
Abstract
The combination of daclatasvir + asunaprevir [Daklinza® + Sunvepra® (Japan)], two direct-acting antiviral agents, has been developed by Bristol–Myers Squibb for the treatment of patients with chronic hepatitis C virus (HCV) genotype 1 infections, including those with compensated cirrhosis. Daclatasvir + asunaprevir has received its first global approval in this indication in Japan. Daclatasvir + asunaprevir is the first all-oral, interferon- and ribavirin-free regimen for this indication. This article summarizes the milestones in the development of daclatasvir + asunaprevir leading to this first approval for the treatment of chronic HCV genotype 1 infections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chronic hepatitis C virus (HCV) infection occurs in an estimated 130–150 million people worldwide, (WHO fact sheet) and is a leading cause of liver transplantation and hepatocellular cancer (HCC) [1–3]. There are at least six major HCV genotypes [3, 4], with genotype 1 being the most common worldwide [5]. The goal of antiviral treatment for chronic HCV is to achieve a sustained virological response (SVR), defined as the absence of HCV in the serum, after the completion of treatment [6–8]. Achieving SVR reduces the risk of long-term complications of chronic HCV infection, including liver-related death, cirrhosis, HCC and the need for liver transplantation [6].
The current standard of care for chronic HCV infection is the combination of pegylated interferon (peginterferon)-α plus ribavirin [4, 9–13]. The efficacy of this combination depends on the patient’s previous treatment history (whether or not they have been treated before and their response to this therapy), the HCV genotype and the extent of liver fibrosis [4]. Genotype 1 HCV infections are the most difficult to treat with peginterferon-α/ribavirin, with a SVR rate of approximately 40–50 % in clinical trials [5, 10, 14, 15]; the SVR rate is lower still among the unselected patient population in the clinical practice setting [14, 15]. Furthermore, a significant proportion of patients, in particular those with advanced liver disease and elderly patients, are unable to tolerate treatment with peginterferon- α plus ribavirin or have contraindications to its use [4, 12, 16]. Treatment options for patients with chronic HCV infections who are unable to receive peginterferon-α/ribavirin therapy or have not responded to this combination are limited, and new therapies are urgently required.
Direct-acting antivirals (DAAs) are a new class of treatments for chronic HCV infection that have shown efficacy in patients who have not responded to interferon-based therapy [9, 17]. DAAs target different parts of the HCV life cycle to prevent the replication of the virus, including the HCV non-structural (NS) proteins NS3, NS5A and NS5B. NS3 is a serine protease that forms complexes with NS4A and cleaves HCV structural proteins into their active forms [4, 9, 18–20]. NS5B is a viral RNA-dependent RNA polymerase required to copy and transcribe HCV mRNA, and is modulated by NS3 and NS5A [19–21]. NS5A forms a replication complex with NS-3-NS5B. DAAs are typically used in combinations of two or more drugs to avoid the development of viral resistance. In addition to potentially increasing the efficacy of the standard peginterferon-α/ribavirin regimen when used in combination, DAAs offer the possibility of interferon-free therapy and the convenience of an all-oral regimen [11, 12].
Features and properties of drug name
Alternative names | Asunaprevir + daclatasvir; BMS 790052 + BMS 650032; BMS-790052 + BMS-650032; Daklinza® + Sunvepra®; DCV Dual regimen (daclatasvir and asunaprevir combination therapy); DCV + ASV Dual regimen |
Class | Amides, Biphenyl-compounds, Carbamates, Cyclopropanes, Imidazoles, Isoquinolones, Pyrrolidines, small-molecules, Sulfonamides |
Mechanism of Action | Hepatitis C Virus NS 5 protein inhibitors; Hepatitis C virus NS3 protein inhibitors |
Route of Administration | Oral |
Pharmacokinetics | Daclatasvir: mainly faecal elimination, tmax 1–2 h, t1/2 13–15 h, 99 % protein bound; AUC and Cmax decreased with hepatic impairment, increased with renal impairment |
Asunaprevir: mainly faecal elimination, tmax 2–3 h, t1/2 15–21 h, >99 % protein-bound; AUC and Cmax increased with hepatic impairment, AUC decreased and Cmax increased in patients with end-stage renal disease | |
Co-administration of daclatasvir + asunaprevir increases AUC and Cmax of daclatasvir, decreases AUC and Cmax of asunaprevir | |
Adverse events | |
Most frequent | Nasopharyngitis, elevated liver enzymes, headache, fever |
Occasional | Diarrhoea |
Rare | Elevated bilirubin levels |
ATC codes | |
WHO ATC code | J05A (Direct acting antivirals) |
EphMRA ATC code | J5B1 (Viral hepatitis products) |
Chemical name | Daclatasvir: Dimethyl N,N′-([1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-[(2S)-pyrrolidine- |
2,1-diyl][(1S)-3-methyl-1-oxobutane-1,2-diyl]])dicarbamate dihydrochloride | |
Asunaprevir: 1,1-Dimethylethyl [(2S)-1[-(2S,4R)-4([7-chloro-4-methoxyisoquinolin-1-yl]oxy)-2- | |
([(1R,2S)-1[(cyclopropanesulfonyl)carbamoyl]-2-ethenylcyclopropyl]carbamoyl) pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]carbamate | |

Chemical structures of daclatasvir and asunaprevir
Daclatasvir is a small-molecule HCV NS5A inhibitor that has been extensively studied as a foundational agent for multiple DAA-based combination regimens (including with sofosbuvir and simeprevir) [22], while asunaprevir is a small-molecule NS3 protease inhibitor that has been studied as a component of daclatasvir-based treatment regimens. The combination of daclatasvir + asunaprevir [Daklinza® + Sunvepra® (Japan)] received its first global approval in Japan on 7 July 2014 for the oral treatment of adult patients with chronic HCV genotype 1 infections, including those with compensated cirrhosis [23]. The indications for which daclatasvir + asunaprevir is approved are the improvement of viraemia in patients with chronic HCV genotype 1, with or without compensated cirrhosis, who are either ineligible for or intolerant to interferon-based therapy or have not responded to interferon-based therapy. The approved dosage of the combination is daclatasvir 60 mg once daily plus asunaprevir 100 mg twice daily, for 24 weeks [23–25]. In patients requiring dose interruption due to adverse events, both drugs should be stopped simultaneously and resumed at the same time [24, 25]. Discontinuation of treatment should be considered if virological breakthrough (increase in HCV RNA levels of >1 log10 of the lowest value) occurs.
Bristol–Myers Squibb (BMS) has submitted new drug applications (NDAs) to the US Food and Drug Administration (FDA) seeking approval for daclatasvir, asunaprevir and their use in the dual combination regimen for the treatment of chronic HCV genotype 1b infections [26]. The FDA has granted priority review status for the NDAs, and has also granted a breakthrough therapy designation to the daclatasvir + asunaprevir dual combination regimen [27]. A marketing authorisation application (MAA) for daclatasvir, for use in combination with other antiviral agents including sofosbuvir, in the treatment of chronic HCV infections in adults has been submitted in the EU and received a positive opinion from the Committee for Medicinal Products for Human Use in June 2014 [28]; however, no MAA has been submitted for asunaprevir or the daclatasvir + asunaprevir combination.
The approval of daclatasvir + asunaprevir in Japan was based on the results of an open-label phase III study evaluating a 24-week daclatasvir + asunaprevir treatment regimen in 224 Japanese patients with chronic HCV genotype 1b infections who were non-responsive to treatment with interferon + ribavirin or were ineligible for, naïve or intolerant to interferon-based therapy (NCT01497834). Results of the multinational phase III HALLMARK-DUAL study of daclatasvir + asunaprevir in patients with chronic HCV genotype 1b infections who were treatment-naïve or intolerant to or ineligible for treatment with peginterferon-α and ribavirin (NCT01581203) were similar to those of the Japanese phase III study and are expected to support regulatory filings in countries with a high prevalence of genotype 1b HCV infection, including Korea and Taiwan. The second multinational trial in the HALLMARK programme, HALLMARK-QUAD (NCT01573351) evaluated daclatasvir + asunaprevir in combination with standard therapy in 398 patients with chronic HCV genotype 1 or 4 infections.
BMS is also developing a triple-drug fixed-combination product, comprising daclatasvir, asunaprevir and a third DAA, beclabuvir, for the treatment of HCV infections. Beclabuvir is a non-nucleoside inhibitor of HCV NS5B polymerase.
1.1 Patent Information
Daclatasvir, asunaprevir and the combination regimen are being developed by BMS. In its Form 10-K SEC filing for the year ending December 2013, BMS reported that it owned composition of matter patents covering daclatasvir and asunaprevir in the US [29]. These patents will expire in the years 2028 and 2023, respectively.
2 Scientific Summary
2.1 Viral Resistance
The presence of L31 and Y93H NS5A polymorphisms, but not NS3 polymorphisms, prior to treatment with daclatasvir + asunaprevir was predictive of virological outcomes in a phase III Japanese trial [30]. Baseline NS5A resistance-associated variant data were available for 214 patients. Of the 38 patients who had resistance-associated variants at baseline, 22 (58 %) subsequently had virological failure; these variants included NS5A-L31M/F (5/8 patients), NS5A-Y93H (16/29) and NS5A-L31V-Y93H (1/1). NS3 resistance-associated variants were detected at baseline for 82 of 221 patients for whom these data were available. Fifteen of these patients later experienced virological failure, including patients with NS3-T54S (1/2 patients), Q80L (4/19), NS3-Q80L-S122G (1/4), S122G (8/52) and D168E (1/2). Significant proportions of the 34 patients with virological failure did not have any NS5A or NS3 resistance-associated variants at baseline (32 and 56 %, respectively). However, at or near the time of virological failure, 33 of 34 patients were found to have NS5A resistance-associated variants, including NS5A-L31 M/V/I-Y93H/N (n = 28), NS5A-R30H/Q-Y93H (n = 2), delP32 (n = 1), L31 M/V (n = 1) and NS5A-Y93H (n = 1). A NS3 resistant-associated variant at D168 (D168A/E/N/T/V/Y) was present in 28 patients with virological failure. Among the 29 patients with data from week 24 follow-up, 12 had no detectable NS3-D168 resistance-associated variants and one had no detectable NS5A L31 and/or Y93 or P32 resistance-associated variants.
An analysis of genotype 1a patients receiving daclatasvir + asunaprevir in a phase IIa study who had virological breakthrough (n = 6) or a relapse (n = 1) showed the emergence of resistance variants to daclatasvir and asunaprevir [31]. Levels of resistance-associated variants in the NS5A domain for daclatasvir (Q30E/R, L31V/M and Y93C/N) and the NS3 protease domain for asunaprevir (R155 K and D168A/E/T/V/Y) were above detectable limits in these patients. SVR was observed at 48 weeks post-treatment in two of six patients with virological breakthrough who received additional treatment with peginterferon-α-2a plus ribavirin; changes in NS3 resistance-associated variants (from D168Y to D168T and from R155K to V36M-R155K) were observed in two of the patients not responding to the intensified treatment. Daclatasvir-resistance variants persisted at week 48 post-treatment, whereas most asunaprevir-resistance variants did not.
Genotypic and phenotypic analyses of another phase IIa study (n = 43) found that all three patients with virological breakthrough and two of four with relapse had NS5A-Y93H polymorphisms at baseline [32]. After virological failure, NS3D168A/V and NS5A-L31M/V-Y93H resistance-associated variants were detected together. Resistance substitutions for daclatasvir generally remained at 48 weeks post-treatment, but asunaprevir-resistance substitutions did not. Virological failure occurred in five of ten patients with a NS5A-Y93H polymorphism at baseline.
Analysis of the relationship between the presence of drug-resistant variants at baseline and clinical outcomes in patients treated with daclatasvir + asunaprevir found these variants to have limited predictive value [33]. Data on resistance variants were obtained from 10 patients with chronic HCV genotype 1b infections who received daclatasvir + asunaprevir for 24 weeks. Eight of these patients achieved SVR, and the other two had relapses of HCV RNA and viral breakthrough after stopping treatment. Three patients with SVR had daclatasvir-resistant variants (L31V/M and/or Y93H) prior to treatment. One patient who had a relapse had L31M and Y93H variants at baseline and one with virological breakthrough had the Y93H variant; no asunaprevir-resistant variants were detected at baseline.
Comparison of resistance-associated variants in Japanese versus US and EU patients with chronic HCV genotype 1b infections in three phase II trials of daclatasvir + asunaprevir identified differences in polymorphisms that were present before treatment, but found that emergent resistance-associated variants were similar [34]. Patients in these studies received daclatasvir 60 mg once daily plus asunaprevir 200 or 200/600 mg twice daily for 24 weeks. At baseline, resistance-associated variants were detected for NS5A but not NS3.The most common polymorphism was NS5A-Y93H, was present in 23 % of the 43 Japanese patients and 10 % of the 40 US/EU patients. Differences were also noted in the prevalence of other polymorphisms at baseline, with NS3-Q80L and NS5A-Q54Y only present in Japanese patients (21 and 7 %, respectively). In patients who did not have SVR (seven Japanese and seven US/EU), the emergence of NS3-D168 variants was similar in Japanese and US/EU patients (5 vs 4 patients); most patients had NS5A-31-93 variants (7 Japanese, 6 US/EU) and one EU patient had a P32 variant. Five Japanese and 4 US/EU patients with treatment failure had NS5A-Y93H polymorphisms before treatment; however, SVR was achieved in 5 of 14 patients with this polymorphism at baseline.
2.2 Pharmacokinetics
2.2.1 Daclatasvir
Daclatasvir is primarily metabolized by cytochrome P450 (CYP) 3A4; metabolites accounted for <5 % of unchanged drug in the plasma [24]. In a study in healthy volunteers who received a single oral dose of [14C] daclatasvir, 88 % of the total radioactivity was recovered in the faeces (53 % unchanged), with another 6.6 % detected in the urine (mainly unchanged).
Daclatasvir had dose-dependent pharmacokinetics in a single-dose study in healthy Japanese male volunteers [24]. In volunteers receiving daclatasvir doses of 1, 10, 50, 100 and 200 mg under fasting conditions, maximum plasma concentration (Cmax) ranged from 18.7 to 2,929.3 ng/mL and AUC∞ from 170.6 to 34,030.8 ng·h/mL. The time to Cmax (tmax) of daclatasvir was 1.00–2.00 h. The oral clearance (CLT/F) was 79.3–98.0 mL/min and the elimination half-life (t1/2) was 8.76–10.19 h.
In a multiple-dose study in healthy Japanese male volunteers, daclatasvir 1, 10 and 100 mg doses were taken on an empty stomach for 14 days [24]. Cmax and AUCτ increased with dose, with Cmax ranging from 9.8 to 1,559.0 ng/mL on day 1 and 13.2–1,853.4 ng/mL on day 14 and AUCτ ranging from 73.1 to 13,026.1 ng·h/mL on day 1 and 110.9 to 17,115.4 ng·h/mL on day 14. Minimum concentrations (Cmin) ranged from 0.8 to 167.2 ng/mL on day 1 and 1.5 to 245.8 ng/mL on day 14. The tmax of daclatasvir was 1.50–2.00 h on day 1 and 1.25–1.75 h on day 14.
In a study in non-Japanese volunteers, the AUC and Cmax of daclatasvir 60 mg decreased by 23 and 28 %, respectively, when taken after a high-fat meal compared with the fasted state [24]. No significant differences in AUC and Cmax were observed after a low-fat meal compared with the fasted state.
When multiple oral doses of daclatasvir were administered to patients with chronic HCV, the elimination t1/2 was 13–15 h [24]. In a study in patients with chronic HCV, daclatasvir (1–100 mg) was 99 % bound by plasma proteins, independent of dose. In healthy volunteers, the steady state volume of distribution (Vdss) of a 100 μg IV dose of daclatasvir was 47L.
2.2.2 Asunaprevir
Elimination of asunaprevir is primarily faecal, with 84 % of the radioactivity from [14C]-asunaprevir recovered in the faeces, and <1 % in the urine [25]. Most of the circulating radioactivity in the plasma was the unchanged drug.
In a study in healthy Japanese male volunteers who received a single oral dose of asunaprevir 200, 400, 600, 900 or 1,200 mg on an empty stomach, the Cmax ranged from 68.3 to 811.5 ng/mL and AUC∞ from 570.6 to 2,793.5 ng·h/mL, although these increases were not dose-proportional [25]. CLT/F ranged from 160.5 to 440.2 L/h. Asunaprevir had a tmax of 2.75–4.00 h and t1/2 of 15.43–21.16 h.
In a multiple-dose study in healthy Japanese male volunteers, the Cmax and AUCτ of asunaprevir increased with dose [25]. Volunteers in this study received asunaprevir 200, 400 or 600 mg twice daily on an empty stomach for 14 days. Cmax ranged from 123.0 to 277.3 ng/mL on day 1 and 310.2 to 889.0 ng/mL on day 14; AUCτ ranged from 450.1 to 1,028.4 ng·h/mL on day 1 and 804.1 to 2,229.6 ng·h/mL on day 14. The Cmin was 12.0–19.4 ng/mL on day 1 and 9.4–14.9 ng/mL on day 14. The tmax was 2.25–3.00 h.
When administered after a high-fat meal, the AUC and Cmax of a 100 mg dose of asunaprevir in healthy volunteers increased by 20 and 34 %, respectively, compared with in the fasted state [25]. The tmax decreased from approximately 2.5 h when taken on an empty stomach to 1.5 h after the high-fat meal. Geometric mean ratios for the AUC and Cmax of post-prandial versus fasting doses of asunaprevir 200 mg were 2.227 and 4.087, respectively.
Asunaprevir was at least 99 % protein-bound in patients with chronic HCV who received twice-daily doses of 200–600 mg [25]. The Vdss of a single IV dose of asunaprevir 100 μg was 194L in healthy volunteers.
2.2.3 Effect of Hepatic Impairment
Hepatic impairment did not have a clinically important effect on the plasma concentration of non-protein-bound daclatasvir in a study in which volunteers with different degrees of hepatic impairment received a single oral dose of daclatasvir 30 mg [24]. However, in volunteers with mild, moderate and severe hepatic impairment (Child–Pugh classes A, B and C, respectively) the AUC of total (protein-bound and unbound) daclatasvir decreased by 42.7, 37.6 and 36.2 %, respectively, compared with volunteers with normal hepatic function; the Cmax decreased by 45.5, 45.2 and 54.6 %, respectively.
The effects of hepatic impairment on the pharmacokinetics of asunaprevir (200 mg twice daily for 7 days) were evaluated in a study in volunteers with normal liver function or mild, moderate or severe hepatic impairment (Child–Pugh classes A, B and C, respectively) [25]. Little difference in Cmax and AUC were observed between volunteers with mild hepatic impairment and those with normal liver function. However, in volunteers with moderate and severe hepatic impairment, Cmax increased to 5.0 and 22.9 times that of volunteers with normal hepatic function, respectively; AUC increased to 9.8 and 32.1 times normal, respectively.
2.2.4 Effect of Renal Impairment
The AUC of protein-bound and unbound daclatasvir increased by 26.4, 59.8 and 79.6 % in patients with HCV with creatinine clearance (CrCl) of 60, 30 and 15 mL/min, compared with patients with normal renal function (CrCl 90 mL/min) after a single oral dose of daclatasvir 60 mg [24]. AUC values for unbound daclatasvir for patients with CrCl of 60, 30 and 15 mL/min were 18.0, 39.2 and 51.2 % higher than for those with normal renal function. In patients with end-stage renal disease (ESRD) on haemodialysis, the AUC values of total and unbound daclatasvir increased by 26.9 and 20.1 %, respectively.
In subjects who did not have HCV infection who received asunaprevir 100 mg twice-daily for 7 days, the AUC decreased by 10.1 % and Cmax increased by 28.6 % in those with ESRD compared with volunteers with normal renal function [25].
2.2.5 Co-Administration of Daclatasvir and Asunaprevir
Compared with monotherapy, concomitant administration of daclatasvir 30 mg once daily and asunaprevir 200 mg twice daily increased the AUC [geometric mean ratio (GMR) 1.20; 90 % confidence interval (CI) 1.11–1.30] and Cmax (1.07; 90 % CI 0.97–1.18) of daclatasvir, and decreased the AUC (0.87; 90 % CI 0.73–1.04) and Cmax (0.58; 90 % CI 0.45–0.76) of asunaprevir [24, 25].
No clinically meaningful pharmacokinetic interaction was observed between daclatasvir and asunaprevir in a multiple-dose study in healthy volunteers [35]. In this open-label, randomized, volunteers received either daclatasvir 60 mg once daily or asunaprevir 600 mg twice daily during a 7-day lead-in period, then daclatasvir 30 mg once daily and asunaprevir 200 mg twice daily for 14 days. Exposure to both drugs was comparable after co-administration to historical data for each drug as monotherapy. GMRs for daclatasvir and asunaprevir AUCτ were 1.156 and 1.025, respectively.
A population pharmacokinetic model was developed based on interim data from three phase II studies of asunaprevir 200–1,200 mg/day in combination with peginterferon-α-2a and ribavirin (A1447016) or with daclatasvir plus peginterferon-α-2a and ribavirin [36]. Modest time-dependent increases in the clearance of asunaprevir were identified. A 37 % increase in steady state AUC was seen in Japanese compared with non-Japanese patients. Higher AUCs of asunaprevir correlated with an increased risk of grade 2 or higher hepatic adverse events, although the relative risk of such events was similar to that with placebo for asunaprevir 200 mg twice-daily in Japanese patients [odds ratio (OR) 1.09 vs 1.06 for non-Japanese] and patients aged >60 years (OR 1.08 vs 1.06 for younger patients). The risk and timing of hepatic adverse events were not affected by concomitant administration of daclatasvir or peginterferon-α-2a and ribavirin. The SVR rate for patients with genotype 1b HCV who were ineligible or unresponsive to interferon therapy was predicted to be lower with once- versus twice-daily administration of asunaprevir 200 mg with only daclatasvir (70 vs 76 %), whereas the predicted SVRs were similar for once- and twice-daily dosing of asunaprevir in combination with peginterferon-α-2a and ribavirin in addition to daclatasvir. Based on these results, a 100 mg twice-daily dose of asunaprevir was selected for phase III trials of daclatasvir + asunaprevir.
2.2.6 Drug Interactions
Daclatasvir is a substrate of P-glycoprotein (P-gp) and CYP3A4. It inhibits breast cancer resistance protein (BCRP), organic anion transporter (OATP) 1B3 and 1B1, and P-gp [24]. Asunaprevir is a substrate of CYP3A, P-gp, OATP 1B1 and 2B1 [25]. It inhibits OATP 1B1, 1B3 and 2B1, P-gp and CYP2D6, and induces CYP3A4.
Daclatasvir and asunaprevir are both contraindicated in patients taking strong CYP3A4 inducers, such as rifampicin, rifabutin, phenytoin, carbamazepine, phenobarbital, systemic dexamethasone and products, including foods, containing Hypericum perforatum (St. John’s Wort) [24, 25]. Asunaprevir is also contraindicated in patients taking systemic azole antifungal agents, clarithromycin, erythromycin, diltiazem, verapamil hydrochloride, cobicistat, HIV protease inhibitors, non-nucleoside reverse transcriptase inhibitors (except rilpivirine hydrochloride), modafinil, bosentan hydrate, flecainide and cyclosporine [25].
Caution is advised when using daclatasvir in combination with other inhibitors of CYP3A4, including azole antifungal agents (including ketoconazole and itraconazole), HIV protease inhibitors (including ritonavir and atazanavir), cobicistat, telaprevir, efavirenz and clarithromycin [24]. Daclatasvir should also be used with caution in patients taking digoxin, rosuvastatin and other statins. Asunaprevir should be used with caution in patients who are taking dextromethorphan, digoxin, midazolam, statins, ethinylestradiol and ethinylestradiol/norgestimate [25].
The pharmacokinetic effects of co-administration with daclatasvir and asunaprevir have been evaluated for a number of drugs [24, 25]. GMRs for the AUC and Cmax for co-administration versus when given alone showed that there were pharmacokinetic interactions with some of these drugs, as noted below.
Increases in the AUC and Cmax of daclatasvir were observed when it was co-administered with atazanavir 300 mg plus ritonavir 100 mg once daily (with dose-correction to 60 mg: 2.10; 90 % CI 1.95–2.26 and 1.35; 90 % CI 1.24–1.47, respectively), ketoconazole 400 mg once daily (10 mg dose: 3.00; 90 % CI 2.62–3.44 and 1.57; 90 % CI 1.31–1.88, respectively), simeprevir 150 mg once daily (1.96; 90 % CI 1.84–2.10 and 1.50; 90 % CI 1.39–1.62, respectively), telaprevir 500 mg twice daily (20 mg dose: 2.32; 90 % CI 2.06–2.62 and 1.46; 90 % CI 1.28–1.66, respectively) and 750 mg three times daily (20 mg dose: 2.15; 90 % CI 1.87–2.48 and 1.22; 90 % CI 1.04–1.44, respectively), tenofovir disoproxil fumarate 300 mg once daily (1.10; 90 % CI 1.01–1.21 and 1.06; 90 % CI 0.98–1.15, respectively), escitalopram 10 mg once daily (1.12; 90 % CI 1.01–1.26 and 1.14; 90 % CI 0.98–1.32, respectively), tacrolimus (1.05; 90 % CI 1.03–1.07 and 1.07; 90 % CI 1.02–1.12) and cyclosporine 400 mg (1.40; 90 % CI 1.29–1.53 and 1.04; 90 % CI 0.94–1.15, respectively) [24].
Reductions in the AUC and Cmax of daclatasvir occurred with concomitant administration with efavirenz 600 mg once daily (corrected for 60 mg dose: 0.68; 90 % CI 0.60–0.78 and 0.83; 90 % CI 0.76–0.92, respectively), omeprazole 40 mg once daily (0.84; 90 % CI 0.73–0.96 and 0.64; 90 % CI 0.54–0.77), famotidine 40 mg (0.82; 90 % CI 0.70–0.96 and 0.56; 90 % CI 0.46–0.67, respectively) and rifampicin 600 mg once daily (0.21; 90 % CI 0.19–0.23 and 0.44; 90 % CI 0.40–0.48, respectively) [24].
Co-administration of daclatasvir increased the AUC and Cmax of digoxin 0.125 mg once daily (1.27; 90 % CI 1.20–1.34 and 1.65; 90 % CI 1.52–1.80, respectively), simeprevir 150 mg once daily (1.44; 90 % CI 1.32–1.56 and 1.39; 90 % CI 1.27–1.52, respectively) and rosuvastatin 10 mg (1.58; 90 % CI 1.44–1.74 and 2.04; 90 % CI 1.83–2.26, respectively), and the AUC of tenofovir disoproxil fumarate 300 mg (1.10; 90 % CI 1.05–1.15) [24].
Marked increases in the AUC and Cmax of asunaprevir were observed when a 200 mg dose was co-administered with ketoconazole 200 mg twice daily (9.65; 90 % CI 8.64–10.77 and 6.92; 90 % CI 5.92–8.09, respectively) or rifampicin 600 mg single dose (14.81; 90 % CI 11.22–19.53 and 21.11; 90 % CI 14.27–31.24, respectively) [25]. The AUC and Cmax of asunaprevir 10 mg were increased when taken with ritonavir 100 mg (4.81; 90 % CI 4.01–5.77 and 5.22; 90 % CI 2.83–9.61, respectively).
Co-administration with asunaprevir 200 mg twice daily increased the AUC and Cmax of dextromethorphan 30 mg (3.94; 90 % CI 3.09–5.03 and 2.72; 90 % CI 2.10–3.53, respectively), rosuvastatin 10 mg (1.41; 90 % CI 1.26–1.57 and 1.95; 90 % CI 1.47–2.58, respectively) and digoxin 0.5 mg (1.30; 90 % CI 1.21–1.40 and 1.09; 90 % CI 0.97–1.22, respectively), and increased the Cmax of losartan 25 mg (1.63; 90 % CI 1.35–1.97) but decreased its AUC (0.89; 90 % CI 0.81–0.98) [25]. Co-administration of asunaprevir 100 mg twice daily and daclatasvir 60 mg once daily increased the AUC and Cmax of digoxin 0.25 mg (1.29; 90 % CI 1.20–1.39 and 1.77; 90 % CI 1.50–2.07, respectively). Reductions in the AUC and Cmax of midazolam 5 mg (0.71; 90 % CI 0.67–0.75 and 0.79; 90 % CI 0.73–0.87, respectively) and the AUC of omeprazole 40 mg (0.80; 90 % CI 0.69–0.94) were observed when administered with asunaprevir 200 mg twice daily; a 100 mg twice daily-dose of asunaprevir reduced the AUC and Cmax of sertraline (0.79; 0.67–0.94 and 0.81; 90 % CI 0.67–0.97, respectively) and the AUC of escitalopram 10 mg once daily (0.95; 90 % CI 0.91–0.98).
Concomitant administration of asunaprevir 600 mg twice daily with ethinylestradiol 0.035 mg and norgestimate 0.180–0.250 mg once daily reduced the AUC and Cmax of ethinylestradiol (0.72; 90 % CI 0.67–0.78 and 0.75; 90 % CI 0.67–0.85, respectively) and norelgestromin (0.66; 90 % CI 0.62–0.70 and 0.71; 90 % CI 0.65–0.77, respectively) [25]. In contrast, no clinically significant pharmacokinetic interaction was observed between daclatasvir and ethinylestradiol/norgestimate [24, 37]. In a study in which the approved 100 mg twice daily dose of asunaprevir was administered with daclatasvir 60 mg once daily and ethinylestradiol 0.030 mg plus norethisterone 1.5 mg once daily, the AUC and Cmax of ethinylestradiol were reduced (0.86; 90 % CI 0.83–0.89 and 0.93; 90 % CI 0.86–0.99) but the AUC and Cmax of norethisterone were similar to when administered as monotherapy (1.02; 90 % CI 0.94–1.11 and 0.93; 90 % CI 0.85–1.01, respectively) [24, 25].
2.3 Therapeutic Trials
2.3.1 Phase III Trials
The primary endpoint of SVR (HCV RNA <15 IU/mL) at 24 weeks post-treatment was achieved in most patients in a phase III Japanese study of daclatasvir + asunaprevir (NCT01497834) [38]. This multicentre, open-label study enrolled a total of 259 patients with chronic HCV genotype 1b infection, 222 of whom received daclatasvir 60 mg once daily and asunaprevir 100 mg twice daily for 24 weeks; 135 of the patients were ineligible for interferon or had intolerance to interferon and 87 patients had not responded to previous interferon treatment. Interferon-ineligible/intolerant patients were more likely to have the IL28B CC genotype (69.6 % of this subgroup), whereas the non-CC genotype was more common in the interferon non-responder subgroup (81.6 %).
Rapid reductions in HCV RNA levels were observed after the initiation of daclatasvir + asunaprevir, with a mean reduction of 5.2 log10 IU/mL from baseline at week 2, and 75.2 and 91.0 % of patients having undetectable HCV RNA levels at weeks 4 and 12, respectively [38]. At 24 weeks post-treatment, the SVR rates among interferon-ineligible/intolerant patients and interferon non-responders were 87.4 and 80.5 %, respectively. SVR rates for patients with and without cirrhosis were 90.9 and 84.0 %, respectively. Similar rates of SVR were observed in patients with IL28B CC and non-CC genotype HCV (84.5 and 84.8 %, respectively).
As per study protocol, nine patients in the interferon-nonresponder subgroup who had virological breakthrough (increase in viral load of ≥1 log10 or confirmed detectable HCV RNA ≥15 IU/mL on or after week 8) with daclatasvir + asunaprevir received additional treatment with peginterferon-α and ribavirin; one of these patients had undetectable HCV RNA levels at week 24 after starting quadruple therapy [38]. Fifteen patients, 11 of whom were in the interferon non-responder subgroup, discontinued treatment with daclatasvir + asunaprevir due to lack of efficacy. During the post-treatment follow-up period, viral relapse occurred in 8.5 % of interferon ineligible/intolerant patients and 7.9 % of interferon nonresponders who had undetectable HCV RNA levels at the end of treatment. Virological failure occurred in 34 patients, 29 of whom had resistance-associated substitutions to daclatasvir and asunaprevir at the time of failure (predominantly NS5A-L31M/V-Y93H and NS3-D168 variants). NS5A polymorphisms (L31M/V and/or Y93H) had been present in 22 of the patients with virological failure prior to starting treatment with daclatasvir + asunaprevir.
Daclatasvir + asunaprevir was effective in patients with chronic HCV genotype 1b infections with or without cirrhosis in the phase III HALLMARK DUAL study (NCT01581203) [39]. This study enrolled a total of 747 patients, 223 of whom had cirrhosis at study entry. The subgroup of 307 patients who were treatment-naïve were randomized 2:1 to double-blind treatment with daclatasvir 60 mg once daily + asunaprevir 100 mg twice daily (n = 205) or placebo (n = 102) for 12 weeks, after which patients in the daclatasvir + asunaprevir group continued treatment for a further 12 weeks and those in the placebo group entered another clinical trial. The subgroups of patients who were ineligible for or intolerant to peginterferon/ribavirin (n = 235) and patients who had a null or partial response to prior peginterferon/ribavirin therapy (n = 205) received daclatasvir + asunaprevir for 24 weeks.
The primary endpoint, SVR at week 12 post-treatment, was achieved in 90 % of treatment-naïve patients who received daclatasvir + asunaprevir, 82 % of interferon-ineligible or intolerant patients and 82 % of partial or null responders [39]. In the overall population SVR rates 12 weeks post-treatment were similar for patients with and without cirrhosis (84 and 85 %, respectively). SVR rates 12 weeks after the end of treatment for patients with and without cirrhosis were 91 and 89 % for treatment-naïve patients, 87 vs 80 % for partial or null responders, and 79 vs 84 % for interferon-ineligible/intolerant patients. SVR rates were not affected by age, sex, ethnicity or IL28B genotype. Among a subset of 77 patients who were ineligible/intolerant to peginterferon/ribavirin who had advanced liver fibrosis/cirrhosis and thrombocytopenia, the SVR rate 12 weeks post-treatment was 73 %. After 4 weeks of treatment with daclatasvir + asunaprevir, rapid virological responses were observed in 83 % of treatment-naïve patients, 73 % of null/partial responders to peginterferon/ribavirin and 68 % of interferon-ineligible/intolerant patients. Rates of virological breakthrough in these three subgroups were 4, 13 and 9 %, respectively. Post-treatment relapse occurred in 2 % of treatment-naïve patients, 4 % of null/partial responders and 6 % of interferon ineligible/intolerant patients.
2.3.2 Phase II Trials
SVRs were achieved in patients with chronic HCV genotype 1 infections who had not responded to previous treatment with peginterferon plus ribavirin in a phase IIa study of daclatasvir + asunaprevir (NCT01012895) [40]. This open-label proof-of-concept study enrolled 21 patients who had not had a reduction in HCV RNA levels of ≥2 log10 after ≥12 weeks of treatment with peginterferon plus ribavirin who were randomized to receive daclatasvir 60 mg once daily plus asunaprevir 600 mg twice daily alone (n = 11) or in combination with peginterferon-α-2a and ribavirin (n = 10). The primary endpoint was SVR at 12 weeks post-treatment. Rapid virological responses occurred in 64 % of patients receiving daclatasvir + asunaprevir and 50 % of those receiving quadruple therapy at week 4; virological response rates in the two subgroups at week 24 of treatment were 45 and 100 %, respectively. SVR was achieved in four (36 %) patients receiving daclatasvir + asunaprevir at both 12 and 24 weeks after the end of treatment (two of nine with genotype 1a and two of two with genotype 1b). Six patients with HCV genotype 1a had virological breakthrough during treatment and one had a viral response at the end of treatment but relapsed post-treatment. In the quadruple therapy subgroup, all 10 patients had SVR at 12 weeks post-treatment, which was maintained at 24 weeks post-treatment in nine. Virological response rates at week 48 post-treatment were 27 and 90 %, respectively, in the daclatasvir + asunaprevir and quadruple therapy groups.
Responses to daclatasvir + asunaprevir-based antiviral treatment were analyzed according to HCV genotype in a phase IIa study in 101 patients with a prior null response to ≥12 weeks of treatment with peginterferon/ribavirin [41]. Patients in this randomized, open-label study all received daclatasvir 60 mg once daily; 38 genotype 1b patients also received asunaprevir 200 mg once or twice daily, 36 genotype 1a and five genotype 1b patients also received asunaprevir once or twice daily in addition to peginterferon/ribavirin (quadruple therapy) and 18 genotype 1a and four genotype 1b patients received asunaprevir twice daily plus ribavirin (triple therapy). SVR rates at 12 weeks post-treatment were 65 and 78 %, respectively, for patients receiving once- and twice-daily asunaprevir in addition to daclatasvir, and 95 % for both quadruple therapy regimens. Virological breakthrough occurred in 10 of 18 genotype 1a patients in the triple therapy group by week 12; peginterferon-α was added to the treatment regimen for nine of these patients and another seven genotype 1a patients received pre-emptive rescue treatment with peginterferon-α to prevent virological breakthrough. All five patients who continued treatment with triple therapy had SVR at week 4 post-treatment.
Daclatasvir + asunaprevir produced high SVR rates in difficult-to-treat patients with chronic HCV genotype 1b infection in an open-label phase IIa study [42]. This study enrolled a total of 43 patients, 36 of whom completed 24 weeks of treatment with daclatasvir 60 mg once daily + asunaprevir 200 mg twice daily; 22 patients were medically ineligible or intolerant to peginterferon/ribavirin therapy and 21 were null responders to 12 weeks of prior peginterferon/ribavirin treatment. HCV RNA levels decreased by a mean of 5.4 log10 IU/mL in interferon-ineligible/intolerant patients and 5.6 log10 IU/mL in null-responders at week 4. Serum HCV RNA levels were undetectable at week 8 in all patients who remained on treatment. At 12 weeks post-treatment, the SVR rate was 76.7 % (63.6 % for interferon-ineligible/intolerant patients and 90.5 % for null responders); at 24 weeks post-treatment, SVR rates were the same overall and in the two subgroups. Virological breakthrough occurred in three interferon-ineligible/intolerant patients and four patients in this subgroup had post-treatment relapses; there were no virological failures among the null responder subgroup.
Treatment with daclatasvir + asunaprevir was associated with SVR at 12 weeks post-treatment in all nine patients who completed 24 weeks of treatment in a phase IIa study [43]. This study enrolled 10 patients with HCV genotype 1b infections who had a null response (reduction in HCV RNA of <2 log10 after 12 weeks) with previous peginterferon and ribavirin therapy. Patients received daclatasvir 60 mg once daily and asunaprevir 600 mg twice daily, with the asunaprevir dosage later reduced to 200 mg twice daily after 12–21 weeks due to cases of hepatic transaminase elevations in another study. One patient discontinued treatment after 2 weeks due to grade 4 bilirubin elevation. All nine patients who completed 24 weeks of treatment had SVR at 12 and 24 weeks post-treatment, and the patient who discontinued treatment also had undetectable HCV RNA levels. Virological breakthrough did not occur in any of the patients in this study.
2.3.3 Outcomes Studies
Long-term clinical outcomes were analyzed for a cohort of 1,000 Japanese patients aged ≥70 years with chronic HCV genotype 1b infections treated with daclatasvir + asunaprevir versus no treatment over a lifetime horizon using a published Markov model based on data from a phase III trial (NCT01497834) [44]. Among patients who were ineligible for interferon therapy, treatment with daclatasvir + asunaprevir was estimated to produce reductions of 494 cases of compensated cirrhosis, 81 cases of decompensated cirrhosis and 314 cases of hepatocellular carcinoma (HCC); this corresponded to numbers need to treat (NNTs) to prevent one event of 2, 12 and 3 patients, respectively. Among patients who had not responded to previous interferon-based treatment, daclatasvir + asunaprevir reduced the number of cases of compensated cirrhosis, decompensated cirrhosis and HCC by 455, 74 and 289, respectively; NNTs to prevent one of these events were 2, 13 and 3, respectively. Liver-related mortality rates were 5.1 and 7.9 %, respectively, in interferon-ineligible patients and interferon non-responders treated with daclatasvir + asunaprevir, compared with 40.3 % in untreated patients (NNT of 3 for both interferon-ineligible and non-responder cohorts).
Treatment with daclatasvir + asunaprevir provided value quality of life gains and avoided life-threatening hepatic complications, according to an analysis based on data from the phase III Japanese trial (NCT01497834) [45]. Outcomes were predicted for a hypothetical cohort of 1,000 Japanese patients aged 70 years with chronic HCV genotype 1b infection or compensated cirrhosis over a lifetime horizon, using Japan-specific disease transition rates. The effects of daclatasvir + asunaprevir were compared with current standard treatment (telaprevir, peginterferon-α, ribavirin triple therapy). SVR rates for daclatasvir + asunaprevir in interferon-ineligible/intolerant patients and interferon partial responders and null responders were 87, 85 and 74 %, respectively; SVR rates for interferon partial and null responders receiving telaprevir-based triple therapy were 59 and 42 %, respectively. When treatment was started while patients had chronic hepatitis C, this resulted in gains of 0.77 and 0.96 quality-adjusted life years (QALYs) for partial- and null-responders treated with daclatasvir + asunaprevir instead of telaprevir-based triple therapy, and a gain of 2.61 QALYs for interferon-ineligible/intolerant patients treated with daclatasvir + asunaprevir versus no treatment. When treatment was not started until patients had compensated cirrhosis, daclatasvir + asunaprevir was associated with gains of 0.90, 1.11 and 3.05 QALYs versus comparators in the three subgroups. The projected incidences of decompensated cirrhosis, HCC and liver-related deaths decreased by 66, 115 and 128 cases, respectively, in patients treated with daclatasvir + asunaprevir.
Key clinical trials of daclatasvir + asunaprevir
Drugs(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor |
|---|---|---|---|---|---|---|
DCV + ASV | HCV genotype 1b | III | Completed | Japan | NCT01497834; AI447-206 | Bristol–Myers Squibb |
DCV + ASV, Peginterferon-α2a, ribavirin | HCV genotype 1b | III | Active, not recruiting | Multinational | NCT01581203; EudraCT2011-005446-35; AI447-028; Hallmark DUAL | Bristol–Myers Squibb |
DCV + ASV, Peginterferon-α2a, ribavirin | HCV genotype 1 or 4 | III | Completed | Multinational | NCT01573351; EudraCT2011-005422-21; Hallmark QUAD | Bristol–Myers Squibb |
DCV + ASV | HCV genotype 1b | III | Recruiting | China, Korea, Taiwan | NCT01995266; AI447-036 | Bristol–Myers Squibb |
DCV + ASV vs telaprevir, peginterferon-α2b, ribavirin | HCV genotype 1b | III | Active, not recruiting | Japan | NCT01718145; AI447-031 | Bristol–Myers Squibb |
DCV + ASV, peginterferon-α2a, ribavirin | HIV-HCV genotype 1 and 4 co-infection | II | Active, not recruiting | France | NCT01725542; EudraCT2012-002589-11; ANRS HC30 QUADRIH | French National Institute for Health and Medical Research-French National Agency for Research on AIDS and Viral Hepatitis (Inserm-ANRS); Bristol–Myers Squibb |
DCV + ASV, peginterferon-α2a, ribavirin | HCV genotype 1 | II | Completed | Multinational | NCT01012895; EudraCT2010-024637-23; AI447-011 | Bristol–Myers Squibb |
DCV + ASV | HCV genotype 1 | II | Completed | Japan | NCT01051414; AI447-017 | Bristol–Myers Squibb |
DCV + ASV | HIV-HCV genotype 1b co-infection | II | Recruiting | US | NCT02124044; 140065; 14-CC-0065 | National Institutes of Health Clinical Center (CC) |
DCV + ASV, peginterferon-α2a, ribavirin | HCV genotype 4 | II | Recruiting | France | NCT02107365; ANRS HC32 QUATTRO | French National Institute for Health and Medical Research-French National Agency for Research on AIDS and Viral Hepatitis (Inserm-ANRS); Bristol–Myers Squibb |
DCV ± ASV, peginterferon-α2a, ribavirin | HCV genotype 1b | II | Active, not recruiting | Multinational | NCT01428063; EudraCT2011-000836-28; AI444-026 | Bristol–Myers Squibb |
DCV and/or ASV | HCV (long-term) | N/A | Active, not recruiting | Multinational | NCT01492504; EudraCT2011-005287-21; AI444-046 | Bristol–Myers Squibb |
DCV + ASV, peginterferon, ribavirin | HCV (genotype 1a and 1b) with and without cirrhosis | II | Recruiting | US | NCT01888900; 130150; 13-DK-0150 | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK); Bristol–Myers Squibb |
2.4 Adverse Events
The combination of daclatasvir + asunaprevir is contraindicated in patients with decompensated liver disease or Child–Pugh class B or C liver dysfunction due to cases of hepatic dysfunction reported in clinical trials [24, 25]. Liver function tests should be performed at least every 2 weeks up to week 12 and every 4 weeks thereafter in patients treated with daclatasvir + asunaprevir.
Daclatasvir + asunaprevir should not be taken during pregnancy as teratogenic effects were observed in animal studies and there is potential for fatal adverse foetal outcomes to occur in humans [24, 25]. Women of childbearing age are required to use appropriate contraception when taking daclatasvir + asunaprevir (the pharmacokinetic interaction with ethinylestradiol-based oral contraceptives should be taken into consideration). If pregnancy is suspected, daclatasvir + asunaprevir should be discontinued immediately. Daclatasvir + asunaprevir should not be taken during breastfeeding.
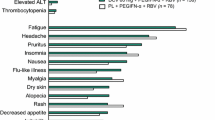
Adverse events occurred in 158 of 255 patients (62.0 %) in Japanese clinical trials of daclatasvir + asunaprevir [24, 25]. The most common adverse events were increased alanine aminotransferase (ALT; 17.6 % of patients), increased aspartate aminotransferase (AST; 14.1 %), headache (12.9 %) and fever (11.8 %). Other adverse events reported in >5 % of patients were eosinophilia, diarrhoea, increased gamma-glutamyl transpeptidase and alkaline phosphatase levels, and nasopharyngitis. Major hepatic adverse effects reported with daclatasvir + asunaprevir were increases in ALT and AST to ≥2.5 times the upper limit of normal (ULN) in 8.2 and 5.9 % of patients, respectively, and elevation of bilirubin to ≥5 times ULN in 0.8 % of patients.
The most common adverse event in a Japanese phase III trial of daclatasvir + asunaprevir (NCT01497834) was nasopharyngitis, which occurred in 30.2 % of patients [38]. Other adverse events occurring in >10 % of patients in this study were headache (15.8 %), increased ALT (15.8 %), increased AST (12.6 %) and pyrexia (12.2 %). Serious adverse events occurred in 5.9 % of patients during treatment. Grade 3–4 laboratory abnormalities reported in this study included elevations in ALT (7.2 % of patients), AST (5.4 %) and bilirubin (0.9 %) levels. None of the patients developed decompensated liver disease. Discontinuation rates due to adverse events were 6.7 % for interferon-ineligible/intolerant patients and 2.3 % for the non-responder subgroup.
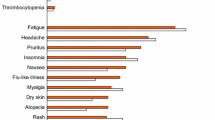
In the HALLMARK DUAL study, the most common adverse events among the 645 patients receiving daclatasvir + asunaprevir were headache (24.7 % of patients), fatigue (21.7 %), diarrhoea (16.0 %), nausea (11.6 %) and asthenia (6.4 %) [39]. Grade 3–4 elevations of ALT, AST and bilirubin levels occurred in 2.3, 1.9 and 0.5 % of patients, respectively. A total of 1.6 % of patients discontinued treatment due to adverse events. Reversible elevations of ALT and AST were the most frequent adverse events leading to discontinuation.
Neither daclatasvir 60 mg nor asunaprevir 200 mg twice daily had clinically important effects on QTc interval in studies in healthy volunteers [24, 25].
2.5 Ongoing Clinical Trials
There are three clinical trials of daclatasvir + asunaprevir that are currently recruiting patients:
-
A phase III trial in patients with HCV genotype 1b infections in China, Korea and Taiwan (NCT01995266) [46];
-
A phase II trial of daclatasvir + asunaprevir with peginterferon + ribavirin in patients with chronic HCV genotype 1a and 1b infections, with or without cirrhosis, in the US (NCT01888900) [47];
-
A phase II trial in patients with HIV and HCV genotype 1b co-infection in the US (NCT02124044) [48].
The long-term effects of daclatasvir + asunaprevir are being evaluated in an ongoing 3-year follow-up study in patients with HCV who were enrolled in previous trials of daclatasvir + asunaprevir (NCT01492504) [49].
In addition, several clinical trials of daclatasvir + asunaprevir remain active but are not enrolling new patients. These include the multinational phase III HALLMARK DUAL study (NCT01581203) and a phase III Japanese study comparing daclatasvir + asunaprevir with telaprevir, peginterferon-α-2b plus ribavirin in patients with chronic HCV genotype 1b infections (NCT01718145).
3 Current Status
The combination of daclatasvir + asunaprevir received its first global approval on 7 July 2014 for the treatment of genotype 1 chronic HCV infection in adults in Japan. It is indicated for the improvement of viraemia in patients with or without compensated cirrhosis who are either ineligible for or intolerant to interferon-based therapy or have not responded to previous interferon-based therapy.
References
Gonzalez SA. Management of recurrent hepatitis C following liver transplantation. Gastroenterol Hepatol. 2010;6(10):637–45.
Perz JF, Armstrong GL, Farrington LA, et al. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45(4):529–38.
Gottwein JM, Jensen SB, Li Y-P, et al. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob Agents Chemother. 2013;57(3):1291–303.
Shah N, Pierce T, Kowdley KV. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin Investig Drugs. 2013;22(9):1107–21.
Wendt A, Adhoute X, Castellani P, et al. Chronic hepatitis C: future treatment. Clin Pharmacol. 2014;6:1–17.
Ghany MG, Strader DB, Thomas DL, et al. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49(4):1335–74.
Ghany MG, Nelson DR, Strader DB, et al. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54(4):1433–44.
European Association for the Study of the L. EASL Clinical Practice Guidelines: management of hepatitis C virus infection. J Hepatol. 2011;55(2):245–64.
Chae HB, Park SM, Youn SJ. Direct-acting antivirals for the treatment of chronic hepatitis C: open issues and future perspectives. Sci World J. 2013;2013:704912.
De Clercq E. Current race in the development of DAAs (direct-acting antivirals) against HCV. Biochem Pharmacol. 2014;89(4):441–52.
Martel-Laferrière V, Bichoupan K, Dieterich D. Interferon-free regimens for hepatitis C: combine and conquer. BioDrugs. 2014;28(2):161–9.
Lange CM, Zeuzem S. Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J Hepatol. 2013;58(3):583–92.
Stedman CAM. Current prospects for interferon-free treatment of hepatitis C in 2012. J Gastroenterol Hepatol. 2013;28(1):38–45.
Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–82.
Kramer JR, Kanwal F, Richardson P, et al. Gaps in the achievement of effectiveness of HCV treatment in national VA practice. J Hepatol. 2012;56(2):320–5.
Ferenci P. Treatment of chronic hepatitis C–are interferons really necessary? Liver Int. 2012;32(Suppl 1):108–12.
Pockros PJ. Interferon-free hepatitis C therapy: how close are we? Drugs. 2012;72(14):1825–31.
Dubuisson J. Hepatitis C virus proteins. World J Gastroenterol. 2007;13(17):2406–15.
Pockros PJ. New direct-acting antivirals in the development for hepatitis C virus infection. Ther Adv Gastroenterol. 2010;3(3):191–202.
Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int. 2013;33:93–104.
Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 2007;132(5):1979–98.
Sulkowski M, Gardiner D, Rodriguez-Torres M, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370(3):211–21.
Bristol–Myers Squibb. Japan approves first all-oral, interferon- and ribavirin-free hepatitis C treatment, Daklinza® (daclatasvir) and Sunvepra® (asunaprevir) Dual Regimen [media release]. 7 July 2014 http://news.bms.com/press-release/japan-approves-first-all-oral-interferon-and-ribavirin-free-hepatitis-c-treatment-dakl.
Bristol–Myers Squibb Company. Daklinza Tablets (tavaborole) Japanese Prescribing Information. 2014.
Bristol–Myers Squibb Company. Sunvepra Capsules (asunaprevir) Japanese Prescribing Information. 2014.
Bristol–Myers Squibb Company. Bristol–Myers Squibb Submits NDAs for Daclatasvir and Asunaprevir to US FDA for the Treatment of Hepatitis C. [media release]. 2014. http://www.bms.com.
Bristol–Myers Squibb. Bristol–Myers Squibb Receives U.S. FDA Breakthrough therapy designation for all-oral daclatasvir dual investigational regimen for chronic hepatitis C [media release]. 24 Feb 2014. http://news.bms.com/press-release/bristol-myers-squibb-receives-us-fda-breakthrough-therapy-designation-all-oral-daclata.
Bristol–Myers Squibb. Bristol–Myers Squibb Receives Positive CHMP Opinion for Daklinza® (daclatasvir) for Treatment of Chronic Hepatitis C in the European Union [media release]. 27 June 2014. http://news.bms.com/press-release/bristol-myers-squibb-receives-positive-chmp-opinion-daklinza-daclatasvir-treatment-chr.
United States securities and exchange commission. Form 10-K: annual report pursuant to section 13 OR 15(d) of the securities exchange act of 1934 for the fiscal year ended December 31, 2012. Commission File Number 1–1136 (Bristol–Myers Squibb Company). 2013. http://www.sec.gov/Archives/edgar/data/14272/000119312513061678/d450621d10k.htm. Accessed 22 July 2014.
McPhee F, Toyota J, Chayama K, et al. Analysis of HCV resistance variants in a phase 3 trial of daclatasvir combined with asunaprevir for Japanese patients with genotype 1b infection. Hepatology. 2013;58(4 suppl 1):749A.
McPhee F, Hernandez D, Yu F, et al. Resistance analysis of hepatitis C virus genotype 1 prior treatment null responders receiving daclatasvir and asunaprevir. Hepatology. 2013;58(3):902–11.
Karino Y, Toyota J, Ikeda K, et al. Characterization of virologic escape in hepatitis C virus genotype 1b patients treated with the direct-acting antivirals daclatasvir and asunaprevir. J Hepatol. 2013;58(4):646–54.
Kosaka K, Imamura M, Hayes CN, et al. Emergence of resistant variants detected by ultra-deep sequencing after asunaprevir and daclatasvir combination therapy in patients infected with hepatitis C virus genotype 1. J Viral Hepat. 2014;. doi:10.1111/jvh.12271.
McPhee F, Hernandez D, Zhou N, et al. Comparison of pre-existing and emerging resistance-associated variants in US, EU and Japanese HCV genotype 1b prior interferon alfa (IFN-alpha) non responders and IFN-alpha ineligible patients treated with daclatasvir and asunaprevir. Hepatology. 2012;56:559A–60A.
Bifano M, Sevinsky H, Bedford BR, et al. Co-Administration of Bms-790052 and Bms-650032 does not result in a clinically meaningful pharmacokinetic interaction in healthy subjects [abstract no. 827]. In: 61st Annual Meeting of the American Association for the Study of Liver Diseases; 29 Oct–02 Nov 2010; Boston.
Chan P, Tafoya E, Eley T, et al. Exposure-response analyses of asunaprevir in combination with daclatasvir peginterferon/ ribavirin among patients with genotype 1 chronic Hcv infection: dose selection for phase 3 clinical trials [abstract no. 804]. J Hepatol. 2013;58:S328–9.
Bifano M, Sevinsky H, Hwang C, et al. Effect of the coadministration of daclatasvir on the pharmacokinetics of a combined oral contraceptive containing ethinyl estradiol and norgestimate. Antivir Ther. 2013. doi:10.3851/IMP2718.
Kumada H, Suzuki Y, Ikeda K, et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology. 2014;59(6):2083–91.
Manns M, Pol S, Jacobson IM, et al. All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: a multinational, phase 3, multicohort study. Lancet. 2014. doi:10.1016/S0140-6736(14)61059-X.
Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med. 2012;366:216–24.
Lok A, Gardiner D, Hezode C, et al. Confirmation that quadruple therapy with daclatasvir (NS5A inhibitor), asunaprevir (NS3 inhibitor) and peginterferon/ribavirin results in high rate of SVR4 in HCV genotype 1 null responders. J Hepatol. 2012;56:S557.
Suzuki Y, Ikeda K, Suzuki F, et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol. 2013;58(4):655–62.
Chayama K, Takahashi S, Toyota J, et al. Dual therapy with the non-structural protein 5A inhibitor, daclatasvir, and the non-structural protein 3 protease inhibitor, asunaprevir, in hepatitis C virus genotype 1b-infected null responders. Hepatology. 2012;55(3):742–8.
Kamae I, Ward T, Webster S, et al. Estimating the long-term clinical effectiveness of daclatasvir plus asunaprevir in Japanese patients chronically infected with HCV genotype 1b: a subgroup analysis. Hepatol Int 2014;8(1 suppl.):S156.
McEwan P, Ward T, Webster S, et al. Estimating the long-term clinical and economic outcomes of daclatasvir plus asunaprevir in difficult-to-treat Japanese patients chronically infected with hepatitis C genotype 1b. Value Health Reg Issues. 2014;3:136–45.
Bristol–Myers Squibb Company. Phase III China GT 1b Interferon (IFN) intolerant prev exclude dual [ClinicalTrials.gov identifier NCT01995266] National Institutes of Health, ClinicalTrials.gov. 2013. http://clinicaltrials.gov/ct2/show/NCT01995266?term=NCT01995266&rank=1. Accessed 22 July 2014.
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). New treatment response in people with and without cirrhosis from chronic hepatitis C [ClinicalTrials.gov identifier NCT01888900] National Institutes of Health, ClinicalTrials.gov. 2013. http://clinicaltrials.gov/ct2/show/NCT01888900?term=nct01888900&rank=1. Accessed 22 July 2014.
National Institutes of Health Clinical Center (CC). Safety, tolerability, and efficacy of asunaprevir and daclatasvir in subjects co-infected with HIV-HCV [ClinicalTrials.gov identifier NCT02124044] National Institutes of Health, Clinicaltrials.gov. 2014. http://clinicaltrials.gov/ct2/show/NCT02124044?term=NCT02124044&rank=1. Accessed 22 July 2014.
Bristol–Myers Squibb Company. Three-year follow-up study of subjects who participated in a previous asunaprevir (BMS-650032) and/or daclatasvir (BMS-790052) chronic hepatitis C clinical trial [ClinicalTrials.gov identifier NCT01492504] National Institutes of Health, ClinicalTrials.gov. 2011. http://clinicaltrials.gov/ct2/show/study/NCT01492504?term=NCT01492504&rank=1. Accessed 22 July 2014.
Disclosure
The preparation of this report was not supported by any external funding. During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. R.M. Poole is a contracted employee of Adis, Springer SBM.
Author information
Authors and Affiliations
Corresponding author
Additional information
This profile has been extracted and modified from the Adis R&D Insight drug pipeline database. Adis R&D Insight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch.
Rights and permissions
About this article

Cite this article
Poole, R.M. Daclatasvir + Asunaprevir: First Global Approval. Drugs 74, 1559–1571 (2014). https://doi.org/10.1007/s40265-014-0279-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0279-4