
Abstract
The integrase enzyme facilitates the incorporation of HIV-1 proviral DNA into the host cell genome and catalyses a function vital to viral replication. Inhibitors of this enzyme represent the newest class of antiretroviral drugs in our armamentarium to treat HIV-1 infection. Raltegravir, an integrase strand transfer inhibitor, was the first drug of this class approved by the US FDA; it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance. These qualities have prompted the search for agents with once-daily dosing, a more robust barrier to resistance, and a resistance profile of limited overlap with that of raltegravir. We review a series of integrase inhibitors that are in clinical or advanced pre-clinical studies. Elvitegravir, recently approved by the FDA as part of the elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir. Dolutegravir is the most advanced second-generation integrase inhibitor, and it boasts good tolerability, once-daily dosing with no need for a pharmacological enhancer, and relatively little cross-resistance with raltegravir. S/GSK1265744 has been developed into a long-acting parenteral agent that shows a high barrier to resistance in vitro and the potential for an infrequent dosing schedule. BI 224436 is in early clinical trials, but is unlikely to demonstrate cross-resistance with other integrase inhibitors. The inhibitors of the lens epithelium-derived growth factor (LEDGF)/p75 binding site of integrase (LEDGINs) are extremely early in development. Each of these contributes a new benefit to the class and will extend the treatment options for patients with HIV-1 infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The choices of antiretroviral regimens available to treat HIV-1 infection have improved in number, ease of administration, safety and tolerability. Nonetheless, the development of resistance and persistent toxicities make it desirable to identify additional medications targeting unique and constitutive steps in the HIV-1 viral life cycle. Integrase, an enzyme critical for retroviral replication, with no human homolog, has been identified as an excellent target for drug development.
Integrase is a 288 amino acid protein encoded by the pol gene. The integrase enzyme performs three functions that lead to viral integration into the host cell genome. The first is site-specific endonucleolytic cleavage of the 3′-ends of the viral DNA. Secondly, it participates in the assembly of the pre-integration complex (PIC) on the ends of the viral DNA, which migrates into the host nucleus. Lastly, integrase catalyses the insertion of the viral DNA into host chromosomal DNA (the strand transfer step) [1].
The enzyme is made up of three functional domains. The N-terminal domain contains a Zn2+-binding site, is the site of interaction with the lens epithelium-derived growth factor/transcriptional co-factor 75 (LEDGF/p75), and facilitates multimerization [2]. LEDGF/p75 links the PIC to host cell chromatin prior to integration [3, 4]. The catalytic core domain (CCD) binds integrase to DNA and is essential to its enzymatic activity (via interaction with Mg2+ and Mn2+). Finally, the C-terminal domain (CTD) non-specifically binds DNA [2].
Mutations in the integrase gene affect not only the primary functions of the enzyme itself, but also the actions of reverse transcriptase (RT) [5–7]; in fact, a putative RT-binding site on integrase has been theorized in the CTD [8].
Raltegravir was the first US FDA-approved integrase inhibitor in 2007, followed by the recent approval of elvitegravir as part of the fixed-dose combination pill containing tenofovir disoproxil fumarate (tenofovir), emtricitabine, cobicistat and elvitegravir (Stribild™, Gilead Sciences). All integrase inhibitors in clinical development interfere with the strand transfer step of integration (Fig. 1). Most bind the catalytic site in the CCD; however, the inhibitors of the LEDGF/p75 binding site of integrase (LEDGINs) and BI 224436 bind alternative sites but still inhibit strand transfer. Mass screening has identified numerous compounds that inhibit integrase by various mechanisms [1]. In this review, we focus on compounds in or near clinical development (for a summary, see Table 1).

Proposed mechanism of action of diketo acids (DKAs). DKAs block strand transfer selectively by binding at the interface between integrase and DNA. In part a, two proposed integrase binding sites are shown—the donor site for viral DNA in blue and the acceptor site for host DNA in red. After 3′-processing, part b shows the integrase-DNA complex undergoing a structural change, rendering the acceptor-site competent (red rectangle) to bind host DNA. Part c indicates binding of the host DNA to the acceptor site, leading to strand transfer under normal condition. However, the DKA inhibitor (grey rectangle) can bind to the acceptor site after 3′-processing. Part e demonstrates the hypothetical binding of DKAs at the interface of the integrase-DNA-divalent metal complex (processed 3—DNA end in blue, integrase acidic catalytic residues in red). Reproduced from Pommier et al., [107] with permission
2 First-Generation Integrase Inhibitors
2.1 Raltegravir (MK-0518)
Raltegravir (Fig. 2a), developed by Merck & Co., Inc., is a diketo acid (DKA) that inhibits the strand transfer step of integration. It was approved by the FDA for treatment of HIV in 2007 and, until recently, was the only integrase inhibitor approved for clinical use. Raltegravir has potent antiviral activity with a 95 % inhibitory concentration (IC95) of 33 nM [9] and a more rapid time to viral suppression than efavirenz [10]. Its efficacy as an antiretroviral agent has been supported by several studies.

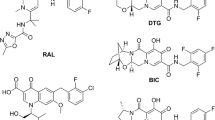
Molecular structures integrase inhibitors. (a) raltegravir; (b) elvitegravir; (c) dolutegravir; (d) S/GSK1265744; (e) BI 224436
2.1.1 Studies in Treatment-Naive Subjects
In Part 1 of the P004 trial [9, 11–14], a multi-centre double-blind study, subjects were given raltegravir monotherapy at various doses (100, 200, 400 or 600 mg twice daily) versus placebo for 10 days. On day 10, the mean change in HIV RNA from baseline was −1.9, −2.0, −1.7 and −2.2 log10 copies/mL, respectively. More than 50 % of subjects in each active group achieved an HIV RNA level below 400 copies/mL. Part 2 of P004 compared raltegravir 100, 200, 400 or 600 mg twice daily with efavirenz 600 mg daily; all groups also received tenofovir 300 mg daily and lamivudine 300 mg daily. Subjects in the raltegravir arms achieved virologic suppression more quickly than did those in the efavirenz arm, but rates of suppression after week 24 were similar. Rates of adverse events were lower in the raltegravir arms. Results from the 96-week, 192-week and 5-year analyses maintain the non-inferiority of raltegravir to efavirenz.
The STARTMRK trial [15–17] was a phase III, double-blind, randomized, non-inferiority trial in antiretroviral-naïve subjects. Subjects received tenofovir and emtricitabine and were randomized to take either raltegravir or efavirenz. Those in the raltegravir group had faster time to virologic suppression, equivalent rates of virologic suppression at week 48, and significantly fewer adverse events. These results were maintained at weeks 96 and 156. At weeks 192 and 240 (5 years), the raltegravir group had statistically superior virologic suppression compared with the efavirenz group; however, a larger number of subjects in the efavirenz arm were counted as non-completers due to clinical adverse events (9 vs. 5 %) [18]. Additionally, raltegravir was equally efficacious as efavirenz in suppressing viraemia regardless of age, sex, race, hepatitis status, baseline viral HIV RNA level, baseline CD4+ T-cell count or HIV-1 subtype [19, 20].
A limitation of raltegravir is its twice-daily dosing, and this led to the QDMRK trial [21, 22]. A total of 770 antiretroviral-naïve HIV-positive subjects were randomized to receive tenofovir and emtricitabine, plus either once-daily or twice-daily administration of raltegravir (800 mg total daily dose). In the once-daily group, 83 % achieved viral suppression, compared with 89 % of the twice-daily group; the difference in these rates was statistically significant and was consistent with inferiority of the once-daily dosing arm. It was noted that the 24-h area under the concentration-time curve (AUC) was the same in both groups, but the trough drug plasma concentration was six times lower in the once-daily group. A low trough drug level and a high baseline viral load (>100,000 copies/mL) were associated with an increased risk of failure.
2.1.2 Studies in Treatment-Experienced Subjects
The P005 study [23, 24] was a multi-centre, randomized, triple-blind, placebo-controlled trial of HIV-positive, antiretroviral-experienced subjects with resistance to multiple classes of antiretrovirals. Subjects were randomized to receive an optimized background regimen (OBR) ± raltegravir (200, 400, 600 mg twice daily) for 24 weeks. At week 24, mean changes from baseline viral load were −1.80 (200 mg), −1.87 (400 mg), −1.84 (600 mg) and −0.35 log10 copies/mL (placebo). Adding raltegravir to an OBR improved the virologic response at all doses studied. Subjects were all offered open-label raltegravir after week 24; at 96 weeks, the improved virologic response was sustained.
BENCHMRK 1 and 2 [24–28] were phase III, randomized, double-blind studies conducted in Europe, Asia, Australia and North and South America. The studies enrolled treatment-experienced HIV-1-infected subjects with resistance to at least one drug in each of three classes; these subjects were randomized to receive an OBR based on resistance testing and treatment history with or without raltegravir. Suppression of HIV RNA levels to <50 copies/mL was achieved in 62.1 % of the subjects in the raltegravir arm versus 32.9 % of those in the placebo arm at week 48. The addition of raltegravir remained virologically superior at weeks 96 and 156. Subgroup analysis showed consistently better outcomes with raltegravir. Of those failures that did occur, 68 % were associated with development of raltegravir drug resistance mutations.
As part of the SHCS (Swiss HIV Cohort Study), the ‘real world’ therapeutic use of raltegravir in treatment-experienced patients was described [29]. The predominant factor associated with a switch to raltegravir in patients with a suppressed baseline viral load was the use of enfuvirtide (odds ratio 41.9, 95 % CI 11.6–151.6). Those with an undetectable baseline viral load had a 95.8 % rate of viral suppression on raltegravir, while those with detectable HIV RNA levels showed 71.8 % viral suppression. Failures were associated with very low genotypic sensitivity of the background regimen, very low plasma raltegravir concentrations, poor adherence, and high baseline viral load. Additionally, higher rates of virologic suppression were seen in raltegravir-based salvage regimens with two nucleoside reverse transcriptase inhibitors (NRTIs), even if genotype testing revealed partial- to high-grade resistance to this class [30]. In Italy, the ISS-NIA (Istituto Superiore di Sanità; New Inhibitors Against HIV) study group conducted an observational analysis of 101 treatment-experienced subjects receiving raltegravir as part of a salvage regimen [31]. Twenty-six subjects experienced virologic failure; this likelihood was found to be independent of sex, age, route of transmission, baseline CD4+ cell count, baseline HIV-1 viral load, or concomitant antiretroviral medications.
The REALMRK study was a phase III, multi-national, single-arm, observational trial that enrolled a diverse cohort of HIV-positive subjects (both sexes, Black/White, treatment-naïve and treatment-experienced) and administered raltegravir 400 mg twice daily in addition to a background antiretroviral therapy (ART) regimen [32]. The open-label design, as well as the variety of patient types and background ART regimens, made it difficult to draw many stringent conclusions. Overall, however, raltegravir had potent efficacy and was generally well tolerated, regardless of sex or race.
2.1.3 Switch Studies
HIV-1 infected subjects with a suppressed viral load receiving boosted lopinavir plus two NRTIs were randomized to switch to raltegravir to improve their lipid profiles in SWITCHMRK 1 and 2 [33]. At week 12, lipids in the raltegravir group improved, while those in the boosted lopinavir group worsened slightly. At week 24, the rate of viral suppression was 84.4 % in the raltegravir group and 90.6 % in the boosted lopinavir group. The study was terminated at this time point because of the failure to establish the non-inferiority of raltegravir to boosted lopinavir. The authors speculate that the raltegravir arm’s more extensive history of virologic failures may have biased this result. The SPIRAL study [34] randomized HIV-infected subjects who virologically suppressed on a ritonavir boosted-protease inhibitor (PI/r)-based regimen to continue this regimen or to switch to a raltegravir-based regimen. At week 48, the raltegravir arm was found to be non-inferior in terms of rates of virologic suppression, and the subjects receiving raltegravir experienced improvements in their lipid profiles. While a similar percentage of subjects of both the SWITCHMRK (33 %) and SPIRAL (38 %) studies had experienced prior virologic failures, the subjects in SWITCHMRK were only required to be virologically suppressed on their entry regimen for 3 months versus 6 months in SPIRAL.
Enfuvirtide, previously a staple of salvage therapy for HIV-1, has the distinct disadvantages of being a twice-daily injectable with a complicated preparation of the dose and adverse reactions that include painful subcutaneous nodules at the site of injection. A small, open-label, uncontrolled trial [35] was conducted, in which 35 HIV-infected treatment-experienced subjects elected to switch from enfuvirtide to raltegravir while leaving the remainder of their regimens intact. After 7 months, 34 subjects maintained virologic suppression with resolution of the enfuvirtide-induced injection site reactions. EASIER ANRS 138 [36, 37] was a randomized, open-label study in which half of the subjects switched from enfuvirtide to raltegravir. At week 24, 88–89 % of subjects in both arms had HIV viral load <50 copies/mL; the control arm was then permitted to switch to raltegravir. At week 48, 90 % of subjects in both arms were virologically suppressed.
There have been a number of smaller studies investigating the incorporation of raltegravir into experimental antiretroviral regimens. One study of eight participants who were virologically suppressed on boosted atazanavir plus two NRTIs switched their regimens to boosted atazanavir plus raltegravir 400 mg once daily; serum levels of raltegravir fell within a range that may be considered adequate [38]. In another, 94 subjects were randomized to receive either boosted atazanavir plus tenofovir/emtricitabine or twice-daily unboosted atazanavir plus raltegravir [39]. While comparable numbers of subjects in each arm achieved viral loads <50 copies/mL, higher rates of hyperbilirubinaemia and development of raltegravir resistance mutations made the atazanavir/raltegravir regimen less desirable. Another small, retrospective study of 18 subjects examined the effect of switching suppressed patients to a regimen of raltegravir plus etravirine; the per-protocol rate of continued virologic suppression at 12 months was 100 % [40].
Raltegravir possesses a modest genetic barrier to resistance. In vitro passaging experiments have revealed the following major mutation pathways in the integrase coding region: [41–43] Y143R/C, Q148H/R/K and N155H. These often appear along with secondary mutations. In STARTMRK, 16 subjects experienced virologic failure with HIV RNA levels >400 copies/mL; of these, four subjects developed integrase mutations (two at codon 143 and two at codon 148) [44]. Of the subjects with virologic failure in the BENCHMRK 1 and 2 studies, 68 % developed integrase mutations (ten subjects with mutations at codon 143, 27 at codon 148, and 38 at codon 155) [27].
Overall, raltegravir is extremely well tolerated [45]. Adverse events in STARTMRK [16] included headache (3.9 %), dizziness (1.4 %), insomnia (3.6 %), fatigue (1.8 %), nausea (2.8 %) and diarrhoea (1.1 %). In BENCHMRK 1 and 2 [24], 46 % of subjects on raltegravir had a drug-related adverse event, including headache (5 %), diarrhoea (2 %), nausea (8 %) and fatigue (4 %). Six patients in the raltegravir group of SWITCHMRK 1 and 2 [33] discontinued treatment early because of adverse events (hypersensitivity n = 1; diarrhoea n = 1; acute stress disorder n = 1; adverse drug reaction n = 1; raised serum concentration of alanine aminotransferase n = 2). Prospective studies have identified rates of grade 3/4 creatine phosphokinase (CPK) elevations in 5–13 % of participants receiving raltegravir, and four reports of possible raltegravir-related rhabdomyolysis have been published [46]. This has led the FDA to advise that raltegravir should be used with caution in patients at risk of myopathy.
Studies support the use of raltegravir as a potent antiretroviral agent in both treatment-naïve patients and as part of salvage regimens in those with multi-class resistance. Notable failures of raltegravir include (a) the mixed results after an attempt to switch to raltegravir from a PI-based regimen and (b) the trial of once-daily raltegravir. This drug is very well tolerated, but limitations of this medication are its twice-daily dosing schedule and its relatively low genetic barrier to viral resistance. These constraints have prompted the search for other integrase inhibitors.
2.2 Elvitegravir (GS-9137/JTK-303)
Elvitegravir (Fig. 2b) is a monoketo acid strand transfer inhibitor [47, 48] developed by Gilead Sciences. It is a potent antiretroviral drug with an in vitro EC90 (90 % effective dose) of 1.7 nM and 30 % oral bioavailability in dogs. Elvitegravir is metabolized primarily by cytochrome P450 (CYP)3A enzymes, with some minor metabolism by glucuronidation [49]. It is excreted predominantly in the faeces. The elimination half-life (t½) is significantly increased from 3.5 h to 9.5 h by co-administration of a potent CYP3A inhibitor. Thus, boosting of elvitegravir with either ritonavir or cobicistat (GS-9350, a non-therapeutic CYP3A inhibitor also developed by Gilead Sciences) allows for once-daily dosing [50, 51]. Though there are no drug-drug interactions between elvitegravir and zidovudine, stavudine, didanosine, abacavir, etravirine, tipranavir or darunavir, the same cannot be said for the fixed-dose combination discussed below [52].
Forty HIV-positive subjects who were not receiving ART were given elvitegravir monotherapy or placebo for 10 days at five dosing regimens (200, 400 or 800 mg twice daily, 800 mg once daily, or 50 mg + 100 mg ritonavir once daily) [50]. Subjects receiving active drug showed at least a 1.91 log10 copies/mL decrease in HIV RNA.
2.2.1 Elvitegravir versus a Boosted Protease Inhibitor
Zolopa et al. [53] described a phase II, non-inferiority trial of ritonavir-boosted elvitegravir (20, 50 or 125 mg) versus a boosted PI (PI/r) added to an OBR (consisting of NRTIs ± enfuvirtide) in treatment-experienced HIV-positive subjects. The study enrolled 278 subjects with an average of 11 protease mutations and three thymidine analogue mutations (TAMs) each. At week 8, the Data Safety Monitoring Board (DSMB) stopped the elvitegravir 20 mg arm due to virologic failures, and allowed subjects in the other groups to electively add a PI/r. The time-weighted change in HIV RNA level from baseline through week 24 was −1.19 log10 copies/mL for the PI/r group, −1.44 log10 copies/mL for the elvitegravir 50 mg arm (non-inferior) and −1.66 log10 copies/mL for the elvitegravir 125 mg arm (superior, p = 0.021). Durability of virologic response was highly dependent upon the activity of the OBR.
Gilead Sciences co-formulated a once-daily combination pill containing elvitegravir, cobicistat, tenofovir and emtricitabine; this combination (previously referred to as Quad) was recently approved by the FDA for treatment of HIV-1 infection in antiretroviral-naïve individuals and is being marketed as Stribild™. The stand-alone formulation of elvitegravir remains investigational, though licensure is anticipated.
GS-236-0103 was a phase III, randomized, multi-national, non-inferiority trial comparing elvitegravir/cobicistat/tenofovir/emtricitabine with boosted atazanavir/tenofovir/emtricitabine (atazanavir/r/tenofovir/emtricitabine) in treatment-naïve, HIV-positive subjects [54]. A total of 708 subjects were randomized; 89.5 % of the elvitegravir/cobicistat/tenofovir/emtricitabine arm and 86.8 % of the atazanavir/r/tenofovir/emtricitabine arm achieved the primary outcome of HIV RNA <50 copies/mL maintained through week 48. Non-inferiority persisted regardless of baseline HIV RNA levels [55].
2.2.2 Elvitegravir versus Efavirenz
Elvitegravir/cobicistat/tenofovir/emtricitabine was first tested against efavirenz/tenofovir/emtricitabine in HIV-positive, antiretroviral-naïve subjects in a phase II study described by Cohen et al. [56] At weeks 24 and 48, 90 % of the elvitegravir/cobicistat/tenofovir/emtricitabine recipients and 83 % of the subjects in the efavirenz/tenofovir/emtricitabine arm had achieved HIV RNA levels <50 copies/mL. This was followed by a phase III study of elvitegravir/cobicistat/tenofovir/emtricitabine versus efavirenz/tenofovir/emtricitabine in 700 HIV-positive, treatment-naïve subjects [57]. An HIV RNA level <50 copies/mL was achieved at week 48 by 87.6 % of subjects in the elvitegravir/cobicistat/tenofovir/emtricitabine arm and by 84.1 % in the efavirenz/tenofovir/emtricitabine arm, demonstrating the non-inferiority of elvitegravir/cobicistat/tenofovir/emtricitabine. This non-inferiority remained true regardless of baseline HIV viral load [58].
2.2.3 Elvitegravir versus Raltegravir
GS-US-183-0145 directly compared elvitegravir versus raltegravir in addition to an OBR (containing a PI/r plus at least one other drug) in subjects who were treatment experienced and/or resistant to at least two classes of antiretrovirals [59]. Fifty-nine percent of subjects in the elvitegravir arm and 58 % in the raltegravir arm achieved and maintained an HIV viral load <50 copies/mL by week 48. At week 96, 47.6 % of the elvitegravir arm and 45.0 % of the raltegravir arm were virologically suppressed [60].
2.2.4 Switch Study
Finally, GS-US-236-0121 is a phase IIIb trial in which HIV-positive subjects currently suppressed on a regimen consisting of a non-NRTI (NNRTI) (efavirenz, nevirapine or rilpivirine) plus tenofovir and emtricitabine are randomized to continue their current regimen or to switch to elvitegravir/cobicistat/tenofovir/emtricitabine [61]. The primary endpoint will be the percentage of subjects with an HIV RNA level <50 copies/mL at week 48. This study is currently recruiting volunteers. A trial with a parallel design is recruiting subjects who are virologically suppressed on a regimen of a PI/r plus tenofovir and emtricitabine [62].
The major resistance mutations in the integrase coding region that affect elvitegravir are T66I, E92Q, Q148H/K/R and N155H; these were selected in vitro and confirmed by in vivo observations [63–65]. There is cross-resistance with raltegravir at the 148 and 155 loci; elvitegravir retains activity against viruses with the Y143R mutation [66]. The dose-ranging monotherapy study revealed no integrase mutations [50]. The study by Zolopa et al. [53] demonstrated the necessity of having at least one other active drug in a regimen to ensure the durability of a virologic response to elvitegravir. In the phase III trial of elvitegravir/cobicistat/tenofivor/emtricitabine versus efavirenz/tenofivor/emtricitabine [57], 14 subjects in the elvitegravir/cobicistat/tenofivor/emtricitabine arm experienced virologic failure and were tested for resistance; eight had resistance mutations (8/8 with NRTI resistance and 7/8 with integrase resistance). The integrase mutations unearthed were E92Q (n = 7), T66I (n = 1), Q148R (n = 1) and N155H (n = 1). Of the 12 subjects taking elvitegravir/cobicistat/tenofovir/emtricitabine in GS-236-0103 with HIV RNA >50 copies/mL, four had resistance mutations detected: two with Q148R, one with N155H and one with T66I. NRTI resistance mutations that arose in the elvitegravir/cobicistat/tenofovir/emtricitabine arm included M184V/I and K65R [54]. In GS-US-183-0145 [59], 20 % (elvitegravir) and 22 % (raltegravir) experienced virologic failure; of the failures, integrase resistance mutations were found in 27 % (elvitegravir) and 20 % (raltegravir). Overall, integrase resistance mutations developed in 6.6 % of the elvitegravir arm and 7.4 % of the raltegravir arm [67].
No serious adverse events were seen in the 10-day monotherapy study of elvitegravir [50]. Subjects in the phase IIb study experienced headache, fatigue, nausea, vomiting, pyrexia, constipation and diarrhoea in a non-dose-dependent fashion [53]. Study drug-related neurological and psychiatric adverse events in the elvitegravir versus efavirenz trial were significantly less frequent in the elvitegravir arm than in the efavirenz arm [56]. Notably though, the estimated glomerular filtration rate (eGFR) decreased by 14 % starting at week 2 through week 48 in the elvitegravir/cobicistat/tenofovir/emtricitabine group; eGFR remained within the normal range during this period. Creatinine was noted to increase by 13 μmol/L (p < 0.001) at week 48 in the elvitegravir/cobicistat/tenofovir/emtricitabine arm of the phase III trial described by Sax et al. [57] When compared with a regimen based on boosted atazanavir [54], the elvitegravir/cobicistat/tenofovir/emtricitabine group had fewer liver function test (LFT) abnormalities and changes to the lipid profile, but a greater increase in creatinine (+11 μmol/L). It should be noted that cobicistat is associated with an observed increase in serum creatinine and eGFR due to inhibition of tubular secretion of creatinine.
Upon direct comparison with raltegravir, the elvitegravir arm showed more diarrhoea, but fewer LFT changes and serious adverse events; it may be preferable in patients with concurrent liver disease [59]. Overall, since earlier elvitegravir trials were conducted with ritonavir, data are more limited regarding the safety profile of cobicistat.
Elvitegravir, both alone and as part of the elvitegravir/cobicistat/tenofovir/emtricitabine fixed-dose combination pill, will be a convenient and potent antiretroviral medication in treatment-naïve and treatment-experienced HIV-positive patients. With its once-daily dosing, it overcomes one of the limitations of raltegravir, and elvitegravir/cobicistat/tenofovir/emtricitabine contributes another complete regimen in one pill once per day in treatment-naïve HIV-infected individuals. However, the cross-resistance with raltegravir may limit its usefulness in heavily treatment-experienced patients. Additionally, the need for a boosting agent raises concerns for drug-drug interactions. Due to these concerns and the lack of data in older patients, women, and patients with creatinine clearance less than 70 mL/min, elvitegravir/cobicistat/tenofovir/emtricitabine has been initially listed as an alternate regimen in Department of Health and Human Services (DHHS) guidelines. There has been a continued search for an integrase inhibitor with a greater genetic barrier to resistance and a resistance profile that does not overlap highly with first-generation integrase inhibitors.
3 Second-Generation Integrase Inhibitors
3.1 Dolutegravir (S/GSK1349572)
This search for a once-daily integrase inhibitor that has minimal cross-resistance with first-generation drugs in this class has led to the discovery and development of dolutegravir (Fig. 2c) by Shionogi-ViiV Healthcare and GlaxoSmithKline (GSK) [68]. It, too, is a potent antiretroviral agent with an EC90 of 2 nM [69] and activity against HIV-2 in addition to all clades of HIV-1 [70].
In a phase IIa, placebo-controlled, dose-ranging study, subjects were randomized to either dolutegravir monotherapy (2, 10 or 50 mg) or placebo for 10 days [71]. Mean change in plasma viral load from baseline in the dolutegravir arms was −1.51 to −2.46 log10 copies/mL, which varied in a dose-dependent fashion. Of the subjects in the dolutegravir 50 mg arm, 70 % achieved an HIV RNA level <50 copies/mL by day 10.
The VIKING-1 cohort [72] consisted of 27 subjects with resistance to raltegravir and at least two other classes of antiretroviral drugs who were on a failing regimen at the time of study entry. They were maintained on their failing background regimen and given dolutegravir 50 mg daily for 11 days, at which point their background regimens were optimized if available. Response varied based on the baseline integrase genotype: 16/16 subjects with single mutations (N155H or Y143H or Q148) achieved a viral load <400 copies/mL or a decrease of ≥0.7 log10 copies/mL versus 3/4 with Q148 plus one mutation versus 0/5 with Q148 plus two or more other mutations. From day 1 to day 11, there was little change in genotypic or phenotypic resistance changes to dolutegravir [73]. The VIKING-2 cohort had the same qualities as cohort 1 and received the same schedule of intervention, except that they were given dolutegravir 50 mg twice daily rather than once daily [74]. A plasma HIV RNA <400 copies/mL or reduction by −0.7 log10 copies/mL was achieved by 96 % of cohort 2 subjects. Additionally, all subjects in this cohort with at least a Q148 integrase mutation responded to twice-daily dolutegravir by day 11. Notably, subjects in cohort 2 were required to have at least one fully active antiretroviral drug available on entry (this was not a requirement for cohort 1). Cohort 2 participants’ virus also had a narrower range in fold-change susceptibility to dolutegravir than did cohort 1.
SPRING-1 was a phase IIb, randomized, dose-finding study comparing dolutegravir (10, 25 or 50 mg daily) with efavirenz (600 mg daily) [75] co-administered with either tenofovir/emtricitabine or lamivudine/abacavir. Subjects (n = 205) received at least one dose of study drug. At week 16, 90 % of the dolutegravir arms and 60 % of the efavirenz arm had achieved a viral load <50 copies/mL. By week 48, 90 % of the dolutegravir arms and 82 % of the efavirenz arm had plasma HIV-1 RNA levels below detection. Dolutegravir remained non-inferior, with a favourable safety profile at the week 96 assessment [76]. At that point, it was noted that fewer subjects withdrew from the study due to drug-related adverse events in the dolutegravir arms (3 %) than in the efavirenz arm (10 %). The results of SPRING-1 were used to support a dolutegravir 50 mg daily dosing schedule for SPRING-2, a phase III, multi-centre, non-inferiority study of dolutegravir versus raltegravir [77]. Antiretroviral-naïve subjects (n = 827) were randomized to receive either dolutegravir 50 mg daily or raltegravir 400 mg twice daily on top of a backbone of tenofovir/emtricitabine or lamivudine/abacavir. At week 48, 88 % in the dolutegravir group and 85 % in the raltegravir arm achieved a viral load <50 copies/mL. The non-inferiority of dolutegravir was maintained regardless of baseline viral load.
Study ING116529 is a multi-centre, randomized trial in which treatment-experienced subjects with virologic failure on raltegravir or elvitegravir (with evidence of integrase-resistance mutations) are randomized to switch to twice-daily dolutegravir or placebo (plus the non-integrase inhibitor background regimen) for 7 days, then switch to open-label dolutegravir plus an OBR [78]. This trial is actively recruiting at this time.
Dolutegravir is being compared with ritonavir-boosted darunavir on a two NRTI background in HIV-positive, treatment-naïve subjects in ING114915 [79]; the study is on-going, but subject recruitment is complete.
Finally, Shionogi-ViiV Healthcare is continuing a trial of ‘572-Trii’, consisting of dolutegravir plus abacavir and lamivudine in a fixed-dose combination pill (Epzicom®, Kivexa®) [80]. This is a phase III trial (SINGLE) of HIV-positive, treatment-naïve subjects who were randomized to 572-Trii versus the efavirenz/tenofovir/emtricitabine fixed-dose combination; outcomes are safety, tolerability, antiviral potency and development of resistance. The 48-week results [81], presented at the 2012 Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), showed a superiority of 572-Trii to efavirenz/tenofovir/emtricitabine (88 % viral suppression to <50 copies/mL at week 48 versus 81 % suppression [p = 0.003]). This difference in efficacy was statistically maintained for baseline viral loads <100,000 copies/mL, but it was no longer superior at viral loads >100,000 copies/mL. Additionally, subjects taking 572-Trii had larger increases in CD4+ cell count (267 vs. 208 cells/mm3; p = 0.001).
Dolutegravir has less resistance overlap with raltegravir than does elvitegravir. Raltegravir failure infrequently selects for mutation combinations that would cause resistance to dolutegravir [82, 83]. Some viruses containing E138K, G140S or Q148R/H mutations demonstrated diminished susceptibility to dolutegravir in vitro [70, 75, 84–86]. Other in vitro dolutegravir mutations that have arisen are T124A (seen frequently in raltegravir failures [87]), S153F/Y and L101I. Notably, the presence of Q148 plus two or more other integrase mutations or Q148H + G140S significantly hamper the antiviral function of dolutegravir [72, 88]. This illustrates the importance of not maintaining a patient on a failing raltegravir- or elvitegravir-containing regimen, as the accumulation of mutations can hinder future attempts to use the second-generation integrase inhibitors. Dolutegravir appears to have a higher genetic barrier to resistance than raltegravir [69, 85]. This may be due to its longer half-life, or to its slower dissociation from HIV-integrase complexes [89]. Of the three virologic failures identified in SPRING-1, no integrase mutations were detected [75]; this remained true for the subjects who failed therapy with dolutegravir in SPRING-2 [77].
The most common complaints in the dose-ranging study were mild to moderate diarrhoea, fatigue and headache [90]. A cumulative safety profile of VIKING and SPRING-1 noted 11 % nausea, 8 % diarrhoea, 5 % headache, 18 % dizziness, 8 % fatigue, 8 % insomnia and 8 % rash [71]. A small increase in creatinine was noted at week 1, remained stable through week 20, and decreased from week 20 through week 48. The creatinine change is not thought to be due to a change in GFR, but rather non-pathological inhibition of organic cation transport (OCT)-2, decreasing tubular secretion of creatinine [91]. In fact, when GFR was calculated based on measurement of iohexol clearance, there was no difference from placebo [92]. Subjects in the dolutegravir arm of SPRING-2 [77] noted nausea, headache, nasopharyngitis and diarrhoea at rates similar to those in the raltegravir arm.
Should dolutegravir be approved by the FDA for the treatment of HIV, it would provide the advantage of once-daily dosing and a comparatively higher genetic barrier to resistance. Multiple studies have demonstrated the relatively narrow overlap in resistance mutations (notably, the Q148 + secondary mutations) that dolutegravir has with first-generation integrase inhibitors. Finally, the drug has been well tolerated in studies performed to date.
3.2 S/GSK1265744 and 744 Long-Acting Parenteral
S/GSK1265744 (Fig. 2d) is another strand transfer inhibitor originally developed by Shionogi-ViiV Healthcare/GSK. The drug is very potent, with an in vitro half maximal inhibitory concentration (IC50) of 0.34 nM. However, it is highly protein bound to serum albumin and has a protein-adjusted IC90 of 166 ng/mL. It has a plasma half-life of approximately 40 h in its oral formulation, allowing for once-daily dosing. A long-acting parenteral (LAP) nanosuspension preparation was created, which has a t½ of 21–50 days.
Oral S/GSK1265744 has been studied in a three-part series of phase I/IIa trials [93]. In Part A, 18 HIV-negative volunteers received a single dose (5, 10, 25 or 50 mg) of S/GSK1265744 or placebo. Thirty HIV-negative subjects received daily doses of S/GSK1265744 (5, 10, 25 mg) versus placebo for 14 days in Part B. There were no serious adverse events in either of the preceding two parts. In Part C, 11 HIV-positive subjects received 30 mg of S/GSK1265744 versus placebo for 10 days, followed by 3 days of no study drug, then combination ART was begun on day 14. HIV RNA levels decreased by a median of 2.6 log10 copies/mL and suppression of viral load was maintained through day 14 in 88 % of subjects.
A phase IIb, blinded, dose-ranging study [94] of oral S/GSK1265744 is planned; about 200 subjects are to be enrolled. In the induction phase, they will be randomized to one of three doses of S/GSK1265744 (10, 30 and 60 mg) or a control arm of efavirenz; all groups will receive a backbone regimen of lamivudine/abacavir or tenofovir/emtricitabine. The maintenance phase will enrol those subjects who were randomized to receive S/GSK1265744, and who achieved an HIV viral load <50 copies/mL through week 24. These volunteers will be offered the opportunity to simplify their regimens to S/GSK1265744 (at their randomized dose) plus rilpivirine 25 mg daily for 72 weeks. This study is designed to form the basis for parenteral therapy, either intramuscular or subcutaneous, with 744-LAP in combination with rilpivirine, as both agents can be administered in this manner on a monthly schedule.
Phase I studies are also being conducted to characterize 744-LAP. In a randomized, placebo-controlled, double-blind study described by Spreen et al. [95], HIV-negative subjects were given a gluteal intramuscular injection (100, 200, 400 or 400 mg × 2) or a subcutaneous abdominal injection (100, 200, 200 mg × 2) of 744-LAP versus placebo. Fifty-six subjects received an injection; the most common adverse event was a self-limited injection site reaction. There were no systemic serious or grade 3–4 adverse events. The half-life ranged from 21 to 50 days in a dose-dependent manner; split dosing increased the AUC and the half-life. These results led to the planning of LAI115428, a phase I, repeat-dose study of 744-LAP. Healthy subjects are to receive intramuscular or subcutaneous injections of 744-LAP monthly or quarterly versus daily oral S/GSK1265744 alone versus S/GSK1265744 in combination with TMC278-LA (a long-acting form of rilpivirine) [96]. This trial is recruiting subjects.
Thus far, there has not been clinical evidence of viral resistance to S/GSK1265744, although few HIV-positive subjects have been treated. Viral isolates from subjects with high-level raltegravir resistance remained sensitive to S/GSK1265744 in vitro [86]. The maximum fold change in S/GSK1265744 sensitivity occurred in mutants with the Q148K/R (5.6/5.1-fold change, respectively) [97]. Overall, S/GSK1265744 has been well tolerated, and the long-acting formulation displays excellent pharmacokinetics.
4 Dual Reverse Transcriptase and Integrase Inhibitors
The integrase protein of HIV-1 and the RNAse-H domain of RT share structural and functional similarities. Integrase mutations also affect the function of RT, and an RT binding site on integrase has been proposed [5–8]. DKAs have been found that bind both enzymes, and these may be developed as a hybrid class of compounds. These are reviewed in depth by Di Santo [98].
5 Non-Catalytic Site Integrase Inhibitors
5.1 LEDGINs
LEDGINs are small molecules, designed to be potent inhibitors of the integrase-LEDGF/p75 interaction, that allosterically block catalytic integrase activities. This activity is promoted by the stabilization of the dimer interface of integrase upon LEDGIN binding [99–103]. These compounds have been shown to retain potency in vitro against an array of clades and against viruses harbouring mutations against integrase strand transfer inhibitors [101].
5.2 BI 224436
BI 224436 (Fig. 2e) is an integrase inhibitor currently licensed by Gilead (originally developed by Boehringer-Ingelheim) that operates at a non-catalytic site to interfere with the interaction between integrase and the chromatin targeting the LEDGF/p75 protein; it inhibits 3′ processing and HIV replication [104]. This is a potent antiviral with an EC95 of 78 ± 18 nM. Against raltegravir-resistant viral isolates, BI 224436 had a mean fold change in EC50 of 1.4 ± 0.8 relative to a wild-type control virus. The optimal range of human doses was projected using a hollow-fibre infection model and pharmacokinetic modelling [105].
BI 224436 has been tested to date in a phase I, double-blind, placebo-controlled, first-in-human trial [106]. In this study, 48 healthy volunteers were randomized to BI 224436 in oral solution (6.2, 12.5, 25, 50, 100 or 200 mg in consecutive dose cohorts) or placebo. Plasma levels of drug increased proportional to dose, and t½ was 7.11 h. There were four mild adverse events: headache (n = 1, 25-mg dose), upper abdominal pain (n = 1, 25-mg dose) and oral paresthesia (n = 2, 50-mg dose).
6 Conclusion
Since the approval of raltegravir in 2007, its clinical performance has supported the data from its phase III clinical trials. This is a potent, well tolerated antiretroviral medication. Its twice-daily dosing and low genetic barrier to resistance has spurred the search for other integrase inhibitors. Elvitegravir, recently approved as part of the fixed-dose combination pill elvitegravir/cobicistat/tenofovir/emtricitabine and currently in phase IIIb trials, can be administered only once daily when given concurrently with a potent CYP3A inhibitor (ritonavir or cobicistat). However, there is extensive cross-resistance between the two drugs.
Another well tolerated, once-daily integrase inhibitor is dolutegravir. As the second-generation integrase inhibitor with the most advanced development, this drug is also very well tolerated and has only a narrow overlap in resistance profile with raltegravir (Q148 + two or more other mutations). The ‘backup’ drug to dolutegravir, S/GSK1265744, also demonstrates favourable tolerability and a relatively high genetic barrier to resistance in the limited testing to date. The long-acting formulation, 744-LAP, is being studied with monthly or quarterly administration, and this dosing schedule is exciting from both a treatment and an HIV-1 prevention standpoint. Lastly, BI 224436 represents the first integrase inhibitor to bind at a non-catalytic site; viral isolates with high-grade raltegravir resistance retained sensitivity to this drug.
The second-generation integrase inhibitors face the challenges of competing with and/or complementing the currently available arsenal of antiretroviral medications. Those in development seem to have good tolerability, and the fact that integrase has no human homolog will likely limit the toxicities of future candidates. They will need to have a maximum dosing frequency of once-daily administration.
Importantly, increased stability of binding integrase and new binding targets on the enzyme will minimize the level of intra-class cross-resistance. While elvitegravir resistance mutations mirror most of those for raltegravir, dolutegravir has already demonstrated a higher genetic barrier to resistance and a much more narrow overlap with raltegravir resistance mutations. Studies are very early in S/GSK1265744/744-LAP, but no resistance was seen so far in vivo, and in vitro experiments support a high barrier to resistance; raltegravir-resistant viral isolates retained sensitivity to S/GSK1265744. BI 224436 binds a separate site on the integrase protein and retained effectiveness in vitro against isolates with high-level raltegravir resistance.
Of the integrase inhibitors currently in use or in clinical trials, only elvitegravir requires co-administration with ritonavir or cobicistat to act as a pharmacoenhancer to support once-daily dosing. Newer compounds have been identified whose half-lives and potency allow for once-daily dosing in the absence of the need for a pharmacological enhancing agent.
Raltegravir has been a productive addition to our arsenal of antiretroviral drugs since 2007. Its novel target and tolerability have made it a useful drug in both treatment-naïve and heavily treatment-experienced patients with HIV-1. It can also boast the most complete safety profile of the members of this drug class. Raltegravir is best suited for the naïve or experienced patient with the ability to comply with a twice-daily dosing schedule. Stribild™ (elvitegravir/cobicistat/tenofovir/emtricitabine) is a convenient, one-pill, once-daily antiretroviral regimen that will certainly be a welcome addition to treatment options for the antiretroviral naïve, HIV-1-infected patient, now that it has been approved by the FDA. Caution must be used in patients with pre-existing renal disease, dyslipidaemia, or in patient populations in which the drug has yet to be studied (e.g. older patients, women). Dolutegravir and S/GSK1265744 appear to offer higher barriers to resistance, limited cross-resistance with raltegravir and elvitegravir, and once-daily dosing. If they continue to have favourable safety profiles, these agents show promise both as first-line antiretrovirals and in deep salvage regimens for patients with prior integrase inhibitor exposure. BI 224436 binds to a different site in integrase and is unlikely to have cross-resistance with the other integrase inhibitors. The LEDGINs will require further development prior to clinical testing. Integrase is a target that will continue to be exploited in the treatment, and possibly the prevention, of HIV-1 infection.
References
Marchand C, Maddali K, Metifiot M, Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr Top Med Chem. 2009;9(11):1016–37.
Prada N, Markowitz M. Novel integrase inhibitors for HIV. Expert Opin Investig Drugs. 2010;19(9):1087–98.
Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, et al. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007;21(14):1767–78.
Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D, et al. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One. 2007;2(12):e1340.
Wu X, Liu H, Xiao H, Conway JA, Hehl E, Kalpana GV, et al. Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex. J Virol. 1999;73(3):2126–35.
Zhu K, Dobard C, Chow SA. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J Virol. 2004;78(10):5045–55.
Dobard CW, Briones MS, Chow SA. Molecular mechanisms by which human immunodeficiency virus type 1 integrase stimulates the early steps of reverse transcription. J Virol. 2007;81(18):10037–46.
Wilkinson TA, Januszyk K, Phillips ML, Tekeste SS, Zhang M, Miller JT, et al. Identifying and characterizing a functional HIV-1 reverse transcriptase-binding site on integrase. J Biol Chem. 2009;284(12):7931–9.
Markowitz M, Morales-Ramirez JO, Nguyen BY, Kovacs CM, Steigbigel RT, Cooper DA, et al. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr. 2006;43(5):509–15.
Murray JM, Emery S, Kelleher AD, Law M, Chen J, Hazuda DJ, et al. Antiretroviral therapy with the integrase inhibitor raltegravir alters decay kinetics of HIV, significantly reducing the second phase. AIDS. 2007;21(17):2315–21.
Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, et al. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007;46(2):125–33.
Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, et al. Sustained antiretroviral effect of raltegravir after 96 weeks of combination therapy in treatment-naive patients with HIV-1 infection. J Acquir Immune Defic Syndr. 2009;52(3):350–6.
Gotuzzo E, Nguyen BY, Markowitz M, Mendo F, Ratanasuwan W, Lu C, et al. Sustained antiretroviral efficacy of raltegravir after 192 weeks of combination ART in treatment-naive HIV-1-infected patients. 17th Conference on retroviruses and opportunistic infections, San Francisco; 2010.
Gotuzzo E, Markowitz M, Ratanasuwan W, Smith G, Prada G, Morales-Ramirez JO, et al. Sustained efficacy and safety of raltegravir after 5 years of combination antiretroviral therapy as initial treatment of HIV-1 infection: final results of a randomized, controlled, phase II study (protocol 004). J Acquir Immune Defic Syndr. 2012;71(1):73–7.
Lennox JL, DeJesus E, Lazzarin A, Pollard RB, Madruga JV, Berger DS, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet. 2009;374(9692):796–806.
Lennox JL, Dejesus E, Berger DS, Lazzarin A, Pollard RB, Ramalho Madruga JV, et al. Raltegravir versus efavirenz regimens in treatment-naive HIV-1-infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune Defic Syndr. 2010;55(1):39–48.
Rockstroh JK, Lennox JL, Dejesus E, Saag MS, Lazzarin A, Wan H, et al. Long-term treatment with raltegravir or efavirenz combined with tenofovir/emtricitabine for treatment-naive human immunodeficiency virus-1-infected patients: 156-week results from STARTMRK. Clin Infect Dis. 2011;53(8):807–16.
Rockstroh JK, DeJesus E, Saag MS, Yazdanpanah Y, Lennox JL, Rodgers AJ, et al. Long-term safety and efficacy of raltegravir (RAL)-based versus efavirenz (EFV)-based combination therapy in treatment-naive HIV-1-infected patients: final 5-year double-blind results from STARTMRK. Washington, DC: XIX International AIDS conference; 2012.
Lazzarin A, DeJesus E, Rockstroh JK, Lennox JL, Saag MS, Wan H, et al. Durable and consistent efficacy of raltegravir (RAL) with tenofovir (TDF) and emtricitabine (FTC) across demographic and baseline prognostic factors in treatment-naive patients (pts) from STARTMRK at wk 156. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Rockstroh JK, Lazzarin A, Zhao J, Rodgers AJ, DiNubile MJ, Nguyen BY, et al. Long-term efficacy of raltegravir or efavirenz combined with TDF/FTC in treatment-naive HIV-1-infected patients: week-192 subgroup analyses from STARTMRK. Belgrade: European AIDS conference; 2011.
Eron JJ Jr, Rockstroh JK, Reynes J, Andrade-Villanueva J, Ramalho-Madruga JV, Bekker LG, et al. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis. 2011;11(12):907–15.
Rizk ML, Hang Y, Luo WL, Su J, Zhao J, Campbell H, et al. Pharmacokinetics and pharmacodynamics of once-daily versus twice-daily raltegravir in treatment-naive HIV-infected patients. Antimicrob Agents Chemother. 2012;56(6):3101–6.
Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, et al. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet. 2007;369(9569):1261–9.
Gatell JM, Katlama C, Grinsztejn B, Eron JJ, Lazzarin A, Vittecoq D, et al. Long-term efficacy and safety of the HIV integrase inhibitor raltegravir in patients with limited treatment options in a phase II study. J Acquir Immune Defic Syndr. 2010;53(4):456–63.
Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, et al. Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359(4):339–54.
Steigbigel RT, Cooper DA, Teppler H, Eron JJ, Gatell JM, Kumar PN, et al. Long-term efficacy and safety of raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 phase III trials. Clin Infect Dis. 2010;50(4):605–12.
Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359(4):355–65.
Eron JE, Cooper DA, Steigbigel RT, Clotet B, Wan H, Meibohm AR, et al. Sustained antiretroviral effect of raltegravir at week 156 in the BENCHMRK studies, and exploratory analysis of late outcomes based on early virologi responses. 17th Conference on retroviruses and opportunistic infections, San Francisco; 2010.
Scherrer AU, von Wyl V, Fux CA, Opravil M, Bucher HC, Fayet A, et al. Implementation of raltegravir in routine clinical practice: selection criteria for choosing this drug, virologic response rates, and characteristics of failures. J Acquir Immune Defic Syndr. 2010;53(4):464–71.
Scherrer AU, von Wyl V, Boni J, Yerly S, Klimkait T, Burgisser P, et al. Viral suppression rates in salvage treatment with raltegravir improved with the administration of genotypic partially active or inactive nucleoside/tide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr. 2011;57(1):24–31.
Bucciardini R, D’Ettorre G, Baroncelli S, Ceccarelli G, Parruti G, Weimer LE, et al. Virological failure at one year in triple-class experienced patients switching to raltegravir-based regimens is not predicted by baseline factors. Int J STD AIDS. 2012;23(7):459–63.
Squires KE, Bekker LG, Eron JE, Cheng B, Rockstroh JK, Marquez F, et al. Safety, tolerability, and efficacy of raltegravir (RAL) in a diverse cohort of HIV-infected patients: 48-week results from the REALMRK study. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Eron JJ, Young B, Cooper DA, Youle M, Dejesus E, Andrade-Villanueva J, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet. 2010;375(9712):396–407.
Martinez E, Larrousse M, Llibre JM, Gutierrez F, Saumoy M, Antela A, et al. Substitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL study. AIDS. 2010;24(11):1697–707.
Harris M, Larsen G, Montaner JS. Outcomes of multidrug-resistant patients switched from enfuvirtide to raltegravir within a virologically suppressive regimen. AIDS. 2008;22(10):1224–6.
De Castro N, Braun J, Charreau I, Pialoux G, Cotte L, Katlama C, et al. Switch from enfuvirtide to raltegravir in virologically suppressed multidrug-resistant HIV-1-infected patients: a randomized open-label trial. Clin Infect Dis. 2009;49(8):1259–67.
Gallien S, Braun J, Delaugerre C, Charreau I, Reynes J, Jeanblanc F, et al. Efficacy and safety of raltegravir in treatment-experienced HIV-1-infected patients switching from enfuvirtide-based regimens: 48 week results of the randomized EASIER ANRS 138 trial. J Antimicrob Chemother. 2011;66(9):2099–106.
Calcagno A, Tettoni MC, Simiele M, Trentini L, Montrucchio C, D’Avolio A, et al. Pharmacokinetics of 400 mg of raltegravir once daily in combination with atazanavir/ritonavir plus two nucleoside/nucleotide reverse transcriptase inhibitors. J Antimicrob Chemother. Epub 2012 Oct 19.
Kozal MJ, Lupo S, DeJesus E, Molina JM, McDonald C, Raffi F, et al. A nucleoside- and ritonavir-sparing regimen containing atazanavir plus raltegravir in antiretroviral treatment-naive HIV-infected patients: SPARTAN study results. HIV Clin Trials. 2012;13(3):119–30.
Calin R, Paris L, Simon A, Peytavin G, Wirden M, Schneider L, et al. Dual raltegravir/etravirine combination in virologically suppressed HIV-1-infected patients on antiretroviral therapy. Antivir Ther. 2012;17(8):1601–4.
Goethals O, Vos A, Van Ginderen M, Geluykens P, Smits V, Schols D, et al. Primary mutations selected in vitro with raltegravir confer large fold changes in susceptibility to first-generation integrase inhibitors, but minor fold changes to inhibitors with second-generation resistance profiles. Virology. 2010;402(2):338–46.
Kobayashi M, Nakahara K, Seki T, Miki S, Kawauchi S, Suyama A, et al. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antiviral Res. 2008;80(2):213–22.
Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011;203(9):1204–14.
Lennox JL, DeJesus E, Lazzarin A. Raltegravir demonstrates durable efficacy through 96 weeks: results from STARTMRK, a phase III study of raltegravir (RAL)-based vs efavirenz (EFV)-based combination therapy in treatment-naive HIV-infected patients. 49th Interscience conference on antimicrobial agents and chemotherapy, San Francisco; 2009.
Merck&Co. I. ISENTRESS: highlights of prescribing information. 2009. http://www.merck.com/product/usa/pi_circulars/i/isentress/isentress_pi.pdf Accessed 15 August 2012.
Lee FJ, Carr A. Tolerability of HIV integrase inhibitors. Curr Opin HIV AIDS. 2012;7(5):422–8.
Klibanov OM. Elvitegravir, an oral HIV integrase inhibitor, for the potential treatment of HIV infection. Curr Opin Investig Drugs. 2009;10(2):190–200.
Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, et al. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J Med Chem. 2006;49(5):1506–8.
Schafer JJ, Squires KE. Integrase inhibitors: a novel class of antiretroviral agents. Ann Pharmacother. 2010;44(1):145–56.
DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, et al. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr. 2006;43(1):1–5.
Mathias AA, West S, Hui J, Kearney BP. Dose-response of ritonavir on hepatic CYP3A activity and elvitegravir oral exposure. Clin Pharmacol Ther. 2009;85(1):64–70.
Ramanathan S, Shen G, Hinkle J, Enejosa J, Kearney BP. Pharmacokinetics of coadministered ritonavir-boosted elvitegravir and zidovudine, didanosine, stavudine, or abacavir. J Acquir Immune Defic Syndr. 2007;46(2):160–6.
Zolopa AR, Berger DS, Lampiris H, Zhong L, Chuck SL, Enejosa JV, et al. Activity of elvitegravir, a once-daily integrase inhibitor, against resistant HIV Type 1: results of a phase 2, randomized, controlled, dose-ranging clinical trial. J Infect Dis. 2010;201(6):814–22.
DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379(9835):2429–38.
DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, et al. Analysis of efficacy by baseline viral load: phase 3 study comparing elvitegravir/cobicistat/emtricitabine/tenofovir DF (Quad) versus ritonavir-boosted atazanavir plus emtricitabine/tenofovir DF in treatment-naive HIV-1-positive subjects: week 48 results. Washington, DC: XIX International AIDS conference; 2012.
Cohen C, Elion R, Ruane P, Shamblaw D, DeJesus E, Rashbaum B, et al. Randomized, phase 2 evaluation of two single-tablet regimens elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for the initial treatment of HIV infection. AIDS. 2011;25(6):F7–12.
Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379(9835):2439–48.
Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, et al. Analysis of efficacy by baseline HIV RNA: week 48 results from a phase 3 study of elvitegravir/cobicistat/emtricitabine/tenofovir DF (Quad) compared to efavirenz/emtricitabine/tenofovir DF in treatment-naive HIV-1-positive subjects. Washington, DC: XIX International AIDS conference; 2012.
Molina JM, Lamarca A, Andrade-Villanueva J, Clotet B, Clumeck N, Liu YP, et al. Efficacy and safety of once daily elvitegravir versus twice daily raltegravir in treatment-experienced patients with HIV-1 receiving a ritonavir-boosted protease inhibitor: randomised, double-blind, phase 3, non-inferiority study. Lancet Infect Dis. 2012;12(1):27–35.
Elion R, Molina JM, Arribas JR, Cooper DA, Maggiolo F, Wilkins E, et al. Efficacy and safety results from a randomized, double blind, active controlled trial of elvitegravir (once-daily) versus raltegravir (twice-daily) in treatment-experienced HIV-positive patients: long term 96-week data. Washington, DC: XIX International AIDS conference; 2012.
Gilead Sciences. Phase 3b open label study to evaluate switching from regimens consisting of a non-nucleoside reverse transcriptase inhibitor plus emtricitabine and tenofovir df to the elvitegravir/cobicistat/emtricitabine/tenofovir DF single-tablet regimen in virologically suppressed, HIV 1 infected patients [ClinicalTrials.gov identifier NCT01495702]. 2012. http://clinicaltrials.gov/ct2/show/NCT01495702?term=GS-US-236-0121&rank=1 Accessed 17 August 2012.
Gilead Sciences. Phase 3b open label study to evaluate switching from regimens consisting of a ritonavir-boosted protease inhibitor plus emtricitabine/tenofovir fixed-dose combination to the elvitegravir/cobicistat/emtricitabine/tenofovir df single-tablet regimen in virologically suppressed, HIV 1 infected patients. 2012. http://clinicaltrials.gov/ct2/show/NCT01475838?term=elvitegravir&rank=5 Accessed 17 August 2012.
Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol. 2008;82(2):764–74.
Goethals O, Clayton R, Van Ginderen M, Vereycken I, Wagemans E, Geluykens P, et al. Resistance mutations in human immunodeficiency virus type 1 integrase selected with elvitegravir confer reduced susceptibility to a wide range of integrase inhibitors. J Virol. 2008;82(21):10366–74.
McColl D, Fransen S, Gupta S. Resistance and cross-resistance to first generation integrase inhibitors: insights from a phase 2 study of elvitegravir (GS-9137). Bridgetown: XVI International drug resistance workshop; 2007.
Metifiot M, Vandegraaff N, Maddali K, Naumova A, Zhang X, Rhodes D, et al. Elvitegravir overcomes resistance to raltegravir induced by integrase mutation Y143. AIDS. 2011;25(9):1175–8.
Margot N, Rhee SY, Szwarcberg J, Miller MD, Team G-U–S. Low rates of integrase resistance for elvitegravir and raltegravir through week 96 in the phase 3 clinical study GS-US-183-0145. XIX International AIDS conference, Washington, DC; 2012.
Katlama C, Murphy R. Dolutegravir for the treatment of HIV. Expert Opin Investig Drugs. 2012;21(4):523–30.
Sato A, Kobayashi M, Yoshinaga T. S/GSK1349572 is a potent next generation HIV integrase inhibitor. 5th International AIDS Society, Cape Town; 2009.
Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, et al. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55(2):813–21.
Min S, Carrod A, Curtis L, Stainsby C, Brothers C, Yeo J, et al. Safety profile of dolutegravir (DTG, S/GSK1349572), in combination with other antiretrovirals in antiretroviral (ART)-naive and ART-experienced adults from two phase IIb studies. Rome: XIII International AIDS conference; 2011.
Eron JE, Durant J, Poizot-Martin I, Reynes J, Soriano V, Kumar PN, et al. Activity of a next generation integrase inhibitor (INI), S/GSK1349572, in subjects with HIV exhibiting raltegravir resistance: initial results of VIKING study (ING112961). Vienna: XVII International AIDS conference; 2010.
Clotet B, DeJesus E, Lazzarin A, Livrozet J-M, Moriat P, Vavro C, et al. HIV integrase genotypic and phenotypic changes between day 1 and day 11 in subjects with raltegravir (RAL) resistant HIV treated with S/GSK1349572: results of VIKING study (ING112961). Vienna: XVII International AIDS conference; 2010.
Eron JE, Kumar PN, Lazzarin A, Richmond G, Soriano V, Huang J, et al. DTG in subjects with HIV exhibiting RAL resistance: functional monotherapy results of VIKING study cohort II. 18th Conference on retroviruses and opportunistic infections, Boston; 2011.
van Lunzen J, Maggiolo F, Arribas JR, Rakhmanova A, Yeni P, Young B, et al. Once daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naive adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect Dis. 2012;12(2):111–8.
Stellbrink H, Reynes J, Lazzarin A, Voronin E, Pulido F, Felizarta F, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naive adults: 96-week results from SPRING-1. 19th Conference on retroviruses and opportunistic infections, Seattle; 2012.
Raffi F, Rachlis A, Stellbrink H, Hardy WD, Torti C, Orkin C, et al. Once-daily dolutegravir (DTG; S/GSK1345972) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). Washington, DC: XIX International AIDS conference; 2012.
ViiV Healthcare. A phase III randomized, double-blind trial investigating the activity of dolutegravir 50 mg BID vs placebo over 7 days in HIV-1-infected subjects with RAL/ELV resistance, followed by an open-label phase with an optimized background regimen. 2012. http://clinicaltrials.gov/ct2/show/NCT01568892?term=dolutegravir&rank=2 Accessed 17 August 2012.
ViiV Healthcare. A phase IIIb, randomized, open-label study of the safety and efficacy of GSK1349572 (Dolutegravir, DTG) 50 mg once daily compared to darunavir/ritonavir (DRV/r) 800 mg/100 mg once daily each administered with fixed-dose dual nucleoside reverse transcriptase inhibitor therapy over 96 weeks in HIV-1 infected antiretroviral naïve adult subjects. 2012. http://clinicaltrials.gov/ct2/show/NCT01449929?term=dolutegravir&rank=9 Accessed 17 August 2012.
ViiVHealthcare. Shionogi-ViiV Healthcare starts phase III trial for “572-Trii” fixed-dose combination HIV therapy. 2011. http://www.viivhealthcare.com/media-room/press-releases/2011-02-03.aspx?sc_lang=en&p=1 Accessed 2 August 2012.
Walmsley S, Antela A, Clumeck N, Diuiculescu D, Eberhard A, Gutierrez F, et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results - SINGLE (ING114467). 52nd Interscience conference on antimicrobial agents and chemotherapy, San Francisco; 2012.
Saladini F, Meini G, Bianco C, Monno L, Punzi G, Pecorari M, et al. Prevalence of HIV-1 integrase mutations related to resistance to dolutegravir in raltegravir naive and pretreated patients. Clin Microbiol Infect. Epub 2012 May 28.
Underwood MR, Johns BA, Sato A, Martin JN, Deeks SG, Fujiwara T. The Activity of the Integrase Inhibitor Dolutegravir Against HIV-1 variants isolated from raltegravir-treated adults. J Acquir Immune Defic Syndr. Epub 2012 Aug 8.
Yoshinaga T, Kanamori-Koyama M, Seki T, Ishida K, Akihisa E, Kobayashi M, et al. Strong inhibition of wild-type and integrase inhibitor (INI)-resistant HIV integrase (IN) strand transfer reaction by the novel INI S/GSK1349572. Dubrovnik: International HIV and hepatitis virus drug resistance workshop; 2010.
Seki T, Wakasa-Morimoto C, Yoshinaga T, Sato A, Fujiwara T, Underwood M, et al. S/GSK1349572 is a potent next generation HIV integrase inhibitor and demonstrates a superior resistance profile substantiated with 60 integrase mutant molecular clones,. 17th Conference on retroviruses and opportunistic infections, San Francisco; 2010.
Underwood M, St Clair M, Johns B, Sato A, Fujiwara T, Spreen W. S/GSK1265744: a next generation integrase inhibitor (INI) with activity against raltegravir-resistant clinical isolates. XVIII International AIDS conference, Vienna; 2010.
Malet I, Wirden M, Fourati S, Armenia D, Masquelier B, Fabeni L, et al. Prevalence of resistance mutations related to integrase inhibitor S/GSK1349572 in HIV-1 subtype B raltegravir-naive and -treated patients. J Antimicrob Chemother. 2011;66(7):1481–3.
Canducci F, Ceresola ER, Boeri E, Spagnuolo V, Cossarini F, Castagna A, et al. Cross-resistance profile of the novel integrase inhibitor dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J Infect Dis. 2011;204(11):1811–5.
Hightower KE, Wang R, Deanda F, Johns BA, Weaver K, Shen Y, et al. Dolutegravir (S/GSK1349572) exhibits significantly slower dissociation than raltegravir and elvitegravir from wild-type and integrase inhibitor-resistant HIV-1 integrase-DNA complexes. Antimicrob Agents Chemother. 2011;55(10):4552–9.
Min S, Sloan L, DeJesus E, Hawkins T, McCurdy L, Song I, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS. 2011;25(14):1737–45.
Koteff J, Borland J, Chen S, et al. An open label, placebo-controlled study to evaluate the effect of dolutegravir (DTG, S/GSK1349572) on iohexol and para-aminohippurate clearance in healthy subjects. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Koteff J, Borland J, Chen S. An open-label, placebo-controlled study to evaluate the effect of dolutegravir (DTG, S/GSK1349572) on iohexol and para-aminohippurate clearance in healthy subjects. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Min S, DeJesus E, McCurdy L. Pharmacokinetics (PK) and safety in healthy and HIV-infected subjects and short-term antiviral efficacy of S/GSK1265744, a next generation once daily HIV integrase inhibitor. 49th Interscience conference on antimicrobial agents and chemotherapy (ICAAC), San Francisco; 2009.
ViiV Healthcare. Dose ranging study of GSK 1265744 plus nucleoside reverse transcriptase inhibitors for induction of human immunodeficiency virus-1 (HIV-1) irologic suppression followed by virologic suppression maintenance by GSK 1265744 plus rilpivirine. 2012. http://clinicaltrials.goc/ct2/show/NCT01641809?term=viiv&rank=6 Accessed 15 August 2012.
Spreen W, Ford SL, Chen S, Gould E, Wilfret D, Subich D, et al. Pharmacokinetics, safety and tolerability of the HIV integrase inhibitor S/GSK1265744 long acting parenteral nanosuspension following single dose administration to healthy adults. Washington, DC: XIX International AIDS conference; 2012.
ViiV Healthcare. A study to investigate the safety, tolerability and pharmacokinetics of repeat dose administration of long-acting GSK1265744 and long-acting TMC278 intramuscular and subcutaneous injections in healthy adult subjects. 2012. http://clinicaltrials.gov/ct2/show/NCT01593046 Accessed 16 August 2012.
Yoshinaga T, Kobayashi M, Seki T, Kawasuji T, Taishi T, Sato A, et al. Antiviral characteristics of S/GSK1265744, an HIV integrase inhibitor (INI) dosed by oral or long-acting parenteral injection. 52nd Interscience conference on antimicrobial agents and chemotherapy, San Francisco; 2012.
Di Santo R. Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain. Curr Med Chem. 2011;18(22):3335–42.
De Luca L, Ferro S, Morreale F, Chimirri A. Inhibition of the interaction between HIV-1 integrase and its cofactor LEDGF/p75: a promising approach in anti-retroviral therapy. Mini Rev Med Chem. 2011;11(8):714–27.
Meehan AM, Saenz DT, Morrison J, Hu C, Peretz M, Poeschla EM. LEDGF dominant interference proteins demonstrate prenuclear exposure of HIV-1 integrase and synergize with LEDGF depletion to destroy viral infectivity. J Virol. 2011;85(7):3570–83.
Christ F, Pickford C, Demeulemeester J, Shaw S, Desimmie BA, Smith-Burchnell C, et al. Pre-clinical evaluation of HIV replication inhibitors that target the HIV-integrase-LEDGF/p75 interaction. Washington, DC: XIX International AIDS conference; 2012.
Christ F, Pickford C, Shaw S, Demeulemeester J, Desimmie BA, Smith-Burchnell C, et al. Preclinical evaluation of HIV replication inhibitors that target the HIV integrase-LEDGF/p75 interaction. 19th Conference on retroviruses and opportunistic infections, Seattle; 2012.
Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, et al. Small molecule inhibitors of the LEDGF/p75 binding site of integrase (LEDGINs) block HIV replication and modulate integrase multimerization. Antimicrob Agents Chemother. Epub 2012 Jun 4.
Fenwick C, Bethell R, Cordingley M, Edwards P, Quinson A-M, Robinson P, et al. BI 224436, a non-catalytic site integrase inhibitor, is a potent inhibitor of the replication of treatment-naive and raltegravir-resistant clinical isolates of HIV-1. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Brown AN, McSharry J, Kulawy R. Pharmacodynamics of BI 224436 for HIV-1 in an in vitro hollow fiber infection model system. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Aslanyan S, Ballow CH, Sabo JP, Habeck J, Roos D, Macgregor TR, et al. Safety and pharmacokinetics (PK) of single rising oral doses of a novel HIV integrase inhibitor in healthy volunteers. 51st Interscience conference on antimicrobial agents and chemotherapy, Chicago; 2011.
Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4(3):236–48.
Acknowledgments
Martin Markowitz receives research grants from Gilead Sciences, Tobira and GlaxoSmithKline. He is a consultant to Gilead Sciences, Merck and Janssen; and he is a speaker for Gilead Sciences, Bristol-Myers Squibb and Janssen. Sharon Karmon reports no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karmon, S.L., Markowitz, M. Next-Generation Integrase Inhibitors. Drugs 73, 213–228 (2013). https://doi.org/10.1007/s40265-013-0015-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-013-0015-5