sensitive to the Akt inhibitor with an EC50 of less than 1 μm. ALL, non-Hodgkin lymphomas, and Burkitt lymphomas exhibited 89, 73, and 67 % sensitivity to GSK690693, respectively. Importantly, GSK690693 did not inhibit the proliferation of normal human CD4+ peripheral T lymphocytes as well as mouse thymocytes.
GSK2141795 is a GSK Akt inhibitor under development. It is reported by GSK to be an oral, pan-Akt inhibitor which shows activity in various cancer models, including blood cancer and solid tumor models. In addition, it is reported by GSK to delay tumor growth in solid tumor mouse xenograft models. It has been investigated further in clinical trials.
13.4.9.4 KP372-1
KP372-1 inhibits PDK1, Akt, and Fms-like tyrosine kinase 3 (Flt-3) signaling and induces mitochondrial dysfunction and apoptosis in AML cells but not normal hematopoietic progenitor cells [124]. It also suppressed colony formation of primary AML patient sample cells but not normal hematopoietic progenitor cells. It has also been investigated in other cancer types, including squamous cell carcinomas of the head and neck, thyroid cancers, and glioblastomas.
13.4.9.5 Enzasturin
Enzasturin (LY317615) is a Protein Kinase C-β (PKC-β) and Akt inhibitor developed by Lilly. It has been investigated in clinical trials either by itself or in combination with other agents in various types of cancer patients including brain [125] and NSC [126], CRC [127] as well as other cancer types. It is reported to be in approximately 48 clinical trials on the ClinicalTrials.gov website.
13.4.9.6 Perifosine
Perifosine (KRX-0401, Keryx/AOI Pharmaceuticals, Inc., and licensed to AEterna Zentaris) is an alkylphospholipid that can inhibit Akt [128]. The effects of perifosine have been examined on many different tumor types. Perifosine induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multi-drug-resistant T-ALL cells by a JNK-dependent mechanism [104]. Perifosine is or has been in at least 43 clinical trials to treat various cancer patients, with either blood cancers or solid tumors, either by itself, or in combination with other agents. It has advanced to phase III clinical trials for CRC and MM. In the USA, it has orphan drug status for the treatment for MM and neuroblastoma.
13.4.9.7 Erucylphosphocholine (ErPC) and Erucylphosphohomocholine (ErPC3)
Erucylphosphocholine (ErPC) and Erucylphosphohomocholine (ErPC3) have been shown to inhibit Akt and induce apoptosis in malignant glioma cell lines which are normally resistant to the induction of apoptosis. They are structurally related to perifosine [129]. ErPC enhanced radiation-induced cell death and clonogenicity [130]. These effects on the induction of apoptosis were correlated with increased Bim levels and decreased Bad and Foxo-3 phosphorylation, potentially consequences of decreased Akt activity. ErPC3 is the first intravenously applicable alkylphosphocholine. ErPC3 was cytotoxic to AML cells through JNK2- and PP2-dependent mechanisms [131].
13.4.9.8 PBI-05204
PBI-05204 (oleandrin) is an Akt inhibitor. PBI-05024 is a botanical drug candidate derived from Nerium oleander and developed by Phoenix Biotechnology. It also has other targets including FGF-2, NF-κB, and p70S6K. PBI-05204 is in clinical trials for cancer patients with advanced solid tumors [132]. Interestingly, PBI-05204 also provides significant neuroprotection to tissues damaged by glucose and oxygen deprivation which occurs in ischemic stroke [133].
13.4.9.9 RX-0201
RX-0201 (Akt1AO, Rexahn Pharmaceuticals, Inc.) is an Akt-1 antisense oligonucleotide molecule. RX-0201 downregulated Akt-1 expression at nanomolar concentrations in multiple types of human cancer cells. RX-0201 also inhibited tumor growth in mice xenografted with U251 human glioblastoma and MIA human pancreatic cancer cells [134]. RX-021 is in a clinical trial in combination with gemcitabine for patients with metastatic pancreatic cancer [135].
13.4.9.10 XL-418
XL-418 is reported to be a dual Akt/p70S6K inhibitor by developed by Exelixis/GSK. It was in clinical trials for patients with advanced cancer; however, those trials were suspended.
13.4.10 mTORC1 Inhibitors
Rapamycin (Rapamune, Pfizer) was approved by the FDA in 1999 to prevent transplant rejection in organ transplant patients. Rapamycin/rapalogs act as allosteric mTORC1 inhibitors and do not directly affect the mTOR catalytic site [15]. They associate with the FK506-Binding Protein 12 (FKBP-12) and by so doing, they induce disassembly of mTORC1, resulting in the repression of its activity [136, 137]. The rapalogs have been examined in clinical trials of various cancers including brain, breast, HCC, leukemia, lymphoma, MM, NSCLC, pancreatic, prostate, and RCC [138, 139]. The rapalogs Torisel (Pfizer) and Afinitor (Novartis) were approved in 2007 and 2009 (respectively) to treat RCC patients [140]. In 2008, Torisel was approved to treat Mantle cell lymphoma patients. In 2010, Afinitor was approved to treat Subependymal Giant cell Astrocytoma (SEGA) tumors in Tuberous Sclerosis (TS) patients. In 2011, Afinitor was approved to treat patients with pancreatic neuroendocrine tumors [141]. Ridaforolimus (also known as AP23573 and MK-8669; formerly known as deforolimus) is a rapalog developed by ARIAD and Merck. Ridaforolimus has been evaluated in clinical trials with patients having metastatic soft-tissue or bone sarcomas where it displays promising results in terms of the risk of progression or death [142]. Recently, the ability of rapamycin and rapalog to treat various viral infections including AIDS has been considered [143, 144]. Clearly, rapamycin has proven to be a very useful drug.
13.4.11 mTOR Inhibitors
Small molecules designed for inhibiting the catalytic site of mTOR have shown promising effects on the suppression of signaling downstream of mTOR. mTOR kinase inhibitor has been developed which directly inhibits mTORC1 and mTORC2. The mTOR kinase inhibitors have advantages over rapamycin and the rapalogs as mTOR inhibitors will inhibit both mTORC1 and mTORC2, while rapamycin and the rapalogs only inhibit mTORC1. Also, the mTOR kinases inhibitors do not induce the feedback pathways which result in Akt activation. In vitro studies with purified mTOR and PI3K proteins have demonstrated that the mTOR inhibitors selectively bind mTOR more than PI3K.
13.4.11.1 OSI-027
OSI-027 is a pan-TOR inhibitor developed by OSI Pharmaceuticals/Astellas Pharma Inc. OSI-027 has been shown to be effective in inducing apoptosis in different types of cancer, including breast and leukemias [145, 146]. OSI-027 has been shown to inhibit the growth of imatinib-resistant CML cells which contain the BCR-ABL T315I mutation that are resistant to all BCR-ABL inhibitors [147]. OSI-027 has been evaluated in clinical trials with patients with advanced solid tumors and lymphoma [148].
13.4.11.2 Intelllikine mTOR Inhibitors
PP-242 is a potent inhibitor of both mTORC1 and mTORC2. INK-128 is a derivative of PP-242 which has shown anti-tumoral effects on multiple cancer types including RCC, MM, NHL, and prostate [149, 150]. INK-128 is in phase I clinical trials for patients with relapsed or refractory multiple myeloma or Waldenstrom’s macroglobulinemia or patients with solid malignancies.
13.4.11.3 AstraZenica mTOR Inhibitors
AZD8055 and AZD2014 are pan-mTOR inhibitors with potent anti-tumor activity [151]. They are being evaluated in clinical trials patients with gliomas who have not responded to standard glioma therapies as well as patients with other types of cancer.
13.4.11.4 Palomid 529
Palomid 529 (Paloma Pharmaceuticals) is a pan-mTOR inhibitor which has potent anti-tumor affects and reduces tumor angiogenesis and vascular permeability [152]. Palomid 529 is undergoing phase I clinical trials for patients with macular degeneration.
13.4.11.5 Pfizer mTOR Inhibitors
WAY600, WYE353, WYE687, and WYE132 were developed by Wyeth (Pfizer). These inhibitors were derived from WAY001 which was more specific for PI3Kα than either mTORC1 or mTORC2. These inhibitors were modified which resulted in WYE132 (WYE125132)/WYE132 has 5000-fold greater selectivity for mTOR over PI3K. It caused tumor regression in breast, glioma, lung, renal tumors [153].
13.4.11.6 Other mTOR Inhibitors
Many other TOR inhibitors have been described which include Ku0063794 (KuDOS Pharmaceuticals) [154] and OXA-01 (OSI Pharmaceuticals) [155]. Torin2 has been developed by optimizing from Torin1 [156]. TORKiCC223 is a pan-TOR inhibitor developed by Celgene. Other companies are developing mTOR inhibitors; clearly, this is a very competitive but important research and clinical area.
13.5 Increasing the Effectiveness of Targeting the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Pathways by Simultaneous Treatment with Two Pathway Inhibitors
In the following section, we discuss the potential of combining inhibitors that target two pathways to more effectively limit cancer growth. Treatment for inducible murine lung cancers containing KRAS and PIK3CA mutations with PI3K/mTOR (NVP-BEZ235) and MEK (selumetinib) inhibitors led to an enhanced response [157]. Synergistic responses between sorafenib and mTOR inhibitors were observed in xenograft studies with a highly metastatic human HCC tumor [158]. Some recent studies in thyroid cancer have documented the benefit of combining Raf and PI3K/mTOR inhibitors [159].
Intermittent dosing of MEK and PI3K inhibitors has been observed to suppress the growth of tumor xenografts in mice [74]. This study demonstrated that continuous administration of MEK and PI3K inhibitors is not required to suppress xenograft growth. These important results were obtained by performing washout studies in vitro and alternate dosing schedules in mice with MEK and PI3K inhibitors with cancer cells having mutations at BRAF and KRAS.
The combined effects of inhibiting MEK with PD-0329501 and mTOR with rapamycin or its analog, the rapalog AP-23573 (ARIAD Pharmaceuticals/Merck) were examined in human NSCLC cell lines, as well as in animal models of human lung cancer. PD-0325901 and rapamycin demonstrated synergistic inhibition of proliferation and protein translation. Suppression of both MEK and mTOR inhibited ribosomal biogenesis and was associated with a block in the initiation phase of translation [160]. The pan-TOR inhibitor AZD-8055 has been examined as a single agent and in combination with the MEK inhibitor selumetinib in a NSCLC xenograft and increased cell death and tumor regression [151, 161]. These preclinical results support the suppression of both the MEK and mTOR pathways in lung cancer therapy and indicate that both pathways converge to regulate the initiation of protein translation. ERK phosphorylates Mnk1/2 and p90Rsk, which regulate the activity of the eukaryotic translation initiation factor eIF4E. The phosphorylation of 4EBP1 is altered in cells containing BRAF mutations. It should also be pointed out that 4EBP1 is also regulated by Akt, mTOR, and p70S6K. This may result in the efficient translation of certain mRNAs in BRAF-mutant cells. This could explain how co-inhibition of MEK and PI3K/Akt/mTOR synergizes to inhibit protein translation and growth in certain lung cancer cells.
13.6 Clinical Trials Based on Inhibiting Both the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Pathways
Combinations of Raf and PI3K/Akt/mTOR or MEK and PI3 K/Akt/mTOR inhibitors are in clinical trials. The results of a phase 1 clinical trial on patients with advanced solid tumors indicate that the combined dosing appears to be well tolerated, at least as well as single agent dosing. Some anti-tumor effects were observed, and dose-escalation trials were performed [162]. Clinical trial combining MEK and Akt inhibitors (GSK1120212 and GSK2141795, respectively) is in progress. A clinical trial for patients with advanced cancers combining the PI3K/mTOR inhibitors (PF-04691502 and PF-05212384) with the MEK inhibitor (PD-0325901) or irinotecan is in progress. The study will include patients with metastatic CRC patients who have received previous therapy for their disease and whose cancers have a mutant KRAS gene. The dual PI3 K/mTOR inhibitor NVP-BEZ235 is in a combination clinical trial with RAD001 (everolimus) in patients with advanced solid cancers. A phase 1 clinical trial is in progress combining the MEK1/2 inhibitor MEK162 and the PI3K/mTOR dual inhibitor NVP-BEZ235. This combination will be evaluated in various cancer patients, for example, NSCLC with mutations at EGFR who have progressed after treatment with EGFR inhibitors, triple-negative breast, CRC, melanoma, and pancreatic cancers. In addition, patients with other advanced solid tumors with KRAS, NRAS, and/or BRAF mutations will be included in the study. A trial is underway testing the effects of combining two experimental drugs, SD703026 (MSC1936369B) (a MEK inhibitor) and XL755 SAR245409 (a PI3K/mTOR inhibitor) for the treatment for locally advanced or metastatic solid tumors. Patients with breast, NSCLC, melanoma, and colorectal cancers will be treated with this inhibitor combination. A clinical trial is examining the effects of combining MK-2206 (an Akt inhibitor) with selumetinib (a MEK inhibitor) in cancer patients with advanced solid tumors. A combination clinical trial combining the MEK inhibitor selumetinib and the Akt inhibitor MK-2206 in patients with stage III or stage IV melanoma that previous failed after treatment with vemurafenib or dabrafenib is in progress.
13.7 Trials Based on Combining Raf/MEK, PI3K/Akt/mTOR Inhibitors with Chemotherapy or MoAbs
Treatment of mice xenografted with vemurafenib-resistant BRAF-mutant CRCs with various combinations of vermurafenib and chemotherapeutic drugs (capecitabine, irinotecan), MoAbs [bevacizumab (avastin, targets VEGFα), cetuximab (erbitux, targets EGFR)], the Akt inhibitor MK-2206, or the EGFR inhibitor erlotinib, increased survival [163]. Combination of the Akt inhibitor MK-2206 and either EGFR/HER2-targeted therapy [erlotinib or lapatinib (tykerb, a dual EGFR and HER2 inhibitor from GSK)] or chemotherpapeutic drugs doxorubicin, camptothechin, gemcitabine, 5-flurouracil, docetaxel or carboplatin resulted in synergistic responses in lung (NCI-H460) and ovarian (A2780) cancer cell lines. In some cases, the timing of drug addition was determined to be important as MK-2206 suppressed the Akt activation induced by carboplatin and gemcitabine [164]. The effects of combining the dual PI3K/mTOR inhibitor NVP-BEZ235 and various chemotherapeutic drugs as well as other targeted therapies are being examined (doxorubicin, melphalan, vincristine, bortezomib) [165, 166]. The anti-tumor effects of WYE132 (a mTOR inhibitor) could be enhanced upon combination with bevacizumab in lung and breast xenograft models [153]. A clinical trial with INK-128 in combination with paclitaxel, either in the absence or in the presence of trastuzumab, is in progress in patients with advanced solid malignancies.
Clinical trials are ongoing based on combining the dual PI3K/mTOR inhibitor NVP-BEZ235 with PI3K and MEK inhibitors (BKM120, MEK162) and chemotherapeutic drugs (paclitaxel, trastuzumab) to treat advanced solid cancers and metastatic breast cancers which are difficult to treat (see below). BKM120 is a pan-PI3 K inhibitor. It is being included in some clinical studies since NVP-BEZ235 does not inhibit PI3Kβ [167]. Furthermore, NVP-BEZ235 is not effective in suppressing the growth of tumors which have the KRAS G12D mutation [157]. Thus, to achieve effective suppression of cancer growth in some situations, it may be important to combine PI3K/mTOR inhibitors with pan-PI3K inhibitors.
Palomid 529, a pan-mTOR inhibitor, is in some circumstances is effective as a single agent. However, when Palomid 529 was combined with either cisplatin or docetaxel, it had a better effect on hormone-refractory prostate cancers [168]. It also improved the effects of radiotherapy on prostate cancer cells [169].
The effects of inhibiting Akt in combination with other pathways, inhibitors, and chemotherapy are being evaluated in numerous phase I clinical trials. These trials highlight the importance of targeting multiple molecules to suppress the growth of cancer which are resistant to most therapies. Combination clinical trials with the Akt inhibitor MK-2206 and the dual EGFR/HER2 inhibitor lapatinib are in progress with patients having advanced or metastatic solid tumors or breast cancer patients. The effects of combining MK-2206 and erlotinib, docetaxel, or carboplatin + paclitaxel are being examined in clinical trials in certain patients with advanced cancers. Clinical trials with NSCLC patients are underway to examine the effects of combining MK-2206 with gefitinib (iressa, EGFR inhibitor AstraZenica). Clinical trials with postmenopausal metastatic breast cancer patients are in progress to examine the effects of combining anastrozole, letrozole, exemestane (aromatase-inhibitors), or fulvestrant (an estrogen receptor antagonist). Clinical trials are also underway examining the effects of combining MK-2206 with bendamustin (nitrogen mustard alkylating agent) and Rituximab (chimeric monoclonal antibody targeting CD20 from IDEC Pharmaceuticals/Genentech) on CLL cancer patients who have relapsed or on cancer patients with small lymphocytic lymphoma. Clinical trials combining MK-2206 and various other drugs including dalotuzumab (a MoAb which targets IGF-1R from Merck) and MK-0752 (a Y-secretase inhibitor which inhibits the NOTCH pathway from Merck) are in progress. The effects of MK-8669 (ridaforolimus an mTORC1 inhibitor from Merck) and dalotuzumab are being examined in patients with advanced cancers. Clinical trials combining MK-2206 and paclitaxel in cancer patients with locally advanced, metastatic solid tumors, or metastatic breast cancers are in progress. The above mentioned clinical trials document the importance of targeting Akt and other signaling molecules as well as critical targets involved in cellular division. Furthermore, the clinical trials document how basic research on these pathways is being translated into clinical therapy for cancer and other types of patients.
13.8 Enhancing the Effectiveness of Raf/MEK and PI3K/mTOR Inhibitors with Radiotherapy
Radiotherapy is a common therapeutic approach for treatment for many diverse cancers. A side effect of radiotherapy in some cells is induction of the Ras/Raf/MEK/ERK cascade [2]. Various signal transduction inhibitors have been evaluated as radiosensitizers. The effects of pre-treatment for lung, pancreatic, and prostate cancer cells with selumetinib were evaluated in vitro using human cell lines and in vivo employing xenografts [170]. The MEK inhibitor treatment radiosensitized the various cancer cell lines in vitro and in vivo. The MEK inhibitor treatment was correlated with decreased Chk1 phosphorylation 1–2 h after radiation. The authors noticed the effects of the MEK inhibitor on the G2 checkpoint activation after irradiation, as the MEK inhibitor suppressed G2 checkpoint activation. Since ERK1/ERK2 activity is necessary for the carcinoma cells to arrest at the G2 checkpoint, suppression of phosphorylated Chk1 was speculated to lead to the abrogated G2 checkpoint, increased mitotic catastrophe, and impaired activation of cell cycle checkpoints. Mitotic catastrophe was increased in cells receiving both the MEK inhibitor and radiation when compared to the solo-treated cells. It was also postulated in this study that the MEK inhibitor suppressed the autocrine cascade in DU145 prostate cancer cells that normally resulted from EGF secretion and EGFR activation. Suppression of this autocrine cascade by the MEK inhibitor may have served as a radiosensitizer. The other two cancer cell lines examined in this study (A549 and MiaPaCa2) had KRAS mutations and both were radiosensitized by the MEK inhibitor. Although these studies document the ability of a MEK inhibitor to radiosensitize certain cells, clearly other cancer cell lines without activating mutations in the Ras/Raf/MEK/ERK pathway or autocrine growth stimulation should be examined for radiosensitization by the MEK inhibitor as the KRAS mutation may also activate the PI3K pathway which could lead to therapy resistance.
PI3K/Akt/mTOR inhibitors will sensitize the tumor vasculature to radiation both in vitro in cell lines and in vivo in xenografts [171, 172]. mTOR and radiation play critical roles in the regulation of autophagy [173, 174]. When mTOR is blocked by rapamycin, there is an increase in autophagy. This is important as apoptotic cell death is a minor component to cell death in many solid tumors. These studies document the potential beneficial use of combining mTOR inhibitors and radiation to improve the induction of autophagy in the treatment for solid tumors.
13.9 Conclusions
Inhibitors to the Ras/Raf/MEK/ERK and Ras/PI3K/PTEN/Akt/mTOR pathways. Initially, MEK inhibitors were demonstrated to have the most specificity. However, these inhibitors may have limited effectiveness in treating human cancers, unless the particular cancer proliferates directly in response to the Raf/MEK/ERK pathway. Moreover, MEK inhibitors are often cytostatic as opposed to cytotoxic; thus, their ability to function as effective anticancer agents in a monotherapeutic setting is limited, and they may be more effective when combined with other small molecule inhibitors, MoAbs, chemo- or radiotherapy. Raf inhibitors have also been developed, and some are being used to treat various cancer patients (e.g., sorafenib and vemurafenib). This particular Raf inhibitor also inhibits other receptors and kinases which may be required for the growth of the particular cancer. This promiscuous nature of sorafenib has contributed to the effectiveness of this particular Raf inhibitor for certain cancers. Raf inhibitors such as vemurafenib, dabrafenib, and GDC-0879 are promising for the treatment for melanoma, CRC, thyroid as well as other solid cancers, and leukemias/lymphomas/myelomas which have mutations at BRAF V600E. However, problems have been identified with certain Raf inhibitors as they will be ineffective if Ras is mutated/amplified, if an exon of BRAF is deleted, if BRAF is amplified, if there are mutations at MEK1 and various other genetic mechanisms that result in resistance [175]. Combination therapies with either a traditional drug/physical treatment or another inhibitor that targets a specific molecule in either the same or a different signal transduction pathway are also key approaches for improving the effectiveness and usefulness of MEK and Raf inhibitors.
Modified rapamycins (rapalogs) are being used to treat various cancer patients (e.g., patients with RCC and some other cancers). While rapalogs are effective and their toxicity profiles are well known, one inherent property is that they are not very cytotoxic when it comes to killing tumor cells. This inherent property of rapamycins may also contribute to their low toxicity in humans.
Mutations at many of the upstream receptor genes or RAS can result in abnormal Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathway activation. Hence, targeting these cascade components with small molecule inhibitors may inhibit cell growth. The usefulness of these inhibitors may depend on the mechanism of transformation of the particular cancer. If the tumor exhibits a dependency on the Ras/Raf/MEK/ERK pathway, then it may be sensitive to Raf and MEK inhibitors. In contrast, tumors that do not display enhanced expression of the Ras/Raf/MEK/ERK pathway may not be sensitive to either Raf or MEK inhibitors, but if the Ras/PI3K/Akt/mTOR pathway is activated, these cancers may be sensitive to specific inhibitors that target this pathway. Finally, it is likely that many of the inhibitors that we have discussed in this review will be more effective in inhibiting tumor growth in combination with MoAb, cytotoxic chemotherapeutic drugs, or radiation. This is documented by the large number of clinical trials combining signal transduction inhibitors with these various therapeutic agents.
Some scientists and clinicians have considered that the simultaneous targeting of Raf and MEK by individual inhibitors may be more effective in cancer therapy than just targeting Raf or MEK by themselves. This is based in part on the fact that there are intricate feedback loops from ERK which can inhibit Raf and MEK. For example, when MEK1 is targeted, ERK1,2 is inhibited, and the negative feedback loop on MEK is broken and activated MEK accumulates. However, if Raf is also inhibited, it may be possible to completely shut down the pathway. This is a rationale for treatment with both MEK and Raf inhibitors. Likewise, targeting both PI3K and mTOR may be more effective than targeting either PI3K or mTOR by themselves. If it is a single inhibitor which targets both molecules, such as the new PI3K and mTOR dual inhibitors, this becomes a realistic therapeutic option. Although it should be pointed out that some studies are examining the effects of combining rapamycins and PI3K/mTOR inhibitors. Finally, an emerging concept is the dual targeting of two different signal transduction pathways, Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR, for example. This has been explored in some preclinical models as discussed in the text. The rationale for targeting of both pathways may be dependent on the presence of mutations in either/or both pathways or in upstream Ras in the particular cancer which can activate both pathways.
It is not always clear why a particular combination of a signal transduction inhibitor and chemotherapeutic drug works in one tumor type but not at all in a different tumor type. This has also been experienced with the development of individual chemotherapeutic drugs, some work in some cells but not others. This may result from many different complex interacting events. Some of these events could include percentage of cells in different phases of the cell cycle, persistence of cancer stem cells, presence of multiple mutated activated oncogenes, or repressed tumor suppressor genes, epigenetic modifications and many other factors. Finally, chemotherapeutic drug therapy and other types of therapy (radiotherapy, antibody therapy) may induce certain signaling pathways (e.g., the reactive oxygen species generated by chemotherapy and radiotherapy induce the Ras/Raf/MEK/ERK pathway). The induction of these signaling pathways may counteract some of the effects of the signal transduction inhibitors. These effects could indicate that the timing of each therapy will be important for effective treatments.
In summary, targeted therapy has advanced from basic research studies to the treatment of cancer patients in less than 25 years. We have learned a lot regarding how specific inhibitors exert their effects and how the Ras/Raf/MEK/ERK and PI3K/Akt/mTOR pathways function; we still need to discover more about resistance mechanisms and how we can overcome therapeutic resistance and improve the effectiveness of targeted therapy.
References
Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA et al (2011) Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2(3):135–164
McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F et al (2011) Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3 K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol 226(11):2762–2781
Hayashi K, Shibata K, Morita T, Iwasaki K, Watanabe M, Sobue K (2004) Insulin receptor substrate-1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem 279(39):40807–40818
Mischak H, Seitz T, Janosch P, Eulitz M, Steen H, Schellerer M et al (1996) Negative regulation of Raf-1 by phosphorylation of serine 621. Mol Cell Biol 16(10):5409–5418
Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA et al (2000) Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J Biol Chem 275(29):22300–22304
Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM et al (2011) A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472(7343):366–369
McKay MM, Ritt DA, Morrison DK (2011) RAF inhibitor-induced KSR1/B-RAF binding and its effects on ERK cascade signaling. Curr Biol 21(7):563–568
Pao W, Girard N (2011) New driver mutations in non-small-cell lung cancer. Lancet Oncol 12(2):175–180
Topisirovic I, Sonenberg N (2011) mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harb Symp Quant Biol 28:28
Xing J, Ginty DD, Greenberg ME (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science 273(5277):959–963
Balan V, Leicht DT, Zhu J, Balan K, Kaplun A, Singh-Gupta V et al (2006) Identification of novel in vivo Raf-1 phosphorylation sites mediating positive feedback Raf-1 regulation by extracellular signal-regulated kinase. Mol Biol Cell 17(3):1141–1153
Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD et al (2005) Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell 17(2):215–224
Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O et al (2009) A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol 16(3):294–303
Sturm OE, Orton R, Grindlay J, Birtwistle M, Vyshemirsky V, Gilbert D et al (2010) The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal 3(153)
Martelli AM, Evangelisti C, Chappell W, Abrams SL, Basecke J, Stivala F et al (2011) Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia 25(7):1064–1079
Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D et al (2012) Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget 3(4):371–394
Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P et al (2011) Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 3(3):192–222
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB et al (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7(4):261–269
Coffer PJ, Woodgett JR (1992) Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem 205(3):1217
Gonzalez E, McGraw TE (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8(16):2502–2508
Du K, Montminy M (1998) CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273(49):32377–32379
Brennan P, Babbage JW, Burgering BM, Groner B, Reif K, Cantrell DA (1997) Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity 7(5):679–689
Kane LP, Shapiro VS, Stokoe D, Weiss A (1999) Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol 9(11):601–604
Buitenhuis M, Coffer PJ (2009) The role of the PI3K-PKB signaling module in regulation of hematopoiesis. Cell Cycle 8(4):560–566
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278(5338):687–689
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378(6559):785–789
Chalhoub N, Baker SJ (2009) PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 4:127–150
Weng LP, Brown JL, Baker KM, Ostrowski MC, Eng C (2002) PTEN blocks insulin-mediated ETS-2 phosphorylation through MAP kinase, independently of the phosphoinositide 3-kinase pathway. Hum Mol Genet 11(15):1687–1696
Mahimainathan L, Choudhury GG (2004) Inactivation of platelet-derived growth factor receptor by the tumor suppressor PTEN provides a novel mechanism of action of the phosphatase. J Biol Chem 279(15):15258–15268
Krymskaya VP, Goncharova EA (2009) PI3K/mTORC1 activation in hamartoma syndromes: therapeutic prospects. Cell Cycle 8(3):403–413
Tamburini J, Green AS, Chapuis N, Bardet V, Lacombe C, Mayeux P et al (2009) Targeting translation in acute myeloid leukemia: a new paradigm for therapy? Cell Cycle 8(23):3893–3899
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137(5):873–886 (Epub 2009 May 14)
Sato T, Nakashima A, Guo L, Tamanoi F (2009) Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem 284(19):12783–12791 (Epub 2009 Mar 19)
Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA et al (2004) The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6(1):91–99
Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA et al (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310(5754):1642–1646
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330(7503):1304–1305
Shackelford DB, Shaw RJ (2009) The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9(8):563–575
Martinelli A, Chiarini F, Evangelisti C, Ognibene A, Bressanin D, Billi AM, Manzoli L, Cappellini A, McCubrey JA (2012) Targeting the liver kinase B1/AMP-dependent kinase pathway as a therapeutic strategy for hematologic malignancies. Expert Opinion Therapeutic Targets (In Press)
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A et al (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 118(9):3065–3074
Hresko RC, Mueckler M (2005) mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem 280(49):40406–40416
Liang C (2010) Negative regulation of autophagy. Cell Death Differ 17(12):1807–1815
Poulikakos PI, Solit DB (2011) Resistance to MEK inhibitors: should we co-target upstream? Sci Signal 4(166)
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H et al (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64(19):7099–7109
Rimassa L, Santoro A (2009) Sorafenib therapy in advanced hepatocellular carcinoma: the SHARP trial. Expert Rev Anticancer Ther 9(6):739–745
Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G (2012) Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget 3(3):236–260
Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R et al (2009) CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene 28(1):85–94
Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H et al (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315):596–599
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA et al (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363(9):809–819
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516
Long G, Kefford RF, Carr PJA, Brown MP, Curtis M, Ma B, Lebowitz P, Kim KB, Kurzrock R, Flachook G(2010) Phase 1/2 study of GSK2118436, a selective inhibitor of V600 mutant (mut) BRAF kinase: evidence of activity in melanoma brain metastases (mets). Annals of Oncology. 21(LBA27 (supplement 8)):viii12
Kefford R, Arkenau H, Brown MP, Millward M, Infante JR, Long GV, et al (2010) Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutant BRAF kinase, in patients with metastatic melanoma and other solid tumors. ASCO Meeting Abstracts 2010 June 14, 28(15_suppl):8503
Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN et al (2012) Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 11(4):909–920
Whittaker S, Menard D, Kirk R, Ogilvie L, Hedley D, Zambon A et al (2010) A novel, selective and efficacious nanomolar pyridopyrazinone inhibitor of V600EBRAF. Cancer Res 70(20):8036–8044
Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J et al (2009) Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res 69(7):3042–3051
Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE et al (2008) Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 68(12):4853–4861
Schwartz GK, Robertson S, Shen A, Wang E, Pace L, Dials H et al (2009) A phase I study of XL281, a selective oral RAF kinase inhibitor, in patients (Pts) with advanced solid tumors. ASCO Meeting Abstracts. 27(15S):3513 (2009 June 8)
Buchholz B, Klanke B, Schley G, Bollag G, Tsai J, Kroening S et al (2011) The Raf kinase inhibitor PLX5568 slows cyst proliferation in rat polycystic kidney disease but promotes renal and hepatic fibrosis. Nephrol Dial Transplant 26(11):3458–3465
Zitzmann K, de Toni E, von Ruden J, Brand S, Goke B, Laubender RP et al (2011) The novel Raf inhibitor Raf265 decreases Bcl-2 levels and confers TRAIL-sensitivity to neuroendocrine tumour cells. Endocr Relat Cancer 18(2):277–285
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G et al (2011) Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 129(1):245–255
Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351(Pt 1):95–105
Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P et al (2004) Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol 11(12):1192–1197
Sebolt-Leopold JS (2008) Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin Cancer Res 14(12):3651–3656
McCubrey JA, Steelman LS, Abrams SL, Chappell WH, Russo S, Ove R et al (2010) Emerging MEK inhibitors. Expert Opin Emerg Drugs 15(2):203–223
Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al (2007) AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Molecular Cancer Therapeutics. 6(8):2209–2219
Tai YT, Fulciniti M, Hideshima T, Song W, Leiba M, Li XF et al (2007) Targeting MEK induces myeloma-cell cytotoxicity and inhibits osteoclastogenesis. Blood 110(5):1656–1663
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ et al (2007) Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase 1/2 inhibitor. Clin Cancer Res 13(5):1576–1583
Friday BB, Yu C, Dy GK, Smith PD, Wang L, Thibodeau SN et al (2008) BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res 68(15):6145–6153
Yoon J, Koo KH, Choi KY (2011) MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res 71(2):445–453
Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G et al (2007) Preclinical Studies of TW-37, a New Nonpeptidic Small-Molecule Inhibitor of Bcl-2, in Diffuse Large Cell Lymphoma Xenograft Model Reveal Drug Action on Both Bcl-2 and Mcl-1. Clin Cancer Res 13(7):2226–2235
Haura EB, Ricart AD, Larson TG, Stella PJ, Bazhenova L, Miller VA et al (2010) A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res 16(8):2450–2457
LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB et al (2010) Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res 16(6):1924–1937
Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A et al (2006) BRAF mutation predicts sensitivity to MEK inhibition. Nature 439(7074):358–362
Iverson C, Larson G, Lai C, Yeh LT, Dadson C, Weingarten P et al (2009) RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res 69(17):6839–6847
Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L et al (2012) Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res 72(1):210–219
Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S et al (2010) Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol 149(4):537–549
Lee TX, Packer MD, Huang J, Akhmametyeva EM, Kulp SK, Chen CS et al (2009) Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. Eur J Cancer 45(9):1709–1720
Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T et al (2011) Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett 21(5):1315–1319
Longoni R, Spina L, Vinci S, Acquas E (2011) The MEK inhibitor SL327 blocks acquisition but not expression of lithium-induced conditioned place aversion: a behavioral and immunohistochemical study. Psychopharmacology 216(1):63–73
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464(7287):427–430
Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B et al (2012) MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia 26(4):778–787
Ricciardi MR, Scerpa MC, Bergamo P, Ciuffreda L, Petrucci MT, Chiaretti S et al (2012) Therapeutic potential of MEK inhibition in acute myelogenous leukemia: rationale for “vertical” and “lateral” combination strategies. J Mol Med 8:8
Aronov AM, Tang Q, Martinez-Botella G, Bemis GW, Cao J, Chen G et al (2009) Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J Med Chem 52(20):6362–6368
Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM et al (2012) ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther 11(5):1143–1154
Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G et al (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3(7):763–772
Burrows N, Babur M, Resch J, Ridsdale S, Mejin M, Rowling EJ et al (2011) GDC-0941 inhibits metastatic characteristics of thyroid carcinomas by targeting both the phosphoinositide-3 kinase (PI3K) and hypoxia-inducible factor-1alpha (HIF-1alpha) pathways. J Clin Endocrinol Metab 96(12):12
Zou ZQ, Zhang LN, Wang F, Bellenger J, Shen YZ, Zhang XH (2012) The novel dual PI3K/mTOR inhibitor GDC-0941 synergizes with the MEK inhibitor U0126 in non-small cell lung cancer cells. Mol Med Report 5(2):503–508
Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F et al (2005) Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 106(3):1063–1066
Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B et al (2006) A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 25(50):6648–6659
Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L et al (2008) Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood 111(1):379–382
Workman P, van Montfort RL (2010) PI(3) kinases: revealing the delta lady. Nat Chem Biol 6(2):82–83
Berndt A, Miller S, Williams O, Le DD, Houseman BT, Pacold JI et al (2010) The p110delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat Chem Biol 6(3):244
Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ et al (2011) CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117(2):591–594
Meadows SA, Vega F, Kashishian A, Johnson D, Diehl V, Miller LL et al (2012) PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood 119(8):1897–1900
Gale S, Croasdell G (2010) 28th Annual JPMorgan healthcare conference–forest laboratories and Icagen. IDrugs 13(3):145–148
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D et al (2012) Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 11(2):317–328
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D et al (2012) Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 30(3):282–290
Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C (2009) PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol 21(2):194–198
Molckovsky A, Siu LL (2008) First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J Hematol Oncol 1:20
Xu CX, Li Y, Yue P, Owonikoko TK, Ramalingam SS, Khuri FR et al (2011) The combination of RAD001 and NVP-BEZ235 exerts synergistic anticancer activity against non-small cell lung cancer in vitro and in vivo. PLoS One 6(6):14
Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D et al (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9(5):341–349
Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM et al (2007) A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res 67(17):7960–7965
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C et al (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7(7):1851–1863
Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A et al (2010) Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res 16(22):5424–5435
Chiarini F, Fala F, Tazzari PL, Ricci F, Astolfi A, Pession A et al (2009) Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res 69(8):3520–3528
Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A et al (2010) Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res 70(20):8097–8107
Schuster K, Zheng J, Arbini AA, Zhang CC, Scaglioni PP (2011) Selective targeting of the mTORC1/2 protein kinase complexes leads to antileukemic effects in vitro and in vivo. Blood Cancer J 1:e34
Shuttleworth SJ, Silva FA, Cecil AR, Tomassi CD, Hill TJ, Raynaud FI et al (2011) Progress in the preclinical discovery and clinical development of class I and dual class I/IV phosphoinositide 3-kinase (PI3K) inhibitors. Curr Med Chem 18(18):2686–2714
Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED et al (2011) Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res 17(10):3193–3203 (Epub 2011 Feb 15)
Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J et al (2011) PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther 10(11):2189–2199
Mallon R, Hollander I, Feldberg L, Lucas J, Soloveva V, Venkatesan A et al (2010) Antitumor efficacy profile of PKI-402, a dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor. Mol Cancer Ther 9(4):976–984
Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA et al (2011) Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol 13(4):384–392
Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K et al (2011) Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med 89(9):877–889
Heffron TP, Berry M, Castanedo G, Chang C, Chuckowree I, Dotson J et al (2010) Identification of GNE-477, a potent and efficacious dual PI3K/mTOR inhibitor. Bioorg Med Chem Lett 20(8):2408–2411
Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L et al (2011) GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther 10(12):2426–2436
Li T, Wang J, Wang X, Yang N, Chen SM, Tong LJ et al (2010) WJD008, a dual phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin inhibitor, prevents PI3K signaling and inhibits the proliferation of transformed cells with oncogenic PI3K mutant. J Pharmacol Exp Ther 334(3):830–838
Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW et al (2004) From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res 64(12):4309–4318
Ding H, Han C, Guo D, Wang D, Duan W, Chen CS et al (2008) Sensitivity to the non-COX inhibiting celecoxib derivative, OSU03012, is p21(WAF1/CIP1) dependent. Int J Cancer 123(12):2931–2938
Falasca M, Chiozzotto D, Godage HY, Mazzoletti M, Riley AM, Previdi S et al (2010) A novel inhibitor of the PI3K/Akt pathway based on the structure of inositol 1,3,4,5,6-pentakisphosphate. Br J Cancer 102(1):104–114
Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI et al (2004) Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 64(13):4394–4399
Garrett CR, Coppola D, Wenham RM, Cubitt CL, Neuger AM, Frost TJ et al (2011) Phase I pharmacokinetic and pharmacodynamic study of triciribine phosphate monohydrate, a small-molecule inhibitor of AKT phosphorylation, in adult subjects with solid tumors containing activated AKT. Invest New Drugs 29(6):1381–1389
Tan S, Ng Y, James DE (2011) Next-generation Akt inhibitors provide greater specificity: effects on glucose metabolism in adipocytes. Biochem J 435(2):539–544
Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F et al (2012) Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 22(10):136
Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ et al (2008) Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 68(7):2366–2374
Zeng Z, Samudio IJ, Zhang W, Estrov Z, Pelicano H, Harris D et al (2006) Simultaneous inhibition of PDK1/AKT and Fms-like tyrosine kinase 3 signaling by a small-molecule KP372-1 induces mitochondrial dysfunction and apoptosis in acute myelogenous leukemia. Cancer Res 66(7):3737–3746
Rampling R, Sanson M, Gorlia T, Lacombe D, Lai C, Gharib M et al (2012) A phase I study of LY317615 (enzastaurin) and temozolomide in patients with gliomas (EORTC trial 26054). Neuro Oncol 14(3):344–350
Vansteenkiste J, Ramlau R, von Pawel J, San Antonio B, Eschbach C, Szczesna A (2012) A phase II randomized study of cisplatin-pemetrexed plus either enzastaurin or placebo in chemonaive patients with advanced non-small cell lung cancer. Oncology 82(1):25–29
Wolff RA, Fuchs M, Di Bartolomeo M, Hossain AM, Stoffregen C, Nicol S et al (2011) A double-blind, randomized, placebo-controlled, phase 2 study of maintenance enzastaurin with 5-fluorouracil/leucovorin plus bevacizumab after first-line therapy for metastatic colorectal cancer. Cancer 27(10):26692
Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK (2003) Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther 2(11):1093–1103
Pal SK, Reckamp K, Yu H, Figlin RA (2010) Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs 19(11):1355–1366
Handrick R, Rubel A, Faltin H, Eibl H, Belka C, Jendrossek V (2006) Increased cytotoxicity of ionizing radiation in combination with membrane-targeted apoptosis modulators involves downregulation of protein kinase B/Akt-mediated survival-signaling. Radiother Oncol 80(2):199–206
Martelli AM, Papa V, Tazzari PL, Ricci F, Evangelisti C, Chiarini F et al (2010) Erucylphosphohomocholine, the first intravenously applicable alkylphosphocholine, is cytotoxic to acute myelogenous leukemia cells through JNK- and PP2A-dependent mechanisms. Leukemia 24(4):687–698
Bidyasar S, Kurzrock R, Falchook GS, Naing A, Wheler JJ, Durand J et al (2009) A first-in-human phase I trial of PBI-05204 (oleandrin), an inhibitor of Akt, FGF-2, NF-Kb, and p70S6K in advanced solid tumor patients. ASCO Meeting Abstracts. 27(15S):3537 (2009 June 8)
Dunn DE, He DN, Yang P, Johansen M, Newman RA, Lo DC (2011) In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice-based stroke models. J Neurochem 119(4):805–814
Yoon H, Kim DJ, Ahn EH, Gellert GC, Shay JW, Ahn CH et al (2009) Antitumor activity of a novel antisense oligonucleotide against Akt1. J Cell Biochem 108(4):832–838
Marshall J, Posey J, Hwang J, Malik S, Shen R, Kazempour K, et al (2007) A phase I trial of RX-0201 (AKT anti-sense) in patients with an advanced cancer. ASCO Meeting Abstracts. 25(18_suppl):3564 (2007 June 21)
Oshiro N, Yoshino K, Hidayat S, Tokunaga C, Hara K, Eguchi S et al (2004) Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 9(4):359–366
Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y et al (2007) Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318(5852):977–980
Fouladi M, Laningham F, Wu J, O’Shaughnessy MA, Molina K, Broniscer A et al (2007) Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol 25(30):4806–4812
Owonikoko TK, Khuri FR, Ramalingam SS (2009) Preoperative therapy for early-stage NSCLC: opportunities and challenges. Oncology 23(10):892
Rini BI, Campbell SC, Escudier B (2009) Renal cell carcinoma. Lancet 373(9669):1119–1132
Benjamin D, Colombi M, Moroni C, Hall MN (2011) Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 10(11):868–880
Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D’Amato GZ et al (2012) Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol 30(1):78–84
Donia M, McCubrey JA, Bendtzen K, Nicoletti F (2010) Potential use of rapamycin in HIV infection. Br J Clin Pharmacol 70(6):784–793
Nicoletti F, Fagone P, Meroni P, McCubrey J, Bendtzen K (2011) mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov Today 16(15–16):715–721
Bhagwat SV, Gokhale PC, Crew AP, Cooke A, Yao Y, Mantis C et al (2011) Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther 10(8):1394–1406
Grimaldi C, Chiarini F, Tabellini G, Ricci F, Tazzari PL, Battistelli M et al (2012) AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia 26(1):91–100
Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H et al (2010) Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A 107(28):12469–12474
Tan DS, Dumez H, Olmos D, Sandhu SK, Hoeben A, Stephens AW et al (2010) First-in-human phase I study exploring three schedules of OSI-027, a novel small molecule TORC1/TORC2 inhibitor, in patients with advanced solid tumors and lymphoma. ASCO Meeting Abstracts. 28(15_suppl):3006 (2010 June 14)
Jessen K, Wang S, Kessler L, Guo X, Kucharski J, Staunton J et al Abstract B148: INK128 is a potent and selective TORC1/2 inhibitor with broad oral antitumor activity. Molecular Cancer Therapeutics. 8(Supplement 1):B148
Hsieh AC, Ruggero D (2010) Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res 16(20):4914–4920
Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE et al (2010) AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70(1):288–298
Xue Q, Hopkins B, Perruzzi C, Udayakumar D, Sherris D, Benjamin LE (2008) Palomid 529, a novel small-molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res 68(22):9551–9557
Yu K, Shi C, Toral-Barza L, Lucas J, Shor B, Kim JE et al (2010) Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res 70(2):621–631
Garcia-Martinez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM et al (2009) Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J 421(1):29–42
Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P et al (2011) Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 71(5):1573–1583
Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T et al (2011) Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem 54(5):1473–1480
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R et al (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14(12):1351–1356
Wang Z, Zhou J, Fan J, Qiu SJ, Yu Y, Huang XW et al (2008) Effect of rapamycin alone and in combination with sorafenib in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res 14(16):5124–5130
Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW (2011) Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin Cancer Res 17(20):6482–6489
Legrier ME, Yang CP, Yan HG, Lopez-Barcons L, Keller SM, Perez-Soler R et al (2007) Targeting protein translation in human non small cell lung cancer via combined MEK and mammalian target of rapamycin suppression. Cancer Res 67(23):11300–11308
Marshall G, Howard Z, Dry J, Fenton S, Heathcote D, Gray N et al (2011) Benefits of mTOR kinase targeting in oncology: pre-clinical evidence with AZD8055. Biochem Soc Trans 39(2):456–459
Shapiro G, LoRusso P, Kwak EL, Cleary JM, Musib L, Jones C et al (2011) Clinical combination of the MEK inhibitor GDC-0973 and the PI3 K inhibitor GDC-0941: A first-in-human phase Ib study testing daily and intermittent dosing schedules in patients with advanced solid tumors. ASCO Meeting Abstracts. 29(15_suppl):3005 (2011 June 9)
Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ et al (2012) Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 72(3):779–789
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K et al (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9(7):1956–1967
Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R (2009) The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Exp Cell Res 315(3):485–497
Manara MC, Nicoletti G, Zambelli D, Ventura S, Guerzoni C, Landuzzi L et al (2010) NVP-BEZ235 as a new therapeutic option for sarcomas. Clin Cancer Res 16(2):530–540
Zhang YJ, Duan Y, Zheng XF (2011) Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today 16(7–8):325–331
Gravina GL, Marampon F, Petini F, Biordi L, Sherris D, Jannini EA et al (2011) The TORC1/TORC2 inhibitor, Palomid 529, reduces tumor growth and sensitizes to docetaxel and cisplatin in aggressive and hormone-refractory prostate cancer cells. Endocr Relat Cancer 18(4):385–400
Diaz R, Nguewa PA, Diaz-Gonzalez JA, Hamel E, Gonzalez-Moreno O, Catena R et al (2009) The novel Akt inhibitor Palomid 529 (P529) enhances the effect of radiotherapy in prostate cancer. Br J Cancer 100(6):932–940
Chung EJ, Brown AP, Asano H, Mandler M, Burgan WE, Carter D et al (2009) In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 kinase. Clin Cancer Res 15(9):3050–3057 (Epub 2009 Apr 14)
Edwards E, Geng L, Tan J, Onishko H, Donnelly E, Hallahan DE (2002) Phosphatidylinositol 3-kinase/Akt signaling in the response of vascular endothelium to ionizing radiation. Cancer Res 62(16):4671–4677
Shinohara ET, Cao C, Niermann K, Mu Y, Zeng F, Hallahan DE et al (2005) Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 24(35):5414–5422
Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B et al (2005) Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res 65(23):11061–11070
Moretti L, Attia A, Kim KW, Lu B (2007) Crosstalk between Bak/Bax and mTOR signaling regulates radiation-induced autophagy. Autophagy 3(2):142–144
Kudchadkar R, Paraiso KH, Smalley KS (2012) Targeting mutant BRAF in melanoma: current status and future development of combination therapy strategies. Cancer J 18(2):124–131
Acknowledgments
MC and GM were supported in part by grants from the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca (Ministry for Education, Universities and Research)—MIUR” PRIN 2008 and FIRB-MERIT n. RBNE08YYBM. MC was also supported in part by a grant to the CNR from the Italian Ministry of Economy and Finance for the Project FaReBio di Qualità. ML was supported in part by a grant from the Italian Ministry of Health, Ricerca Finalizzata Stemness 2008 entitled “Molecular Determinants of Stemness and Mesenchymal Phenotype in Breast Cancer”. AMM was supported in part by grants from Fondazione del Monte di Bologna e Ravenna, MinSan 2008 “Molecular therapy in pediatric sarcomas and leukemias against IGF-IR system: new drugs, best drug–drug interactions, mechanisms of resistance and indicators of efficacy”, MIUR PRIN 2008 (2008THTNLC), and MIUR FIRB 2010 (RBAP10447J-003) and 2011 (RBAP11ZJFA_001). MM was supported in part from the Italian Association for Cancer Research (AIRC), the Cariplo Foundation, and the Italian Ministry of Health. AT was supported in part by grants from the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca (Ministry for Education, University and Research) —MIUR—PRIN 2008 and grant from “Sapienza”, University of Rome 2009–11.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
McCubrey, J.A. et al. (2013). New Agents and Approaches for Targeting the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR Cell Survival Pathways. In: Johnson, D. (eds) Cell Death Signaling in Cancer Biology and Treatment. Cell Death in Biology and Diseases. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-5847-0_13
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5847-0_13
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-5846-3
Online ISBN: 978-1-4614-5847-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)