
Abstract
Dolutegravir, an orally administered HIV-1 integrase strand transfer inhibitor (INSTI), is under development by ViiV Healthcare. Like other drugs belonging in the INSTI class of antiretroviral agents, dolutegravir binds to the integrase site of HIV-1 and blocks the strand transfer integration step, thereby preventing the replication of HIV-1. Dolutegravir is being developed as an unboosted once-daily therapy for use in combination with other antiretroviral agents for the treatment of both treatment-naïve and treatment-experienced patients with HIV-1 infection. Dolutegravir has been approved in the USA for the treatment of HIV-1 infection in combination with other antiretroviral agents and has been filed for approval in the EU and Canada. Phase III development is underway in North America, Europe, South Africa, South America, Australia and Taiwan. This article summarizes the milestones in the development of dolutegravir leading to this first approval for the treatment of HIV-1 infection in both therapy-naïve and -experienced patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The integrase strand inhibitors (INSTIs) are a relatively new class of antiretroviral drugs used in the treatment of HIV-1 infection [1–3]. Drugs in this class block the strand transfer step of DNA integration of the HIV genome in the infected host cell, thereby preventing viral replication [4–6]. The two first-generation INSTIs approved for clinical use as components of combination therapy in some countries are raltegravir and elvitegravir, the latter gaining more recent approval in the US for use as a component of a fixed-dose combination tablet [2, 7]. Antiretroviral regimens currently recommended for adults and adolescents with HIV-1 infection include two nucleos(t)ide reverse transcriptase inhibitors (NRTIs) in combination with a non-nucleoside reverse transcriptase inhibitor (NNRTI), a protease inhibitor (PI) or an INSTI, with raltegravir being the preferred INSTI for treatment-naïve patients in current guidelines [2, 8, 9].
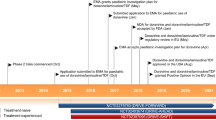
However, although both raltegravir and elvitegravir, as components of combination regimens, have shown good efficacy in the treatment of patients with HIV-1 infection in randomized trials [1], they share common resistance pathways and show cross-resistance, precluding their sequential use [3, 10–12]. Furthermore, raltegravir requires twice-daily administration and elvitegravir, administered once daily, requires administration with a pharmacokinetic booster, to increase its systemic exposure [2]. Limitations relating to the use of both of these first-generation INSTIs have led to the development of dolutegravir (Tivicay®), a new-generation INSTI, administered once-daily, with a high genetic barrier to resistance [13, 14]. In August 2013, dolutegravir gained approval from the US FDA for the treatment of a broad population of patients with HIV-1 infection, including those who are treatment-naïve and those who have previously taken HIV drug therapy, including INSTI inhibitors [15]. Orally administered dolutegravir 50 mg once daily is approved for use in combination with other antiretroviral agents in the treatment of HIV-1 infection in adults and children aged ≥12 years and weighing ≥40 kg [6]. Regulatory approval for the use of dolutegravir in children aged ≥12 years and weighing ≥40 kg (88 lbs) is based on an evaluation of safety, pharmacokinetics, and efficacy over a 24-week period in a multicentre, open-label trial in patients who had not previously been treated with an INSTI [6].
Features and properties of dolutegravir
Alternative names | ′572; 1349572; 572; GSK-1349572; GSK1349572; S-349572; S/GSK 1349572; S/GSK1349572; Tivicay® |
Class | 3-ring heterocyclic compounds |
Mechanism of action | HIV-1 integrase strand transfer inhibitor |
Route of administration | Oral |
Pharmacodynamics | Shows good in vitro activity against a broad range of subtypes of HIV-1 |
Displays a higher genetic barrier to resistance than raltegravir | |
Pharmacokinetics | Well absorbed after oral administration, with maximum plasma dolutegravir concentrations achieved 2–3 h after administration |
Extensively bound (98.9 %) to plasma proteins | |
Terminal half-life ≈14 h | |
Metabolism occurs in the liver mainly via uridine diphosphate glucuronyltransferase (UGT) 1A1; ≈53 % of the total administered dose is excreted in the feces as unchanged drug | |
Adverse events | |
Most frequent | Insomnia and headache (moderate-to-severe intensity and incidence ≥2 %) [treatment-naïve patients] |
Occasional | Nausea (incidence ≤1 %) [treatment-naïve patients]; diarrhoea (incidence 1 %) [treatment-experienced patients] |
Rare | Diarrhoea (incidence <1 %) [treatment-naïve patients] |
ATC codes | |
WHO ATC code | JO5A-X (other antivirals) |
EphMRA ATC code | J5C9 (Other HIV antivirals) |
Chemical name | Sodium (4R,12aS)-9-[(2,4-difluorophenyl)methyl] carbamoyl-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido [1′,2′:4,5] pyrazino [2,1-b] [1, 3] oxazin-7-olate |
Previously, in December 2012, ViiV Healthcare filed dolutegravir for regulatory approval in the USA, the EU and Canada for the treatment of HIV infection in adults and adolescents aged ≥12 years [16]. Regulatory applications are also being considered in Australia and Brazil [17]. Dolutegravir is the first new treatment for HIV-1 infection developed by ViiV Healthcare [17].
1.1 Company Agreements
In October 2012, ViiV Healthcare, a pharmaceutical company specializing in the development of therapies for the treatment of HIV infection, and Shionogi announced a new agreement regarding the development of HIV integrase inhibitors. This agreement, which became effective on 31 October 2012, replaced a joint venture agreement formed in 2001 between GlaxoSmithKline and Shionogi, and later transferred to ViiV Healthcare. Under the revised agreement, ViiV Healthcare acquired exclusive global rights to the assets of the joint venture Shionogi-ViiV Healthcare LLC; these assets included development and commercialisation rights to dolutegravir and other HIV integrase inhibitors. Shionogi will receive a royalty (averaging between 15 and 20 %) on net sales of the HIV integrase inhibitors portfolio and has also acquired a shareholding of 10 % in ViiV Healthcare. For a defined period, Shionogi will be involved in formulating development and commercialisation plans for the HIV integrase inhibitors. The post-transaction equity positions in ViiV Healthcare were: GlaxoSmithKline 76.5 %, Pfizer 13.5 % and Shionogi 10 % [18].
In early November 2009, ViiV Healthcare was established by GlaxoSmithKline and Pfizer. ViiV Healthcare signed a Research Alliance Agreement with GlaxoSmithKline and Pfizer, under which it would invest in research and development of HIV medicines being conducted by both companies. GlaxoSmithKline and Pfizer also agreed to grant ViiV Healthcare a right of first negotiation in relation to any new HIV-related medicine developed by either GlaxoSmithKline or Pfizer [19].

Chemical structure of dolutegravir
2 Scientific Summary
2.1 Pharmacodynamics
2.1.1 Antiretroviral Activity
In cell culture investigations, dolutegravir has shown good activity against laboratory strains of wild-type HIV-1 and a diverse range of clinical isolates of HIV-1, independent of subtype. There was no evidence of antagonism of the activity of dolutegravir when it was combined with various other antiretroviral agents in vitro, including certain NNRTIs, NRTIs and PIs [6].
2.1.2 Resistance
Published data indicate that dolutegravir has a higher genetic barrier to the development of viral resistance than raltegravir [10, 20–24] and resistance is not selected as readily as with raltegravir in vitro [12]. Dolutegravir has a more robust resistance profile than raltegravir and elvitegravir and appears to retain activity against HIV-1 harbouring single mutations [10, 22–25]. However, resistance pathways involving mutations at the G118, R263, S153, N155 and Q148 positions can affect the activity of dolutegravir in vitro and evidence of cross-resistance has been reported for dolutegravir and other INSTIs when multiple integrase mutations are present [3, 6].
Dolutegravir-resistant viruses were selected in cell culture initially from different wild-type HIV-1 strains and clades. Amino acid substitutions E92Q, G118R, S153F or Y, G193E or R263K were identified in different passages and conferred decreased susceptibility to dolutegravir of up to fourfold. Passage of mutant viruses harbouring the Q148R or Q148H substitutions selected for additional substitutions in integrase that conferred decreased susceptibility to dolutegravir (fold-change increase of 13–46). The additional integrase substitutions included T97A, E138K, G140S, and M154I. Passage of mutant viruses containing both G140S and Q148H selected for L74M, E92Q, and N155H [6]. Dolutegravir remained active against clinical isolates from patients who failed raltegravir-based therapy for viral variants carrying the Y143 and N155 resistance mutations. Isolates with Q148 with other integrase mutations had reduced susceptibility to dolutegravir [21].
No evidence of clinically relevant treatment-emergent genotypic or phenotypic resistance to dolutegravir was reported in treatment-naïve patients included in the dolutegravir phase III programme (SPRING-2 and SINGLE), or in the phase IIb SPRING trial over a 96-week treatment period [6, 10, 26]. In treatment-experienced patients, there was no evidence of a decrease in phenotypic susceptibility to dolutegravir in patients with integrase mutations in the SAILING trial [6].
In VIKING 3, of 183 patients for whom baseline data were available, 30 % had virus harbouring a substitution at Q148 and 33 % had no primary INSTI substitutions (T66A/I/K, E92Q/V, Y143C/H/R, Q148H/K/R and N155H) at baseline but did show historical genotypic evidence of substitutions associated with INSTI resistance, phenotypic evidence of resistance to raltegravir or elvitegravir, or genotypic evidence of INSTI-resistance substitutions at screening [6]. In addition, response rates were analysed by baseline integrase genotype in a subset of 124 patients who reached week 24, and for patients with virological rebound or who discontinued treatment before week 24 in VIKING 3. An overall response was reported for 79 (64 %) of 124 patients. By genotype(s) at baseline, the response rate was 80 % (16 of 20 patients) for patients with a genotype of N155H without a Q148 substitution, 56 % (10 of 18) for patients with a genotype of Y143C/H/R without a Q148 substitution, 56 % (10 of 18) for patients with Q148H/R + G140A/S without additional INSTI-resistance substitutions and 18 % (3 of 17) for patients withQ148H/R + ≥2 INSTI-resistance substitutions. Furthermore, according to the US prescribing information, 33 (75 %) of 44 patients with historic evidence of INSTI resistance at baseline had a virological response to dolutegravir-based combination therapy at week 24 in VIKING 3. At week 24, 13 (36 %) of 36 patients with viral isolates harbouring the Q148 substitution at baseline responded to treatment. The Q148 substitution was always present with additional INSTI resistance mutations. A decrease in virological response to dolutegravir-based therapy was seen in 7 (25 %) of 28 patients in the presence of ≥3 of the following resistance substitutions at baseline: L74I/M, E138A/D/K/T, G140A/S, Y143H/R, Q148H/R, E157Q, G163E/K/Q/R/S, or G193E/R [6]. Additional data are reported in the US prescribing information [6].
2.2 Pharmacokinetics
Data on the pharmacokinetics of dolutegravir are reported in the US prescribing information [6]. Completed pharmacokinetic studies on the US National Institutes of Health ClinicalTrials.gov website [27, 28] include an evaluation of the effects of renal impairment on dolutegravir (NCT01353716), an evaluation of the bioequivalence of a combined formulated tablet (NCT01622790) and an evaluation of the distribution of dolutegravir into blood, seminal fluid, and rectal fluid and tissue in healthy HIV-1-negative male volunteers (NCT01459315) and genital tract and colorectum of HIV-1 negative males and females [29]. Studies into the relative bioavailability of a pediatric granule formulation of dolutegravir (NCT01382238) [30] and a relative bioavailability study of two dolutegravir/abacavir/lamivudine fixed-dose combination tablets (NCT01366547) have also been completed [27].
Dolutegravir is readily absorbed [31], with maximum plasma concentrations achieved 2–3 h after oral administration [6]. Steady-state plasma dolutegravir concentrations were achieved within 5 days of initiation of once-daily oral dolutegravir; mean accumulation ratios for the area under the plasma concentration-time curve (AUC), maximum plasma concentration (Cmax) and trough plasma concentration (C24h) ranged from 1.2 to 1.5 [6]. Based on population pharmacokinetic analyses of data from major randomized clinical trials (SPRING-1 and SPRING-2; Sect. 2.3) in adults with HIV-1 infection who received dolutegravir 50 mg once daily, the dolutegravir steady-state mean AUC0–24 value was 53.6 μg · h/mL, the mean Cmax was 3.67 μg/mL and the mean minimum plasma concentration was 1.11 μg/mL. At doses of >50 mg, dolutegravir plasma concentrations increased in a less than dose-proportional fashion. Data are not yet available on the absolute bioavailability of dolutegravir. Although the extent of absorption of dolutegravir is increased and the rate of absorption is decreased when administered with food, this does not occur to a clinically relevant extent; thus, dolutegravir may be taken with or without food [6, 32].
Dolutegravir is extensively bound (≥98.9 %) to human plasma proteins, with binding independent of dolutegravir plasma concentrations [6]. The volume of distribution of dolutegravir was estimated to be 17.4 L after doses of 50 mg once daily, in a population pharmacokinetic analysis. Metabolism of dolutegravir is extensive [33] and occurs in the liver mainly via uridine diphosphate glucuronosyltransferase (UGT) 1A1, with the cytochrome P450 (CYP) 3A enzyme playing a minor role. Following administration of a single oral dose of [14C]dolutegravir, 53 % of the total administered dose was excreted in the feces as unchanged drug. Of the remainder, 31 % of the total administered dose was excreted in the urine as the following: an ether glucuronide of dolutegravir (18.9 %), a metabolite produced by oxidation at the benzylic carbon (3.0 %) and its hydrolytic N-dealkylation product (3.6 %) [6]. Renal elimination of unchanged drug accounted for <1 % of the administered dose. The terminal half-life of dolutegravir of ≈14 h allows for once-daily administration of the drug in the treatment of patients with HIV-1 infection. The apparent clearance of dolutegravir is 1.0 L/h, according to the results of population pharmacokinetic analyses [6].
Gender, race, age (elderly, or paediatric aged 12 to <18 years) had no clinically relevant effects on the pharmacokinetics of dolutegravir in population analyses that used pooled pharmacokinetic data. Although dolutegravir is largely metabolized and eliminated by the liver, dosage adjustment is not required for patients with mild to moderate hepatic impairment (Child-Pugh score A or B). The effect of severe hepatic impairment (Child-Pugh C) on the pharmacokinetics of dolutegravir has not been studied and the drug is therefore not recommended for use in this patient group. Pharmacokinetic data from population analyses indicate that co-infection with hepatitis C virus has no clinically relevant effect on the pharmacokinetics of dolutegravir, but data are limited on the effect of hepatitis B virus co-infection. Data from several studies, including major clinical trials (SAILING and VIKING-3; Sect 2.3), indicate that mild or moderate renal impairment has no clinically relevant effect on dolutegravir systemic exposure [6].
No dosage adjustment is required for treatment-naïve or -experienced and INSTI-naïve patients with mild, moderate, or severe renal impairment or for INSTI-experienced patients (with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance) with mild or moderate renal impairment. However, caution is advised for patients who are INSTI-experienced patients infected with HIV-1 with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance with severe renal impairment, as the reduction in dolutegravir concentrations may lead to a decrease in therapeutic effect and the development of resistance to dolutegravir or other co-administered antiretroviral agents. Dolutegravir has not been studied in patients requiring dialysis [6].
2.2.1 Drug Interactions
Data from in vitro investigations suggest that dolutegravir has a low propensity to interact with other drugs [34]. ViiV Healthcare have completed numerous phase I drug interaction studies, examining possible pharmacokinetic interactions of dolutegravir with other drugs [27, 28]. Drugs include tipranavir/ritonavir (NCT01068925), darunavir plus ritonavir and lopinavir/ritonavir (NCT00774735) [35], etravirine (NCT00774111), a combination of darunavir plus ritonavir or lopinavir/ritonavir with etravirine (NCT00867152) [36], atazanavir/ritonavir and atazanavir (NCT00883935) [37], tenofovir (NCT00726336) [38], omeprazole (NCT00942136), efavirenz (NCT01098526), antacid and multivitamin tablets (NCT00858455) [39], calcium and iron supplements (NCT01762995), rifampicin and rifabutin (NCT01231542) [40], fosamprenavir/ritonavir (NCT01209065), ethiny lestradiol/norgestimate combined oral contraceptive (NCT01498861) [41], prednisone (NCT01425099), methadone (NCT01467518) calcium carbonate and ferrous fumarate (NCT01762995), boceprevir and telaprevir (NCT01563328), rilpivirine (NCT01467531), cenicriviroc and midazolam (NCT01827540).
Dolutegravir did not have a clinically relevant effect on the pharmacokinetics of: tenofovir (an NRTI), atazanavir, darunavir, lopinavir, ritonavir and fosamprenavir (PIs), efavirenz, etravirine and rilpivirine (NNRTIs), methadone, midazolam, and oral contraceptives containing norgestimate and ethinyl estradiol, and telaprevir [6]. Darunavir/ritonavir, lopinavir/ritonavir, rilpivirine, tenofovir, boceprevir, telaprevir, prednisone, rifabutin, and omeprazole had no clinically significant effect on the pharmacokinetics of dolutegravir [6].
2.3 Therapeutic Trials
The therapeutic efficacy of oral dolutegravir, usually administered once daily in combination with two other antiretroviral drugs, in the treatment of adults with HIV-1 infection has been evaluated in several randomized, multicentre trials in the phase III clinical programme. Two of these trials, SPRING-2 (NCT01227824) [42, 43] and SINGLE (NCT01263015) [44] included treatment-naïve patients; a third trial (FLAMINGO; NCT01449929) comparing dolutegravir-based combination therapy with ritonavir-boosted darunavir combination therapy is currently ongoing (Sect. 2.5) [45]. The SAILING (NCT01231516) [46–49] trial included treatment-experienced (but INSTI-naïve) patients, whereas the VIKING-3 trial included treatment-experienced (including INSTI-experienced) patients [50, 51]. Several phase II trials of dolutegravir have also been conducted in treatment-naïve or experienced patients with HIV-1 infection [7, 26, 52, 53]. In most studies, the primary endpoint was the proportion of patients with plasma HIV-1 RNA levels of <50 copies/mL at 24 or 48 weeks.
2.3.1 Phase III Trials
Treatment-Naïve Patients
Dolutegravir 50 mg once daily, administered in combination with a backbone of two NRTIs (either abacavir/lamivudine or emtricitabine/tenofovir) for 48 weeks was an effective treatment for treatment-naïve adults (aged ≥18 years) with HIV-1 infection (HIV-1 RNA levels ≥1,000 copies/mL) in the randomized, double-blind, multicentre SPRING-2 trial (n = 822) that compared a dolutegravir-based regimen with a raltegravir-based regimen [42, 43]. At baseline, median plasma HIV-1 RNA levels were 4.52 and 4.58 log10 copies/mL in the dolutegravir and raltegravir treatment groups, respectively [42]. Corresponding median CD4+ cell counts were 359 and 362 cells/μL. After the start of treatment, plasma HIV-1 RNA levels decreased rapidly and, by week 8, plasma HIV-1 RNA levels of <50 copies/mL were achieved in 350 (85 %) patients treated with dolutegravir and 323 (79 %) patients in the raltegravir group [42].
The dolutegravir-containing regimen was shown to be noninferior to the comparator regimen containing raltegravir 400 mg twice daily instead of dolutegravir. At week 48, the trial met its primary endpoint of demonstrating non-inferiority of dolutegravir versus raltegravir, with HIV-1 RNA levels of <50 copies/mL achieved in 88 % of patients treated with the dolutegravir regimen compared with 85 % of patients on raltegravir [adjusted difference 2.5 %, 95 % confidence interval (CI) −2.2 %, 7.1 %]. As the lower end of the CI (−2.2 %) was above the prespecified −10 % non-inferiority limit, noninferiority was demonstrated. Similar response rates were observed irrespective of the dual NRTI regimen used [42].
In patients who had a high viral load (HIV-1 RNA >100,000 copies/mL) at baseline, the response rate was 82 % versus 75 %, respectively, for the dolutegravir and raltegravir recipients. The non-inferiority of dolutegravir compared with raltegravir was evident also in terms of the proportion of patients without treatment-related discontinuations (93.1 and 91.8 %, respectively, a pre-specified secondary endpoint). Virologic nonresponse occurred in 5 and 8 % of dolutegravir and raltegravir recipients, respectively. None of the dolutegravir recipients with protocol-defined virological failure developed genotypic integrase resistance mutations or NRTI resistance mutations, whereas 1 (6 %) and 4 (21 %) of patients in the corresponding raltegravir treatment group had integrase resistance mutations and NRTI resistance mutations, respectively. Furthermore, immunological improvements were seen in both treatment groups, as evidenced by similar median increases in CD4+ cell counts of 230 cells/μL [42].
Dolutegravir-based combination therapy was shown to produce a durable response over a period of up to 96 weeks in the SPRING-2 trial (reported in an abstract) [43]. At week 96, the dolutegravir once-daily regimen was noninferior to the raltegravir twice-daily regimen and there was no evidence of the emergence of resistance to dolutegravir. Proportions of patients with HIV RNA-1 levels of <50 copies/mL at week 96 were 81 and 76 % for the dolutegravir and raltegravir groups, respectively (difference 4.5 %; 95 % CI −1.1, 10.0 %).
Dolutegravir once daily in combination with abacavir/lamivudine was more effective than a single-tablet regimen of efavirenz/emtricitabine/tenofovir (Atripla®) in the phase III SINGLE trial [44]. The primary endpoint of virological suppression (HIV-1 RNA levels of <50 copies/ mL) at 48 weeks was met, with virological suppression achieved in 88 % of patients taking dolutegravir plus abacavir/lamivudine compared with 81 % of patients receiving the comparator regimen [treatment difference 7.4 % (95 % CI 2.5 %, 12.3 %; p = 0.003)]. Differences in efficacy were primarily due to a higher discontinuation rate due to adverse events amongst patients receiving the active comparator. This trial enrolled 833 adults with HIV-1 infection.
Pooled data from the SPRING-2 and SINGLE trials confirmed that dolutegravir-based combination therapy was effective in patients with high (>100,000 copies/mL) and low (<100,000 copies/mL) viral loads at baseline [54, 55].
Treatment-Experienced Patients
In the SAILING trial, once-daily dolutegravir was compared with twice-daily raltegravir in 715 patients (354 dolutegravir; 361 raltegravir) with HIV-1 infection who were failing on current therapy [48]. Interim results at 24 weeks showed that 79 % of patients receiving dolutegravir-based treatment had virological suppression (HIV-1 RNA level <50 copies/mL), compared with 70 % of patients receiving raltegravir-based treatment (p = 0.003) [47]. At week 48, HIV-1 RNA levels of <50 copies/mL occurred in 71 % of patients treated with dolutegravir compared with 64 % of patients on raltegravir (adjusted difference 7.4%, 95 % CI 0.7, 14.2); therefore, it was concluded that dolutegravir was superior (p = 0.03) to raltegravir [48]. Fewer patients in the dolutegravir arm experienced treatment failure due to INSTI resistance than in the raltegravir arm at week 48 (4 vs. 17 patients; adjusted difference −3.7 %, 95 % CI −6.1 to −1.2; p = 0.003) [48]. In this double-blind, double-dummy, noninferiority, multicentre, phase III trial, antiretroviral-experienced, INSTI-naive patients with HIV-1 infection were randomized 1:1 to receive once-daily dolutegravir 50 mg or twice-daily raltegravir 400 mg, in combination with an investigator-selected background therapy consisting of no more than two other agents (including one fully active agent). Eligible patients had two consecutive plasma HIV-1 RNA assessments of ≥400 copies/mL (unless >1,000 copies/mL at screening) and resistance to at least two classes of antiretroviral drugs [48]. Noninferiority was prespecified with a 12 % margin.
In the VIKING-3 trial, which was open-label and single-arm trial in design, twice-daily dolutegravir was evaluated in 183 HIV-1 infected adults resistant to multiple classes of HIV drugs, including the INSTIs raltegravir and/or elvitegravir [50, 51]. Patients also continued treatment with an optimized background regimen (without raltegravir or elvitegravir). At week 24, virological suppression (an HIV-1 RNA level <50 copies/mL) was achieved in 63 % to 69 % of patients. A treatment benefit was observed after 7 days of treatment with dolutegravir, with mean HIV-1 RNA levels declining by 1.4 log10 copies/mL at this timepoint (p < 0.001). However, a poor virological response occurred in patients with viral isolates with the Q148 plus ≥two additional INSTI-associated mutations.
2.3.2 Phase II and Other Trials
Phase II data from the randomized, dose-ranging SPRING-1 trial have been reported for dolutegravir, administered in combination with 2 NRTIs, in treatment-naïve patients with HIV-1 infection. At week 16, the virological response rate for the dolutegravir-treated patients (all doses) was 93 %, compared with 60 % in patients treated with efavirenz; corresponding results at week 48 were 90 and 82 % [52] and at week 96 were 78–88 and 72 % [26]. In SPRING-1, 205 HIV-1-infected treatment-naive patients were assigned to receive treatment with dolutegravir 10, 25 or 50 mg/day, or efavirenz 600 mg/day, in combination with either abacavir plus lamivudine or tenofovir plus emtricitabine. Virological response was defined as HIV-1 RNA levels of <50 copies/mL. Marked median increases from baseline in CD4+ cell counts of >300 cells/μL were observed with both dolutegravir and efavirenz treatment at week 96 [26].
In a smaller phase IIa trial that was randomized, double-blind and placebo-controlled in design, a significant (p < 0.001) decrease in HIV-1 RNA levels was seen in patients with HIV-1 infection treated with dolutegravir as monotherapy compared with placebo [53]. In this trial, 35 patients who were naïve to previous INSTI therapy received placebo or dolutegravir (2–50 mg/day) for 10 days. On day 11, a mean decrease from baseline in plasma HIV-1 RNA levels of 1.51–2.46 log10 copies/mL occurred in recipients of dolutegravir at all evaluated dosages; this decrease was significantly greater than that reported for the placebo group for whom a mean increase from baseline in 0.05 log10 copies/mL was reported. Undetectable plasma levels (<50 copies/mL) of HIV-1 RNA were achieved in 70 % of the patients treated with dolutegravir 50 mg/day [53].
Dolutegravir in combination with a background antiretroviral regimen also showed efficacy in the VIKING study in treatment-experienced patients with HIV-1 infection. This was the first study to show the clinical efficacy of dolutegravir in patients with HIV-1 infection with genotypic evidence of resistance to raltegravir [7]. At week 24, HIV-1 RNA levels of <50 copies/mL were achieved in 41 % and 75 % of patients treated with dolutegravir 50 mg once-daily or twice-daily, respectively [7]. The Phase I/II IMPAACT P1093 trial was designed primarily to evaluate safety and to assess whether the pharmacokinetic profile of dolutegravir in paediatric patients was similar to that in adults, which was demonstrated [6]. The effectiveness of dolutegravir-based combination therapy, based on virological responses, was a secondary objective of this trial. Interim data from this trial have shown that dolutegravir is also effective in the treatment of paediatric patients (mean age 14 years) with HIV-1 infection. By week 24 of treatment, 70 % of patients treated with once-daily dolutegravir (35 mg: n = 4, 50 mg: n = 19) plus optimized background therapy had HIV-1 RNA levels of <50 copies/mL. The median (percentage) increase from baseline to week 24 in the CD4+ cell count was 63 cells/μL (5 %) [6].
Key clinical trials of dolutegravir, administered as a component of combination antiretroviral therapy, in HIV-1 infection
Drugs | Study phase | Study status | Study location | Trial identifiers | Company |
|---|---|---|---|---|---|
Treatment-naïve patients with HIV-1 infection | |||||
Dolutegravir vs. raltegravir both in combination with fixed-dose dual NRTI regimen | III | Enrolment completed | USA, Canada, France, Germany, Italy, Spain, the UK, Russia & Australia | SPRING-2, ING113086, NCT01227824, EudraCT2009-017950-11 | ViiV Healthcare |
Dolutegravir + abacavir/lamivudine vs. tenofovir/emtricitabine/efavirenz | III | Enrolment completed | USA, Canada, Australia, the UK, Belgium, Denmark, France, Germany, Italy, Netherlands, Hungary, Romania & Spain | SINGLE, ING114467, NCT01263015, EudraCT2010-020983-39 | ViiV Healthcare |
Dolutegravir vs. darunavir/ritonavir, both in combination with fixed-dose dual NRTI regimen | IIIb | Enrolment completed | USA, Italy, France, Germany, Romania, Russia, Spain & Switzerland | FLAMINGO, ING114915, NCT01449929, EudraCT2011-003629-86 | ViiV Healthcare |
Dolutegravir + abacavir/lamivudine | IIIb | Enrolment completed | USA | ING116070; NCT01499199 | ViiV Healthcare |
Dolutegravir + either abacavir/lamivudine or tenofovir/emtricitabine, vs. efavirenz | IIb | Enrolment completed | USA, France, Germany, Italy, Spain & Russia | ING112276, SPRING-1, NCT00951015, EudraCT2009-010269-21 | ViiV Healthcare |
Treatment-naïve or experienced patients with HIV-1 infection | |||||
Dolutegravir | IIa | Completed | USA | 111521, NCT00708110 | GlaxoSmithKline |
Treatment-experienced patients with HIV-1 infection | |||||
Dolutegravir vs. raltegravir, both in combination with a background regimen of one or two active agents | III | Enrolment completed | USA, Canada, Argentina, Brazil, Mexico, Chile, Australia, South Africa, the UK, Belgium, France, Greece, Poland, Hungary, Italy, Netherlands, Romania, Spain, Russia & Taiwan | SAILING, ING111762, NCT01231516, EudraCT2009-018001-51 | ViiV Healthcare |
Dolutegravir + background current failing ART regimen for 7 d, then dolutegravir + optimized background ART regimena | III | Enrolment completed | USA, Belgium, Canada, France, Italy, Portugal & Spain | VIKING-3, ING112574, NCT01328041, EudraCT2009-017951-87, | ViiV Healthcare |
Dolutegravir vs PL, both in addition to the failing regimen for 7 d. From day 8, open-label dolutegravir plus an optimized background regimenb | III | Enrolment completed | USA | VIKING-4, 116529, NCT01568892 | ViiV Healthcare |
Dolutegravir + raltegravir for first 10 d (functional monotherapy phase), then dolutegravir + an optimized background regimen | IIb | Enrolment completed | USA, Canada, France, Italy & Spain | ING112961, VIKING, NCT00950859, EudraCT2009-010270-37 | ViiV Healthcare |
2.4 Adverse Events
Dolutegravir, as a component of combination therapy, was generally well tolerated by patients with HIV-1 infection in phase III trials (Sect. 2.3). Treatment was discontinued by 2 % of patients treated with dolutegravir in the SPRING-2 and SINGLE trials and in 3 % in the SAILING and VIKING-2 trials [6, 48].
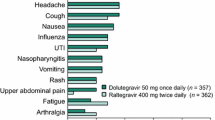
Assessments of the safety of dolutegravir in treatment-naïve patients were conducted in the ongoing SPRING-2 (n = 808) and SINGLE (n = 833) phase III trials in treatment-naïve patients [6]. The most common (incidence ≥2 %) adverse reactions of moderate to severe intensity of treatment were insomnia and headache. Less common adverse events included nausea (incidence ≤1 %) and diarrhea (<1 %) [6].
In treatment-experienced (but INSTI-naïve) patients included in the SAILING trial (n = 719 for safety data), diarrhoea (incidence ≥2 %) was the only adverse drug reaction of moderate to severe intensity in patients treated with dolutegravir or raltegravir combination therapy. Adverse drug reactions occurring in recipients of dolutegravir in this trial were similar to those reported in other phase III trials [6]. Overall, the tolerability of dolutegravir-based therapy was similar to that of raltegravir-based therapy at 48 weeks in the SAILING study [48]. The most commonly reported adverse events for recipients of dolutegravir or raltegravir included diarrhoea (20 and 18 %, respectively) and upper respiratory tract infections (11 and 8 %) [48].
Hypersensitivity reactions have been reported in recipients of dolutegravir and were characterized by rash, and constitutional findings, and sometimes organ dysfunction, including liver injury [6, 17]. The events were reported in ≤1 % of patients receiving dolutegravir in phase III clinical trials [17]. If signs or symptoms of hypersensitivity reactions develop, dolutegravir and other suspect agents should be discontinued immediately as a delay in discontinuing treatment may result in a life-threatening reaction. Dolutegravir should not be administered to patients who have experienced a previous hypersensitivity reaction [6].
In treatment-naïve patients, cholesterol, high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and triglyceride levels (fasted values) increased during 48 weeks of treatment with 2 NRTIs plus dolutegravir or raltegravir in the SPRING-2 trial [6]. In both treatment groups, mean increases from baseline in HDL-C or LDL-C levels at week 48 were ≤2.8 mg/dL. Mean increases from baseline in cholesterol levels for the dolutegravir- and raltegravir-treated patients were 6.7 and 8.3 mg/dL, and corresponding mean increases in triglyceride levels were 7.7 and 9.8 mg/dL, respectively [6]. In the SINGLE trial, increases from baseline in fasted lipid values were also reported; for the dolutegravir plus abacavir/lamivudine and efavirenz/emtricitabine/tenofovir groups, respectively, mean increases from baseline at week 48 were: cholesterol 17.1 and 24.0 mg/dL; HDL-C 5.2 and 7.9 mg/dL; LDL-C 8.5 and 13.1 mg/dL and triglycerides 17.7 and 18.6 mg/dL [6]. Further details are reported in the US prescribing information [6]. Elevated ALT and/or AST levels were reported in ≤3 % of patients treated with dolutegravir in these trials [6].
There is no information in the US prescribing information on the effects of dolutegravir on bone turnover markers or fat tissue. However, the prescribing information states that patients should be informed that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy and that the cause and long-term health effects of these conditions are not known at this time [6].
The adverse reaction profile for dolutegravir in children (in IMPAACT P1093) was similar to that in adults. There have been no reports of Grade 3 or 4 adverse drug reactions [6]. Dolutegravir, administered orally as a single supratherapeutic dose (250 mg) did not have an effect on the QT and corrected QT interval in evaluable healthy adults (n = 38) in a randomized, partial-blind, placebo-controlled, crossover study [56]. At this dose, dolutegravir was generally well tolerated; no serious or severe adverse events were reported.
2.5 Ongoing Clinical Trials
Several clinical trials of dolutegravir-based combination therapy are ongoing. Trials in therapy-naïve patients include the SINGLE trial (NCT01263015) comparing dolutegravir in combination with abacavir and lamivudine with efavirenz/emtricitabine/tenofovir over the long term [57], and a comparison of dolutegravir combination therapy with ritonavir-boosted darunavir, each in combination with dual NRTI therapy (FLAMINGO) [NCT01449929] [45]. SPRING-1, SPRING-2 and SAILING are also ongoing but the control arm or comparator treatment arms have completed. Other ongoing trials include a study evaluating the efficacy of dolutegravir in patients with HIV-1 infection resistant to raltegravir and/or elvitegravir (VIKING-4) [NCT01568892] [58], a study with a 7-day placebo-controlled functional monotherapy phase. The VIKING-3 study of dolutegravir combination therapy in patients with HIV-1 infection with treatment failure on an INSTI-containing regimen (NCT01328041) [59] and a pilot study of dolutegravir in patients with HIV-1 infection resistant to raltegravir (NCT00950859) [60] are also ongoing. A study evaluating the safety and efficacy of dolutegravir in combination with abacavir and lamivudine in therapy-naïve women with HIV-1 infection (ARIA) is planned (NCT01910402) [61]. Dolutegravir is also being evaluated in the ongoing IMPAACT P1093 multicentre, open-label, noncomparative trial in ≈160 HIV-1-infected paediatric patients aged 6 weeks to less than 18 years, including 23 treatment-experienced, INSTI-naïve subjects aged 12 to less than 18 years [6]. An expanded-access study of dolutegravir combination therapy is also ongoing (NCT01536873) [62].
3 Current Status
Dolutegravir received its first global approval on the 12th of August 2013 for the treatment of HIV-1 infection in the USA [15].
References
Messiaen P, Wensing AMJ, Fun A, et al. Clinical use of HIV integrase inhibitors: a systematic review and meta-analysis. PLoS ONE [Electronic Resource]. 2013;8(1):e52562.
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed 23 Aug 2013.
Mesplede T, Quashie PK, Wainberg MA. Resistance to HIV integrase inhibitors. Curr Opin HIV AIDS. 2012;7(5):401–8.
Abram ME, Hluhanich RM, Goodman DD, et al. Impact of primary elvitegravir resistance-associated mutations in HIV-1 integrase on drug susceptibility and viral replication fitness. Antimicrob Agents Chemother. 2013;57(6):2654–63.
Powderly WG. Integrase inhibitors in the treatment of HIV-1 infection. J Antimicrob Chemother. 2010;65(12):2485–8.
ViiV Healthcare. US prescribing information for Tivicay (dolutegravir). 2013. http://www.viivhealthcare.com/media/58599/us_tivicay.pdf. Accessed 23 Aug 2013.
Eron JJ, Clotet B, Durant J, et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis. 2013;207(5):740–8.
Williams I, Churchill D, Anderson J, et al. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy. HIV Med. 2012;13(Suppl. 2):1–85.
EACS European AIDS Clinical Society. Guidelines Version 6.1. 2012. http://www.europeanaidsclinicalsociety.org/images/stories/EACS-Pdf/EacsGuidelines-v6.1-2edition.pdf. Accessed 10 Sep 2013.
Wainberg MA, Mesplede T, Quashie PK. The development of novel HIV integrase inhibitors and the problem of drug resistance. Curr Opin Virol. 2012;2(5):656–62.
Tang MW, Shafer RW. HIV-1 antiretroviral resistance: scientific principles and clinical applications. Drugs. 2012;72(9):e1–25.
Geretti AM, Armenia D, Ceccherini-Silberstein F. Emerging patterns and implications of HIV-1 integrase inhibitor resistance. Curr Opin Infect Dis. 2012;25(6):677–86.
Lenz JCC, Rockstroh JK. S/GSK1349572, a new integrase inhibitor for the treatment of HIV: promises and challenges. Expert Opin Investig Drugs. 2011;20(4):537–48.
Karmon SL, Markowitz M. Next-generation integrase inhibitors: where to after raltegravir? Drugs. 2013;73(3):213–28.
U.S. Food and Drug Administration. FDA approves new drug to treat HIV infection. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm. Accessed 23 Aug 2013.
ViiV Healthcare. ViiV Healthcare announces regulatory submissions for dolutegravir in the EU, US and Canada. 2012. http://www.viivhealthcare.com/media/press-releases/2012/december/viiv-healthcare-announces-regulatory-submissions-for-dolutegravir-in-the-eu-us-and-canada.aspx. Accessed 10 Sep 2013.
ViiV Healthcare. ViiV Healthcare announces U.S. approval of Tivicay(Rm) (dolutegravir) for the treatment of HIV-1. 2013. http://www.viivhealthcare.com/media/press-releases/2013/august/viiv-healthcare-announces-us-approval-of-tivicay%C2%AE-dolutegravir-for-the-treatment-of-hiv-1.aspx. Accessed 10 Sep 2013.
ViiV Healthcare Ltd. Shionogi and ViiV Healthcare announce new agreement to commercialise and develop integrase inhibitor portfolio. 2012. http://www.viivhealthcare.com/media/press-releases/2012/october/shionogi-and-viiv-healthcare-announce-new-agreement-to-commercialise-and-develop-integrase-inhibitor-portfolio.aspx. Accessed 10 Sep 2013.
ViiV Healthcare. ViiV Healthcare launches: A new specialist HIV company dedicated to delivering advances in HIV treatment and care. 2009. http://www.viivhealthcare.com/media/press-releases/2012/october/shionogi-and-viiv-healthcare-announce-new-agreement-to-commercialise-and-develop-integrase-inhibitor-portfolio.aspx. Accessed 10 Sep 2013.
Sato A. In vitro passage of drug resistant HIV-1 against a next generation integrase inhibitor (INI), S/GSK1349572. In: 49th interscience conference on antimicrobial agents and chemotherapy. 2009:abstr. H-932.
Underwood MR, Johns BA, Sato A, et al. The activity of the integrase inhibitor dolutegravir against HIV-1 variants isolated from raltegravir-treated adults. J Acquir Immune Defic Syndr. 2012;61(3):297–301.
Seki T, Kobayashi M, Miki S, et al. High barrier to resistance for dolutegravir (DTG, S/GSK1349572) against raltegravir resistant Y143 mutants: An in vitro passage study. Antivir Ther. 2012;17:A62.
Vavro C, Hasan S, Madsen H, et al. Prevalent polymorphisms in wild-type HIV-1 integrase are unlikely to engender drug resistance to dolutegravir (S/GSK1349572). Antimicrob Agents Chemother. 2013;57(3):1379–84.
Kobayashi M, Seki T, Yoshinaga T, et al. Antiviral activity in vitro of the INI, dolutegravir, against raltegravir-resistant HIV-2 mutants [abstract no. 691]. In: 19th conference on retroviruses and opportunistic infections, Washington, DC, Mar 5–8 2012.
Vavro CL, Dudas KC, Hasan S, et al. Dolutegravir treatment of HIV subjects with raltegravir resistance: integrase resistance evolution in cohort II of the VIKING study. Antivir Ther. 2012;17:A13.
Stellbrink H-J, Reynes J, Lazzarin A, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. AIDS. 2013;27(11):1771–8.
ViiV Healthcare. Dolutegravir studies [ClinicalTrials.gov]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=2. Accessed 26 Aug 2013.
GlaxoSmithKline. GSK1349572 studies [ClinicalTrials.gov]. 2013. http://clinicaltrials.gov/ct2/results?term=GSK1349572&Search=Search. Accessed 26 Aug 2013.
Greener BN, Adams J, Patterson K, et al. Single and multiple dose dolutegravir pharmacokinetics in the genital tract and colorectum of HIV- men and women [abstract no. 531]. In: 20th Conference on Retroviruses and Opportunistic Infections, Atlanta, Mar 3–6 2013.
Patel P, Song I, Borland J, et al. Pharmacokinetics of a dolutegravir pediatric granule formulation in healthy adult subjects [abstract no. 985]. In: 19th conference on retroviruses and opportunistic infections; 2012 Mar 5–8, Seattle, WA, 2012.
Min S, Song I, Borland J, et al. Pharmacokinetics and safety of S/GSK1349572, a next-generation HIV integrase inhibitor, in healthy volunteers. Antimicrob Agents Chemother. 2010;54(1):254–8.
Song I, Borland J, Chen S, et al. Effect of food on the pharmacokinetics of the integrase inhibitor dolutegravir. Antimicrob Agents Chemother. 2012;56(3):1627–9.
Castellino S, Moss L, Wagner D, et al. Metabolism, excretion, and mass balance of the HIV-1 integrase inhibitor dolutegravir in humans. Antimicrob Agents Chemother. 2013;57(8):3536–46.
Reese MJ, Savina PM, Generaux GT, et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013;41(2):353–61.
Song I, Min SS, Borland J, et al. The effect of lopinavir/ritonavir and darunavir/ritonavir on the HIV integrase inhibitor S/GSK1349572 in healthy participants. J Clin Pharmacol. 2011;51(2):237–42.
Song I, Borland J, Min S, et al. Effects of etravirine alone and with ritonavir-boosted protease inhibitors on the pharmacokinetics of dolutegravir. Antimicrob Agents Chemother. 2011;55(7):3517–21.
Song I, Borland J, Chen S, et al. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next-generation HIV integrase inhibitor, S/GSK1349572. Br J Clin Pharmacol. 2011;72(1):103–8.
Song I, Min SS, Borland J, et al. Lack of interaction between the HIV integrase inhibitor S/GSK1349572 and tenofovir in healthy subjects. JAIDS. 2010;55(3):365–7.
Patel P, Song I, Borland J, et al. Pharmacokinetics of the HIV integrase inhibitor S/GSK1349572 co-administered with acid-reducing agents and multivitamins in healthy volunteers. J Antimicrob Chemother. 2011;66(7):1567–72.
Dooley KE, Sayre P, Borland J, et al. Safety, tolerability, and pharmacokinetics of the HIV integrase inhibitor dolutegravir given twice daily with rifampin or once daily with rifabutin: results of a phase 1 study among healthy subjects. J Acquir Immune Defic Syndr. 2013;62(1):21–7.
Song I, Mark S, Borland J, et al. Dolutegravir has no effect on pharmacokinetics of methadone or oral contraceptives with norgestimate and ethinyl estradiol [abstract no. 535]. In: 20th Conference on Retroviruses and Opportunistic Infections; 2013 Mar 3–6; Atlanta, GA.
Raffi F, Rachlis A, Stellbrink H-J, et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet. 2013;381(9868):735–43.
Raffi F, Jaeger H, Motta D, et al. Dolutegravir is non-inferior to raltegravir and shows durable response through 96 weeks: results from the SPRING-2 trial [TULBPE17]. In: 7th International AIDS Society Conference on HIV Pathogenesis and Treatment; 2013 Jun 30–Jul 03, Kuala Lumpur.
Walmsey S, Antela A, Clumeck N, et al. Dolutegravir + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results—SINGLE [abstract H-556b] [abstract no. 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy; 2012 Sept 9–12; San Francisco, CA.
ViiV Healthcare. Dolutegravir compared to darunavir/ritonavir, each in combination with dual nucleoside reverse transcriptase inhibitors (NRTIs) in ART-naive subjects (FLAMINGO) [ClinicalTrials.gov identifier NCT01449929]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=1. Accessed 26 Aug 2013.
Cahn P, Pozniak AL, Mingrone H, et al. Dolutegravir (DTG) is superior to raltegravir (RAL) in ART-experienced, integrase naive subjects: week 48 results from SAILING (ING111762) [WELBB03]. In: 7th International AIDS Society Conference on HIV Pathogenesis and Treatment; 2013 Jun 30–Jul 03, Kuala Lumpur.
Pozniak A, Mingrone H, Shuldyakov A, et al. Dolutegravir vs raltegravir in ART-experienced, integrase-naive subjects: 24-week interim results from SAILING (ING111762) [abstract no. 179LB]. In: 20th Conference on Retroviruses and Opportunistic Infections; 2013 Mar 3–6, Atlanta, GA.
Cahn P, Pozniak AL, Mingrone H, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet. 2013;382(9893):700–8.
Cahn P, Pozniak AL, Shuldyakov A, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase- inhibitor-naive adults with HIV: week 48 results from the randomised, double- blind, non-inferiority SAILING study. World Wide Web [online]. 2013 Jul 10.
Nichols G, Mills A, Grossberg R, et al. Antiviral activity of dolutegravir in subjects with failure on an integrase inhibitor-based regimen: week 24 phase 3 results from VIKING-3 [abstract no. O232]. In: 11th International Congress on Drug Therapy in HIV Infection; 2012 Nov 11–15; Glasgow.
Nichols G, Lazzrin A, Maggiolo F, et al. Phase 3 assessment of dolutegravir (DTG) 50 mg twice daily in HIV-1 infected subjects with raltegravir (RAL) and/or elvitegravir (EVG) resistence in VIKING-3: week 24 results of all 183 patients enrolled [abstract no. TULBPE19]. In: 7th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention; 2013 Jun 30–Jul 3; Kuala Lumpur.
van Lunzen J, Maggiolo F, Arribas JR, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naive adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect Dis. 2012;12(2):111–8.
Min S, Sloan L, DeJesus E, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS. 2011;25(14):1737–45.
Eron Jr J, Rockstroh J, Pozniak A, et al. Dolutegravir treatment response by baseline viral load and NRTI backbone in treatment-naive HIV-infected individuals [poster no. P204]. 11th International Congress on Drug Therapy in HIV Infection. JIAS. 2013; 15(Suppl. 4):18264.
Brinson C, Walmsley S, Arasteh K, et al. Dolutegravir treatment response and safety by key subgroups in treatment-naive HIV + individuals [abstract no. 554]. In: 20th Conference on Retroviruses and Opportunistic Infections; 2013 Mar 3–6; Atlanta (GA), Atlanta.
Chen S, Min SS, Peppercorn A, et al. Effect of a single supratherapeutic dose of dolutegravir on cardiac repolarization. Pharmacother J Human Pharmacol Drug Ther. 2012;32(4):333–9.
ViiV Healthcare. A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial) [Clinicaltrials.gov identifier NCT01263015]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=1. Accessed 26 Aug 2013.
Viiv Healthcare. Study assessing dolutegravir in HIV-1 infected subjects with virus resistant to raltegravir and/or elivitegravir (VIKING-4) [ClinicalTrials.gov identifier NCT01568892]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=1. Accessed 26 Aug 2013.
ViiV Healthcare. A study to assess dolutegravir in HIV-infected subjects with treatment failure on an integrase inhibitor containing regimen (VIKING-3) [ClinicalTrials.gov identifier NCT01328041]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=1. Accessed 26 Aug 2013.
ViiV Healthcare. A pilot study assessing the integrase inhibitor GSK1349572 in HIV-infected persons with virus resistant to raltegravir [ClinicalTrials.gov identifier NCT00950859]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=2. Accessed 26 Aug 2013.
Viiv Healthcare. A study to determine safety and efficacy of dolutegravir/abacavir/lamivudine (DTG/ABC/3TC) in human immunodeficiency virus (HIV)-1 infected antiretroviral therapy (ART) naïve women (ARIA) [ClinicalTrials.gov identifier NCT01910402]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=1. Accessed 26 Aug 2013.
ViiV Healthcare. Dolutegravir Expanded Access Study (DEAP) [ClinicalTrials.gov identifier NCT01536873]. 2013. http://clinicaltrials.gov/ct2/results?term=dolutegravir&pg=1. Accessed 26 Aug 2013.
Author information
Authors and Affiliations
Corresponding author
Additional information
This profile has been extracted and modified from the Adis R&D Insight drug pipeline database. Adis R&D Insight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch.
Rights and permissions
About this article
Cite this article
Ballantyne, A.D., Perry, C.M. Dolutegravir: First Global Approval. Drugs 73, 1627–1637 (2013). https://doi.org/10.1007/s40265-013-0121-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-013-0121-4