
Abstract
The newest class of antiretrovirals for all persons living with HIV are the integrase strand transfer inhibitors (INSTIs). Since 2007, five INSTIs have been introduced: raltegravir, elvitegravir, dolutegravir, bictegravir, and cabotegravir. The INSTIs have favorable pharmacokinetic and pharmacodynamic properties, which contribute to both their effectiveness and their ease of use. With the exception of cabotegravir, each INSTI is US Food and Drug Administration approved for treatment-naïve individuals initiating antiretroviral therapy. All of the INSTIs, except raltegravir, are approved for antiretroviral treatment simplification for virologically suppressed patients without INSTI resistance. Data also support the use of dolutegravir and raltegravir in individuals with antiretroviral resistance as part of an optimized antiretroviral regimen. INSTIs are generally well tolerated by people living with HIV compared with older classes of antiretrovirals, but emerging data suggest that some INSTIs contribute to weight gain. Due to their efficacy, safety, and ease of use, HIV treatment guidelines recommend oral INSTIs as preferred components of antiretroviral therapy for individuals initiating therapy. The newest INSTI, cabotegravir, represents an alternative to oral administration of life-long antiretroviral therapy with the availability of a long-acting injectable formulation. This review summarizes the current use of INSTIs in adults living with HIV, highlighting the similarities and differences within the class related to pharmacodynamics, pharmacokinetics, safety, dosing, and administration that contribute to their role in modern antiretroviral therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bictegravir and dolutegravir are both co-formulated as a single-tablet regimen, and the choice of agent may be driven based on the preferred nucleoside reverse transcriptase inhibitor. |
Elvitegravir/cobicistat is available as a single-tablet regimen, though it has a higher propensity for drug-drug interactions and must be administered with food, increasing complexity compared with other integrase strand transfer inhibitors (INSTIs). |
Raltegravir has the most safety data associated with its use; however, it is the only INSTI not available as a single-tablet antiretroviral regimen, increasing the pill burden associated with a raltegravir-containing regimen. |
Cabotegravir is part of the first long-acting, injectable antiretroviral regimen, recently approved in Canada for treatment of individuals currently virologically suppressed on another antiretroviral regimen. |
1 Introduction
For the nearly 40 million persons living with HIV (PLWH), effective antiretroviral therapy (ART) reduces morbidity and mortality, leading to a life expectancy expected to be similar to age-matched peers [1, 2]. Randomized, clinical trials have demonstrated health benefits associated with early initiation of ART beyond the reduction in HIV-associated morbidity and mortality [3, 4]. Furthermore, effective ART offers a significant public health benefit by preventing transmission of HIV [5,6,7]. Based on these combined benefits, together with improvements in available antiretroviral options, ART is now recommended for all PLWH [8,9,10].
The integrase strand transfer inhibitor (INSTI) class of antiretrovirals contributes to the enhanced safety and efficacy of modern ART regimens. Based on data demonstrating efficacy, safety, and ease of administration, INSTIs are now part of preferred or recommended ART regimens in HIV treatment guidelines throughout the world [8,9,10]. The efficacy of INSTIs has been well established in comparison with both protease inhibitor (PI) and non-nucleoside reverse transcriptase inhibitor (NNRTI)-based ART regimens (see Sect. 5) [8]. The effectiveness of INSTIs is related to their antiviral activity and improved tolerability, which results in fewer treatment discontinuations [11].
Raltegravir was the first drug in the INSTI class and was approved for twice-daily administration by the United States (US) Food and Drug Administration (FDA) in 2007 [12]. A once-daily formulation was approved in 2017, but raltegravir remains the only INSTI not available as a fixed-dose combination (FDC). Elvitegravir was the second INSTI approved by the FDA, in 2012, and was the first once-daily, FDC single-tablet regimen containing an INSTI [13]. The two most commonly used INSTIs are dolutegravir, approved in 2013, and a bictegravir-containing FDC approved in 2018 [16, 17]. Cabotegravir is an INSTI formulated as a long-acting product given intramuscularly in combination with rilpivirine. The combination was recently approved in Canada, and is undergoing regulatory review in the USA and Europe [14].
This review will summarize key information regarding the dosing and administration, pharmacodynamics, pharmacokinetics, efficacy, and safety of INSTIs with an emphasis on treatment of adults with HIV. A detailed review on the use of INSTIs in pediatric patients is addressed by Dehority and colleagues [15]. Similarities and differences between the individual agents will be highlighted to inform the current place in therapy for each INSTI.
2 Dosage and Administration
Table 1 describes the available formulations, dosing, and indications for each INSTI. The companion antiretrovirals included in the INSTI single-tablet regimens vary by agent, as described in Table 1.
3 Pharmacodynamic Properties
3.1 Antiviral Activity
INSTIs inhibit HIV by blocking the strand transfer step of viral DNA integration into the host genome. INSTIs are potent and selective antiretrovirals with sub- to low nanomolar in vitro activity. Against clinical isolates of HIV-1, in vitro 50% inhibitory concentrations (IC50) reported in approved product labels are approximately 0.1 ng/mL for cabotegravir and 0.2 ng/mL for both bictegravir and dolutegravir; and range from 0.04–0.6 ng/mL for elvitegravir and 2.2–5.3 ng/mL for raltegravir [12, 14, 16,17,18]. However, all INSTIs are highly protein bound, which substantially influences clinical IC50 values. For example, in an in vitro assay in peripheral blood mononuclear cells, the IC50 of dolutegravir was 0.21 ng/mL, but was increased 75-fold in the presence of human serum albumin to a protein-adjusted IC50 of 16 ng/mL [19]. The protein-adjusted 90% or 95% inhibitory concentration (IC90 or IC95) values (ng/mL) for the current INSTIs are: raltegravir, 15; elvitegravir, 45; dolutegravir, 64; bictegravir, 162; cabotegravir, 166 [20].
3.2 Resistance
To date, resistance to all antiretrovirals has been documented [21]. With regard to INSTIs, the first-generation agents raltegravir and elvitegravir share similar clinical resistance profiles, including cross-resistance. The second-generation INSTIs bictegravir and dolutegravir are characterized by similar resistance profiles and retention of potency against resistant mutants selected by first-generation INSTIs. There is evidence the resistance barriers of bictegravir and dolutegravir are higher than raltegravir and elvitegravir [22, 23]. Resistance has emerged during therapy with long-acting, intramuscular cabotegravir plus rilpivirine [24, 25]. In the combined results of two phase 3 trials, six of 586 participants (1.02%) had confirmed virologic failure with INSTI-resistance mutations. Characteristics common among all six were cabotegravir and rilpivirine plasma concentrations in the lowest quartile, despite receipt of all intramuscular injections, and HIV-1 subtypes A/A1 or AG; resistance emerged before or at week 28 of therapy in five of the six subjects. Whether clinical resistance emerges to an INSTI (or any antiretroviral) is dependent upon a variety of factors including the drug’s inherent genetic barrier to resistance, the drug’s structure, inhibitory quotient, therapeutic index, and pharmacokinetic forgiveness/adherence [26]. The consequences of resistance are virologic failure and reduced options for future ART regimens. Current treatment guidelines therefore contain specific recommendations for resistance testing in both naïve- and treatment-experienced PLWH, and recommendations for the use of ART regimens to maximally and durably suppress plasma HIV-RNA to minimize the emergence of resistance [8].
3.3 Clinical Pharmacodynamic Characteristics
Explicit pharmacodynamic relationships have been described between the decline in plasma HIV-RNA and trough concentrations (Ctrough) of cabotegravir, dolutegravir, and elvitegravir [27,28,29]. For cabotegravir, pharmacodynamic analyses found the HIV-RNA change from baseline was associated with cabotegravir Ctrough as a maximum effect (Emax) relationship: Emax was 2.56 log10 and 50% effective concentration (EC50) was 82 ng/mL [27]. The reduction in plasma HIV-RNA with dolutegravir monotherapy of 2, 10, and 50 mg once daily for 10 days was associated with dolutegravir Ctrough in an Emax relationship with an estimated EC50 of 36 ng/mL [28]. The Ctrough of elvitegravir in antiretroviral-naïve and -experienced persons not currently on therapy were strongly associated with the log10 change in plasma HIV-RNA in an Emax relationship, with an EC50 of 14 ng/mL and 90% effective concentration (EC90) of 126 ng/mL [29]. Less quantitative, exposure–response relationships have clearly been demonstrated for bictegravir and raltegravir [30, 31]. A 10-day monotherapy study of bictegravir found the mean change in HIV-RNA from baseline to day 11 was − 1.45, − 2.06, − 2.08, and − 2.43 log10 copies/mL for 5, 25, 50, and 100 mg doses, respectively, clearly demonstrating that higher doses, and therefore higher concentrations, were associated with a greater anti-HIV response [30]. A trial of raltegravir 800 mg once daily compared with 400 mg twice daily found Ctrough raltegravir with once-daily dosing correlated with virologic response [31]. Participants who had a raltegravir Ctrough in the lowest quartile (median Ctrough 12.5 ng/mL) had a clear fall-off in virologic response, with < 80% achieving HIV-RNA < 50 copies/mL compared to ≥ 90% achieving HIV-RNA < 50 copies/mL if the Ctrough was > 44 ng/mL. The recently completed phase 3 trials of intramuscular cabotegravir also provide qualitative pharmacodynamic information. The geometric mean cabotegravir concentrations at weeks 8 and 48 were approximately 1500 ng/mL and 3000 ng/mL, respectively, which are ninefold and 18-fold greater than the protein binding-adjusted IC90 of 166 ng/mL [24, 25]. At weeks 4–8 after the start of intramuscular cabotegravir therapy, which is where the lowest concentrations are found, the fifth percentile cabotegravir concentration is approximately 450 ng/mL, which is 2.7-fold greater than the protein binding-adjusted IC90. As noted above, INSTI-resistance mutations emerged before or at week 28 of therapy in the phase 3 trials of injectable cabotegravir. Collectively, for the INSTI class with regard to the anti-HIV response, data support relationships with dose/plasma concentrations.
The inhibitory quotient (IQ) is an intuitive concept for predicting clinical drug activity. The IQ is the ratio of drug concentration in any biologic fluid (e.g., plasma, cerebral spinal fluid) divided by an in vitro inhibitory concentration (i.e., how much drug you have to how much drug you need). The IQ has utility in antiretroviral drug development, as discussed by the FDA [32], because a high IQ indicates sufficient drug concentrations can be achieved that may minimize the emergence of viral resistance and inform the selection of doses for phase 3 and 4 studies, as well as for select patient populations. For the available INSTIs, the hierarchy of IQ values, where IQ is the ratio of typical trough plasma concentration achieved with approved oral dose divided by the protein-binding adjusted IC90 or 95, is cabotegravir > dolutegravir > bictegravir > elvitegravir > raltegravir [20].
4 Pharmacokinetic Properties
The pharmacokinetics of INSTIs have recently been reviewed by Podany and colleagues, and readers can find detailed pharmacokinetic parameters there [20]. All oral INSTIs may be given with or without food (Table 1), except elvitegravir, where one study found that a low- versus high-fat meal increased the area under the concentration time curve (AUC) by 34% and 87%, respectively, compared with fasting [18]. The dolutegravir/rilpivirine FDC must also be administered with food due to the rilpivirine component. As a class, oral absorption can be impaired if taken with divalent or trivalent cations, as discussed further in Sect. 4.2.
INSTIs are not extensively renally cleared and primarily undergo hepatic metabolism [20]. Raltegravir is primarily conjugated via uridine 5′diphosphate glucuronosyltransferase (UGT) 1A1 [12]. Elvitegravir is primarily metabolized via the cytochrome P450 (CYP) 3A4 enzyme and is the only INSTI that must be co-administered with a pharmacokinetic-enhancer, cobicistat [8]. Dolutegravir is primarily conjugated by UGT1A1, with secondary metabolism by CYP3A4 (10–15%) [17], while bictegravir is metabolized by both CYP3A4 and UGT1A [16]. Finally, cabotegravir is metabolized by both UGT1A1 (primarily) and UGT1A9, with minimal CYP involvement [33]. The metabolic pathway of each INSTI informs the likelihood of metabolism-related drug-drug interactions, as discussed in Sect. 4.2.
The oral INSTIs have half-lives ranging from 9 to 38.8 h [12, 16,17,18, 34]. Oral cabotegravir has the longest half-life of the INSTIs, 38.8 h, and intramuscularly administered cabotegravir exhibits an exceptionally long and variable half-life, estimated as 2.3–14.7 weeks [35, 36]. Considering the prolonged time to maximum concentration (Cmax), and the observed elimination half-life, cabotegravir is expected to accumulate over the first several months of administration, and is detectable for several months after discontinuation [37]. Landovitz and colleagues recently described that cabotegravir remained detectable in 23% of males and 63% of females 1 year after discontinuing cabotegravir for pre-exposure prophylaxis [37].
4.1 Special Populations
Overall, population pharmacokinetic studies have shown that oral INSTIs do not have any clinically relevant pharmacokinetic differences based upon race or sex in adults [12, 16, 17, 38, 39].
Ontogenic enzymatic changes that occur in pediatric patients inform INSTI dosing in infants and children. For example, activity of the enzyme responsible for glucuronidation (i.e., UGT) is low at birth and increases dramatically during the first 4–6 weeks of life in full-term neonates [40]. This can lead to increased clearance of INSTIs that primarily undergo UGT enzyme metabolism (i.e., dolutegravir, raltegravir, cabotegravir). Other potential differences in enzymatic activity of specific age groups in pediatric patients should be considered when selecting and dosing agents [41]. Recommendations on specific INSTIs in pediatric patients vary by agent (Table 1), with raltegravir and dolutegravir allowing dosing with formulations suitable for the smallest children. Some INSTIs may be used to treat pediatric patients weighing 20 kg or more using standard, adult doses, while many FDCs and newer agents do not yet have information for use in patients less than 18 years of age.
For many currently available INSTIs, the manufacturers recommend caution in geriatric use (≥ 65 years) given the greater frequency of decreased hepatic, renal, or cardiac function in this population [12, 13, 17]. However, a population pharmacokinetic analysis of participants in phase 3 trials of bictegravir showed age did not have a clinically relevant effect on bictegravir exposures up to 74 years of age [16]. Similarly, clinical trials of elvitegravir included participants over 65 years and found no differences in safety or efficacy compared with participants aged 18–65 years [18]. A pharmacokinetic analysis of dolutegravir in PLWH who were ≥ 60 years of age did find that the Cmax for dolutegravir was significantly higher in this age group compared with a historical control population aged ≤ 50 years (geometric mean 4246 ng/mL vs. 3402 ng/mL, p = 0.005) [42]. Despite the increased Cmax, there was no excess risk of adverse events associated with dolutegravir. No recommendations are available for cabotegravir in geriatric populations. Despite warnings in product labeling, available data suggest INSTIs may be used in geriatric patients. Given the aging PLWH, more pharmacokinetic and pharmacodynamic studies in geriatric persons at conventional FDA-approved doses are needed.
4.1.1 Pregnancy and Lactation
The US Department of Health and Human Services (DHHS) Perinatal HIV Treatment Guidelines recommend ART therapy for all pregnant PLWH [43]. However, pregnancy can cause physiological changes that have the potential to impact ART pharmacokinetics, which may require dose considerations. For example, induction of UGT1A1 and CPY3A4 during pregnancy may influence INSTI metabolism [44, 45]. Detailed reviews of INSTI pharmacokinetics during pregnancy were recently undertaken by van der Galien et al. and Podany et al. [20, 46]; generally, INSTI exposure is lower during pregnancy, resulting in variable clinical recommendations (Table 1).
Though the exposure of dolutegravir during pregnancy was decreased between 10% and 50% compared to post-partum concentrations in several studies [47, 48], it still may be given without dose adjustment during pregnancy as long as it is taken with food to increase absorption [43]. This is based on the high rate of virologic suppression observed, and because the median AUC during pregnancy was similar to nonpregnant adults in clinical trials. Raltegravir concentrations were 30–50% lower during pregnancy, though highly variable, and the effectiveness during pregnancy was not affected by the reduced exposure [49, 50]. Therefore, twice-daily raltegravir is recommended; the once-daily raltegravir high-dose (HD) formulation should not be used in pregnant women due to the lower Ctrough observed with this formulation compared with twice-daily administration [43].
Three INSTIs have insufficient evidence to support their use or are not recommended during pregnancy. Elvitegravir is not recommended due to significantly lower plasma concentrations during pregnancy with associated cases of virologic failure [43, 44, 51]. For example, Momper and colleagues performed a study in 30 pregnant women taking elvitegravir/cobicistat once daily and found that compared to postpartum data, elvitegravir AUCs were 24% lower in the second trimester (n = 14, geometric mean ratio [GMR] = 0.76, 90% confidence interval (CI) 0.57, 1.0) and 44% lower in the third trimester (n = 24, GMR = 0.56, 90% CI 0.42, 0.73) [51]. The reduced elvitegravir exposure during pregnancy is related to a decrease in cobicistat exposure during pregnancy; cobicistat was 44% lower during the second trimester (GMR 0.56, 90% CI 0.37, 0.85) and 59% lower in the third trimester (GMR 9.41, 90% CI 0.30, 0.85) of pregnancy, leading to less pharmacokinetic enhancement of elvitegravir [51]. No pharmacokinetic studies are available for bictegravir during pregnancy, but studies in pregnant women are ongoing. Cabotegravir pharmacokinetic data during pregnancy are limited to three participants who became pregnant during clinical trials [52]. All three individuals had adequate cabotegravir concentrations prior to pregnancy, throughout pregnancy, and postpartum.
It is also important to consider fetal exposure throughout the antepartum period to ensure prevention of HIV transmission from mother to fetus. Pharmacokinetic studies of dolutegravir [47, 48, 53], elvitegravir [51, 54], and raltegravir [50] have shown placental transfer with mean/median cord-to-maternal plasma ratios ranging from 0.09 to 1.5. A comprehensive review of antiretroviral placental transfer is available [55].
The DHHS Perinatal HIV Treatment Guidelines recommend that women with HIV refrain from breastfeeding due to the risk of HIV transmission [43], but this recommendation is not consistent with international guidelines [56]. Data from a recent study found dolutegravir passes to milk in pregnant women (milk-to-plasma ratio of 0.03) [48]. Dolutegravir was detectable in the plasma of the breastfed infants with a mean age of 10 days (range 7–18 days) with a mean Cmax of 66.7 (range 21–654) ng/mL, representing an infant to maternal plasma ratio of 0.03 and a mean minimum concentration of 60.9 (range 16.3–479) ng/mL (infant:maternal ratio 0.08).
Based on pharmacokinetic results, dolutegravir and raltegravir are appropriate for use during pregnancy for treatment of maternal HIV disease and for prevention of HIV transmission to the infant [43]. There were no adverse effects observed due to infant exposure to dolutegravir during breastfeeding in one clinical trial [48].
4.1.2 Renal/Hepatic Impairment
As a class, no clinically relevant differences in INSTI pharmacokinetics have been observed between patients with renal impairment and those with normal renal function. As such, no dose adjustments are required for the INSTI component of regimens (Table 1) [57, 58]. However, plasma concentrations of dolutegravir have been found to be decreased in persons with severe renal impairment [17]; therefore, caution is advised in certain populations (e.g., those with INSTI-associated resistance). INSTIs are highly bound to plasma proteins (albumin and alpha-1-acid glycoprotein, 83–99%) and are not significantly removed by dialysis (hemodialysis or peritoneal dialysis); therefore, no dose adjustments are required in patients who are receiving hemodialysis or peritoneal dialysis [12, 16,17,18, 33]. Because most nucleoside reverse transcriptase inhibitors (NRTIs) are renally excreted, the use of INSTIs containing FDCs in patients with renal insufficiency may be limited by the concurrent NRTIs (Table 1).
Liver disease has the potential to alter the pharmacokinetics of drugs due to changes in hepatic blood flow, altered plasma protein levels, and changes to CYP enzymes and/or glucuronidation [59,60,61]. However, no significant differences have been seen in the exposures of INSTIs in persons with mild to moderate hepatic impairment (Child–Pugh Class A or B) and no dose adjustments are recommended (Table 1) [57, 62,63,64,65,66]. Given the lack of data available in persons with severe hepatic impairment (Child–Pugh Class C), INSTIs are not recommended [12, 16,17,18, 33].
4.2 Drug Interactions
Most INSTIs are not potent inducers or inhibitors of drug metabolizing enzymes, and so infrequently cause metabolism-related drug-drug interactions [8]. The exception is elvitegravir, which is an inducer of CYP2C9, and must be co-administered with cobicistat, a potent CYP3A4 and a weak CYP2D6 inhibitor [13, 18]. Therefore, elvitegravir/cobicistat is commonly associated with metabolism-related drug-drug interactions that effect the exposure of co-administered CYP substrates. Any drug that is a strong inducer or inhibitor of CYP3A and/or UGT1A1 may substantially influence the plasma concentrations of INSTIs [8]. Of the INSTIs, elvitegravir is the most susceptible to adverse drug-drug interactions because it is primarily metabolized by CYP3A4 [13, 18]. Bictegravir and dolutegravir are metabolized by both CYP3A4 and UGT, and, comparatively, bictegravir is more susceptible to drug-drug interactions via CYP3A4 compared to dolutegravir [16, 17]. Raltegravir and cabotegravir have a lower risk of metabolism-related drug-drug interactions as they do not undergo CYP3A4 metabolism [12, 14].
Co-administration of rifampin with each INSTI illustrates the potential for metabolism-related drug-drug interactions. A phase 1 drug interaction study evaluated the exposure of dolutegravir 50 mg once or twice daily when co-administered with rifampin [67]. Twice-daily dolutegravir exposures were 54–72% lower in participants receiving rifampin compared with twice-daily dolutegravir without rifampin. However, participants receiving dolutegravir twice daily with rifampin had higher dolutegravir exposure than those receiving standard-dose dolutegravir once daily without rifampin [GMR (90% CI): AUC0–24h: 1.33 (1.15, 1.5); Ctrough: 1.22 (1.01, 1.48)]. Clinical data in patients being treated for both HIV and tuberculosis support the efficacy of dolutegravir given twice daily when combined with rifampin or other enzyme-inducing agents [68]. Some guidelines recommend an increase in the adult dose of raltegravir from 400 mg to 800 mg twice daily based on reduced raltegravir exposure of 40–61% when combined with rifampin; once-daily raltegravir HD is not recommended [8]. Alternatively, some guidelines do not recommend the increased dose based on one study that demonstrated similar virologic response among participants receiving either raltegravir 400 mg or 800 mg twice daily in combination with rifampin (n = 51 per group) [virologic suppression: 76% (95% CI 65, 88) vs. 78% (95% CI 67, 90), respectively] [69]. In contrast to dolutegravir and raltegravir, co-administration of rifampin and bictegravir, cabotegravir, or elvitegravir is contraindicated [13, 14, 16, 18]. When rifampin was combined with bictegravir 50 mg daily, the bictegravir exposure decreased by 46–61% and could not be overcome when bictegravir was increased to twice daily [8]. Oral cabotegravir has been investigated in one healthy-volunteer study that found that co-administration with rifampin reduced cabotegravir AUC by nearly 60% [70]. Finally, a physiologically based pharmacokinetic model predicted that oral rifampin will decrease the exposure of intramuscular cabotegravir by 41–46% [71].
A common drug-drug interaction encountered with INSTI therapy is co-administration of polyvalent cations, which may decrease INSTI absorption due to chelation. The specific management of these interactions is dependent upon the polyvalent cation and its dose, the INSTI, and how the combination was evaluated in pharmacokinetic studies [8]. For example, bictegravir may be co-administered with antacids containing calcium when taken together with food, but not on an empty stomach because food increases the exposure of bictegravir [16]. In contrast, any antacids containing aluminum or magnesium should be given at least 2 h after or 6 h before bictegravir administration. Once-daily raltegravir should not be co-administered with calcium-containing antacids, but the 400 mg twice-daily dose may be co-administered with calcium antacids irrespective of timing because the twice-daily formulation achieves higher Ctrough compared with the once-daily formulation [12]. Raltegravir should not be combined with aluminum- or magnesium-containing antacids. Interactions with oral cabotegravir, dolutegravir, and elvitegravir may be managed by dose separation of the calcium-, aluminum-, or magnesium-containing antacid and the INSTI [13, 14, 17, 18]. Beyond antacid interactions, consideration of INSTI dosing is required with supplements or laxatives containing polyvalent cations. Specific recommendations exist for calcium and iron supplements, and are often extrapolated to other polyvalent cation-containing products [8].
Drug transporters are a cause of some drug-drug interactions involving some INSTIs. In vitro studies have shown that bictegravir and dolutegravir inhibit the organic cation transporter 2 (OCT2) and multidrug and toxin extrusion transporter 1 (MATE1) [16, 17, 72]. Co-administration of bictegravir or dolutegravir with other drugs that are substrates of OCT2 and MATE1, such as metformin or dofetilide, may increase concentrations of the co-administered medications. Due to the narrow therapeutic index of dofetilide, co-administration with either dolutegravir or bictegravir is contraindicated [16, 17]. Dolutegravir and bictegravir increase metformin exposure 66–79% and 39%, respectively [17]. Case reports of dolutegravir plus metformin report the occurrence of lactic acidosis when metformin was combined with dolutegravir, as well as case reports of loss of glycemic control when the dose of metformin was empirically reduced in combination with dolutegravir [73]. Therefore, the risk/benefit of combining metformin with either bictegravir or dolutegravir should be evaluated on a case-by-case basis.
INSTIs are involved in bi-directional drug-drug interactions to a varying degree based on their unique pharmacology. An up-to-date drug-drug interaction resource is recommended when evaluating INSTI-related interactions, including the DHHS Adult and Adolescent Treatment Guideline drug-drug interaction tables [8] or the University of Liverpool’s database (http://hiv-druginteractions.org).
5 Clinical Efficacy
The INSTIs are a key component of modern ART regimens for treatment-naïve and treatment-experienced PLWH. One unique attribute of the INSTIs is rapid virologic suppression compared with other antiretroviral classes. All of the oral INSTIs demonstrate a rapid virologic decay and potent suppression as early as 4 weeks after initiation in treatment-naïve individuals [8, 43, 74,75,76]. For example, in a study comparing dolutegravir- to bictegravir-containing regimens, 76–80% of participants demonstrated virologic suppression within 4 weeks of treatment initiation [74]. Table 2 summarizes the difference (95% CI) in virologic suppression for each clinical trial discussed in this section.
5.1 Raltegravir
Raltegravir efficacy was compared against an NNRTI regimen containing efavirenz in the STARTMRK study [77]. With raltegravir twice daily plus two NRTIs, virologic suppression rates at week 48 were 86.1% compared with 81.9% in the efavirenz arm. Raltegravir efficacy remained largely durable, with 71% of raltegravir participants and 61.3% of efavirenz participants maintaining virologic suppression through week 240 [78]. The long-term success of raltegravir compared with efavirenz was largely driven by efavirenz treatment discontinuations.
Two switch studies, SWITCHMRK-1 and -2, investigated raltegravir efficacy in treatment-experienced, virologically suppressed adults receiving a lopinavir/ritonavir-based ART regimen [79]. Participants were randomized to continue the PI-based regimen or to switch to a raltegravir twice-daily regimen. The study was terminated at week 24 due to lower than expected virologic efficacy of raltegravir: 84% of raltegravir recipients and 91% of lopinavir/ritonavir recipients achieved virologic suppression. These results failed to establish non-inferiority of raltegravir as a switch strategy [79].
The BENCHMRK trials explored efficacy of raltegravir in treatment-experienced participants with triple drug-class resistance [80]. Both studies were randomized studies, comparing raltegravir twice daily versus placebo in addition to optimized background therapy. Week 48 results demonstrated the efficacy of twice-daily raltegravir as part of a salvage regimen (virologic suppression: raltegravir, 62.1%; placebo, 32.9%). Weeks 96 and 156 results largely mimic week 48 outcomes, with 51% of raltegravir recipients and 22% of placebo recipients sustaining virologic suppression through week 156 [81, 82].
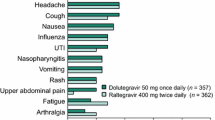
Given the necessity of twice-daily dosing of the original raltegravir formulation, the QDMRK study investigated the possibility of once-daily dosing in treatment-naïve individuals [83]. Comparing raltegravir once daily (two 400 mg tablets taken together every 24 h) versus twice daily (one 400 mg tablet every 12 h), both in combination with emtricitabine/tenofovir DF, the once-daily regimen had lower virologic suppression at week 48. Subsequently, the raltegravir HD 600 mg tablet was developed and ONCEMRK again investigated the possibility of once-daily raltegravir dosing with the new formulation [84]. The ONCEMRK study found the once-daily raltegravir 1200 mg was non-inferior to standard twice-daily raltegravir in virologic suppression at week 48 [84]: 89% of those receiving once-daily raltegravir versus 88% of the twice-daily group. These results continued through week 96, where 81.5% of the once-daily and 80.1% of the twice-daily participants with HIV-RNA < 40 copies/mL [85].
In summary, raltegravir was the first-in-class INSTI with demonstrated efficacy, given as twice-daily dosing, for treatment-naïve and -experienced individuals. The raltegravir HD formulation offers an alternative, once-daily dosing regimen for treatment-naïve individuals. Raltegravir is the only INSTI not approved for antiretroviral switch in virologically suppressed individuals.
5.2 Elvitegravir
Elvitegravir efficacy has been demonstrated in a number of studies, beginning with the FDC containing tenofovir DF in GS102 and GS103 in treatment-naïve individuals. When compared against efavirenz/tenofovir DF/emtricitabine in treatment-naïve individuals, 87.6% of participants receiving elvitegravir had virologic suppression compared with 84.1% for efavirenz at week 48 [86]. Similar rates of virologic suppression were observed through week 144 (elvitegravir, 80.2% vs. efavirenz, 75.3%) [87, 88]. Similar non-inferior performance was observed when the elvitegravir FDC was compared against boosted atazanavir-based regimens in treatment-naïve individuals [89,90,91]. At week 48, 89.5% of elvitegravir recipients and 86.8% of PI recipients were virologically suppressed [89]; and 77.6% and 74.6%, respectively, remained suppressed through week 144 [91].
The elvitegravir FDC containing tenofovir DF was evaluated in three switch studies for treatment-experienced, virologically suppressed individuals. In participants randomized to continue on NNRTI-based ART or switch to the elvitegravir FDC, no difference was found in virologic suppression at 48 or 96 weeks [92, 93]. Similarly, in individuals randomized to continue a PI-containing ART regimen or switch to the elvitegravir FDC no difference was found in virologic suppression at week 48; at week 96, however, the elvitegravir FDC was superior (87% vs. 70%) [94, 95], driven by virologic failures and discontinuations for non-virologic reasons [94]. Finally, in individuals on twice-daily raltegravir-based ART, 100% of 48 study participants remained virologically suppressed at week 48 after switching to the elvitegravir FDC [96].
Two studies, GS104 and GS111, compared the efficacy of the newer elvitegravir FDC containing tenofovir AF with the elvitegravir FDC containing tenofovir DF in treatment-naïve individuals [97,98,99]. In GS104, 93% versus 92% of participants in each of the arms met the study-defined endpoint of virologic suppression at week 48, while GS111 reported 92% (tenofovir AF arm) versus 89% (tenofovir DF arm) of participants meeting virologic suppression at week 48 [97]. No differences were found between regimens with respect to baseline CD4 + , age, or race. Combined long-term efficacy data from both studies found similar rates of virologic suppression between groups at weeks 96 and 144 [98, 99].
The tenofovir AF-containing elvitegravir FDC was also studied in treatment-experienced patients [100]. In GS109, virologically suppressed adults receiving a tenofovir DF-containing regimen (combined with efavirenz, elvitegravir/cobicistat, or boosted-atazanavir) for ≥ 96 weeks were switched to the elvitegravir FDC containing tenofovir AF, or continued their tenofovir DF-containing ART. At week 48, switching to the tenofovir AF regimen was non-inferior to remaining on the tenofovir DF-containing regimens with virologic suppression rates of 97% versus 93%, respectively; at week 96, the tenofovir AF arm was statistically favored (93% vs. 89%) [100, 101].
In summary, both elvitegravir FDCs have demonstrated efficacy in clinical trials of antiretroviral-naïve individuals or treatment-experienced, virologically suppressed individuals who have not previously received INSTIs. The available data suggest the newer FDC formulation of elvitegravir containing tenofovir AF results in similar treatment efficacy compared to the original FDC containing tenofovir DF. Therefore, if elvitegravir is used, clinicians may choose between the formulations based upon clinical considerations related to pill size, cost, or adverse effects.
5.3 Dolutegravir
Dolutegravir efficacy has been demonstrated in studies of treatment-naïve individuals as well as in ART-experienced individuals. In the SINGLE trial, a once-daily regimen of dolutegravir/abacavir/lamivudine was compared against a once-daily regimen of efavirenz/tenofovir DF/emtricitabine in treatment-naïve individuals. At week 48, superiority of the dolutegravir-based regimen was realized with 88% of participants in the dolutegravir arm and 81% in the efavirenz arm achieving virologic suppression [102]. The superiority of dolutegravir continued to week 144, with 71% of dolutegravir versus 63% (p < 0.01) of efavirenz recipients maintaining virologic suppression [103]. Interestingly, the dolutegravir arm had lower study drug discontinuation rates at weeks 48 and 144 (2% vs. 10% at week 48 and 3% vs. 11% at week 144), likely due to central nervous system adverse events associated with efavirenz [102, 103]. Dolutegravir was also compared with ritonavir-boosted darunavir in the FLAMINGO trial. In this study of ART naïve-individuals, dolutegravir-based ART was superior to darunavir-containing regimens at both week 48 and week 96 [104, 105].
Dolutegravir was also compared with a first-generation INSTI, raltegravir, for treatment-naïve individuals [106]. SPRING-2 randomized participants to either dolutegravir once daily or raltegravir twice daily, both with two NRTIs. At week 48, 88% of dolutegravir and 85% of raltegravir recipients achieved virologic suppression, meeting the prespecified non-inferiority margin of 10%. Non-inferiority was confirmed at the week 96 analysis with 81% of the dolutegravir group and 76% of the raltegravir group maintaining virologic suppression [107]. In the SAILING study, dolutegravir was again compared against a raltegravir-based regimen, only this time in ART-experienced participants who were INSTI naïve but with at least two-drug class resistance [108]. Participants were randomly assigned to either dolutegravir once daily or raltegravir twice daily in addition to optimized background therapy. At week 48, 71% of dolutegravir and 64% of raltegravir participants were virologically suppressed, demonstrating superiority of dolutegravir in this group of individuals with drug resistance.
The VIKING phase 2b study was the first study to evaluate dolutegravir in participants with prior INSTI resistance, dosed either once (cohort 1) or twice daily (cohort 2) [109]. Participants failing a raltegravir-containing regimen substituted dolutegravir for raltegravir for 10 days, then received an optimized background regimen containing dolutegravir through week 24. More participants receiving dolutegravir twice daily achieved the primary endpoint of at least a 0.7 log10 decline in the HIV-RNA, or HIV-RNA < 400 copies/mL, on day 11 (23/24; 96%) compared with dolutegravir once daily (21 of 27; 78%). The advantage of twice-daily dosing persisted through 24 weeks (virologic suppression: twice daily, 75% vs. once daily, 41%). The VIKING-3 study investigated dolutegravir twice daily in participants failing either a raltegravir- or an elvitegravir-based regimen with INSTI resistance [110]. Dolutegravir twice daily replaced raltegravir or elvitegravir in the failing regimen during the first 7 days of the study, followed by an optimized background regimen according to resistance data. The primary study endpoints were the change in HIV-RNA from baseline to day 8 and the proportion of participants with virologic suppression at week 24. In the intention-to-treat population of 183 participants, the mean change in plasma HIV-RNA from baseline to day 8 was -1.43 log10 and 69% of the participants were suppressed at week 24.
There are currently two FDA-approved FDC products containing dolutegravir plus a second antiretroviral, representing two-drug combination ART. Efficacy of dolutegravir/rilpivirine was demonstrated in two switch studies in participants receiving three-drug ART [111]. In these trials, SWORD-1 and SWORD-2, participants were on a combination of two NRTIs plus a PI, NNRTI, or INSTI for at least 6 months with virologic suppression. Participants were randomized to continue their current three-drug therapy or switch to the dolutegravir/rilpivirine FDC. In a pooled-data analysis, 95% of participants in each treatment arm reached the primary endpoint of virologic suppression at week 48. There were no significant treatment differences by baseline CD4 + count, age, gender, race, or baseline ART regimen. At week 52, 477 of the 511 participants originally randomized to the three-drug arm were eligible to switch to dolutegravir/rilpivirine [112]. An analysis of all participants receiving dolutegravir/rilpivirine at week 100 found 93% of these participants maintained virologic suppression.
Efficacy of dolutegravir/lamivudine was demonstrated in two randomized trials of treatment-naïve adults with baseline HIV-RNA < 500,000 copies/mL [113]. In the GEMINI-1 and GEMINI-2, participants were randomized to receive dolutegravir plus emtricitabine/tenofovir DF or dolutegravir plus lamivudine, each given as two separate tablets once daily. In a pooled analysis, there were no significant differences in virologic suppression at week 48 (91% in the two-drug arm and 93% in the three-drug arm). In each of the trials, lower response rates were observed for individuals with baseline CD4 + counts ≤ 200 cells/mm3 in the two-drug group versus the three-drug group (81 vs. 90%, respectively, GEMINI-1; 78 vs. 96%, respectively, GEMINI-2), although it must be noted only 8% (n = 118) of the total study population had CD4 + ≤ 200 cells/mm3 [114]. Dolutegravir plus lamivudine remained non-inferior to the three-drug regimen at week 96, with 86% and 89.5% of participants in the two- and three-drug arms maintaining virologic suppression [115].
Clinical trials involving dolutegravir have demonstrated its efficacy in treatment-naïve individuals as part of both traditional, three-drug ART regimens, and as a novel, two-drug ART regimen containing lamivudine as a partner drug. Notably, the three drug regimens containing dolutegravir were superior to efavirenz-based ART and non-inferior to raltegravir-based ART, while dolutegravir/lamivudine was non-inferior to dolutegravir-containing, three-drug ART regimens for antiretroviral-naïve individuals. In addition, dolutegravir is an alternative INSTI for individuals who have failed raltegravir- or elvitegravir-based regimens with INSTI resistance. Finally, the dolutegravir/rilpivirine FDC is a switch-strategy for individuals suppressed on another ART combination.
5.4 Bictegravir
Bictegravir efficacy has been demonstrated in two phase 3 randomized trials in HIV treatment-naïve individuals, each comparing the bictegravir FDC with a dolutegravir-based regimen. In study GS1489, the bictegravir FDC was non-inferior to the dolutegravir/abacavir/lamivudine FDC [116, 117]. At week 48, 92.4% of participants in the bictegravir arm and 93% in the dolutegravir arm achieved virologic suppression [116]; non-inferiority was maintained at week 96 (bictegravir: 88%, vs. dolutegravir: 90%) [117]. Similar virologic suppression was observed in study GS1490 where the bictegravir FDC was compared with dolutegravir in combination with emtricitabine/tenofovir AF [118, 119]. At week 48, 89% of bictegravir recipients and 93% of dolutegravir were virologically suppressed [118]; non-inferiority was maintained through week 96 (bictegravir: 84% vs. dolutegravir: 86%) [119]. There were no differences in efficacy in participants with lower (< 100,000 copies/mL) versus higher (> 100,000 copies/mL) HIV-RNA. Nor were there differences in treatment outcomes in a subgroup analysis of those with varying baseline CD4 + cell counts.
In switch studies, the bictegravir FDC was non-inferior to remaining on either a dolutegravir-based regimen or a PI-based regimen. In study GS1844, the bictegravir FDC was compared against dolutegravir/abacavir/lamivudine in persons who were virologically suppressed for at least 3 months [120]. Participants were randomly assigned to continue the dolutegravir regimen or switch to the bictegravir FDC regimen. At week 48, three of 282 (1%) participants in the bictegravir arm and one of 281 (< 1%) in the dolutegravir arm had HIV-RNA of ≥ 50 copies/mL, demonstrating non-inferiority. In a similar study design with virologically suppressed participants receiving a boosted atazanavir- or darunavir-containing ART regimen, switching to the bictegravir FDC was again non-inferior to remaining on the boosted PI regimen [121]. At week 48, each arm had 2% of participants with HIV-RNA ≥ 50 copies/mL. Finally, a switch study looking at participants with pre-existing resistance to NRTIs (e.g., M184V, K65R, thymidine analog mutations (TAMs)) found no significant differences in virologic suppression at week 48 between switching to the bictegravir FDC versus a dolutegravir plus emtricitabine/tenofovir AF [122].
In summary, the bictegravir FDC has demonstrated non-inferiority to dolutegravir FDC for individuals who are ART-naïve and those virologically suppressed on another regimen. Recent data suggest bictegravir may be effective in participants with a history of NRTI resistance [122], but data are needed to confirm the expectation that this second-generation INSTI will also offer an effective alternative for individuals with resistance to first-generation INSTIs.
5.5 Cabotegravir
The cabotegravir/rilpivirine combination has been studied in phase 3 clinical trials as once-monthly intramuscular injections [24, 25]. In the FLAIR trial, treatment-naïve adults were given 20 weeks of oral therapy with dolutegravir/abacavir/lamivudine [24]. Participants who reached virologic suppression by week 16 were randomized 1:1 to either continue dolutegravir-based therapy or receive 1 month of oral cabotegravir and rilpivirine followed by long-acting injectable cabotegravir/rilpivirine. At week 48, 2.1% of the participants in the long-acting arm and 2.5% in the oral therapy arm were virologically suppressed, demonstrating non-inferiority.
A second phase 3 study investigated monthly intramuscular cabotegravir and rilpivirine as maintenance therapy [25]. The ATLAS study enrolled PLWH who were virologically suppressed for at least 6 months while taking a NNRTI-, PI-, or INSTI-based regimen. Dolutegravir/abacavir/lamivudine was excluded in an attempt to broaden the generalizability of the ATLAS study, as dolutegravir-based regimens were the focus of the FLAIR study. Participants were randomized to continue their oral ART or to switch to intramuscular cabotegravir/rilpivirine. Similar to the FLAIR study, participants randomized to the cabotegravir arm of ATLAS were first given a 4-week lead-in of daily oral cabotegravir and rilpivirine, followed by monthly injections of the long-acting cabotegravir/rilpivirine formulations. At week 48, 1.6% of participants in the long-acting ART arm and 1.0% in the continued oral therapy arm were virologically suppressed, meeting the predefined criteria for non-inferiority.
The ideal dosing interval for intramuscular cabotegravir/rilpivirine is being explored in ATLAS-2M [123]. Participants who were virologically suppressed on either oral therapy or in the ATLAS study receiving cabotegravir/rilpivirine intramuscularly were randomized to receive cabotegravir/rilpivirine either every 4 weeks or every 8 weeks. Preliminary data show the two dosing strategies met the non-inferiority criteria, with 1.0% of those receiving every-4-week injections and 1.7% of those receiving every-8-week injections maintaining virologic suppression at week 48 [123].
Efficacy data support injectable cabotegravir/rilpivirine for individuals who attain virologic suppression on oral ART. The optimal dosing frequency of injectable cabotegravir/rilpivirine remains an area of investigation, with the most data to date supporting once-monthly injections. However, emerging data suggest the potential for future dosing every 8 weeks. Cabotegravir is also being investigated as a single agent for injectable pre-exposure prophylaxis, which is beyond the scope of this review [124, 125].
6 Safety and Tolerability
INSTIs are generally associated with lower rates of adverse effects than comparator classes in clinical trials. A summary of safety and tolerability data from phase 3 clinical trials is given in Table 3. Additionally, cohort studies have identified low rates of discontinuation due to adverse effects among INSTIs [11, 126]. Specific concerns related to INSTIs, including neuropsychiatric adverse events (NPAEs), fetal toxicity, as well as weight and metabolic complications, have been identified and are discussed in Sects. 6.1–6.3.
In clinical trials of raltegravir-based ART, mild to moderate rashes were most commonly observed through 96 weeks [81, 127]. Rash was more frequent with raltegravir versus placebo (11.3% vs. 6.3%) but lower in comparison with efavirenz (9.6% vs. 20.9%) [128]. While Stevens-Johnsons syndrome has been reported with raltegravir, no cases were observed in clinical trials [128]. Further, significantly lower rates of dyslipidemia were observed when compared with efavirenz- [77, 129] or PI-based regimens [130, 131]. Creatinine phosphokinase (CPK) elevations occurred in clinical trials with raltegravir, but none of these cases lead to treatment discontinuation [128, 132]. One cross-sectional, cohort study found higher rates of myalgias (19% vs. 3%, p < 0.001) and myopathies (4% vs. 0%, p < 0.001) in persons taking raltegravir compared to a control group [133]. There were no differences in CPK elevations (14% vs. 16%, p < 0.001) nor any cases of rhabdomyolysis observed, but post-marketing reports of rhabdomyolysis have been reported [134,135,136,137,138].
Overall, a higher rate of adverse effects are reported with elvitegravir compared with other INSTIs, which may be related to the co-formulation with cobicistat. Gastrointestinal adverse events (nausea and diarrhea), abnormal dreams, CPK elevations, and headache have been reported in > 5% of individuals receiving elvitegravir-based regimens (Table 3) [86, 89, 96, 98]. Compared to dolutegravir or raltegravir, the elvitegravir FDC resulted in higher rates of fatigue, malaise, and gastrointestinal adverse effects [139]. Despite cobicistat-boosting of elvitegravir, lower rates of dyslipidemias, diarrhea, and hyperbilirubinemias were observed with elvitegravir FDC compared with atazanavir/ritonavir-based regimens [89]. Greater increases in serum creatinine and reductions in glomerular filtration rates were observed with the elvitegravir FDC containing tenofovir DF compared with both NNRTI- and PI-based regimens [86, 89]. This adverse effect is secondary to a well described non-pathologic inhibition of renal tubular secretion of serum creatinine by cobicistat [140]. In contrast, the FDC containing tenofovir AF demonstrated less effect on glomerular filtration rate, as well as lower proteinuria, albuminuria, and tubular proteinuria [100]. Additionally, safe use of the elvitegravir FDC containing tenofovir AF was demonstrated among persons with moderate to severe renal dysfunction (creatinine clearance 30–59 mL/min) and also those on hemodialysis [141].
In clinical trials, dolutegravir has been associated with nausea, headache, and elevation in aspartate aminotransferase and CPK in ≥ 5% of participants (Table 3) [102, 106, 111, 113, 116, 142]. Asymptomatic elevations in CPK across trials with dolutegravir intervention arms occurred in 5% (naïve) and 2% (experienced) of patients [132]. Insomnia was reported more often among naïve participants on dolutegravir compared with other antiretrovirals in a recent meta-analysis (6.1% vs. 4.5%; p = 0.02) [143].
The most common adverse events associated with the bictegravir FDC in clinical trials were gastrointestinal complaints (diarrhea, nausea), headache, and CPK elevations (Table 3) [116, 118]. In comparison with dolutegravir/abacavir/lamivudine, lower rates of nausea were observed with bictegravir/emtricitabine/tenofovir AF among treatment-naïve participants at week 96 (11% vs. 24%; p < 0.001) [117]. Additionally, statistically lower rates of nausea were observed in a trial of participants randomized to switch to bictegravir/emtricitabine/tenofovir AF or remain on dolutegravir/abacavir/lamivudine (0% vs. 2%, respectively; p = 0.03) [120]. The difference in nausea may be related to the NRTIs (abacavir vs. tenofovir AF) rather than to the INSTI component of the FDC, but because bictegravir is not available outside of the FDC, it is difficult to separate related adverse events. One cohort study observed low rates of adverse events (8.9%; n = 18/201) and discontinuations due to adverse events (4%; n = 9/201) in persons treated with bictegravir [144]. Rash was the most commonly reported adverse event (n = 10/18; 56%) and cause for discontinuation (n = 7/9; 78%).
Due to the high-volume intramuscular injection of cabotegravir/rilpivirine, high rates of injection site reactions (ISRs), including pain, nodules, and swelling at the injection site were observed among trial participants. The majority receiving injectable cabotegravir/rilpivirine experienced at least one ISR in FLAIR and ATLAS (83% and 82%, respectively) [24, 25]. ISRs were typically mild to moderate in severity, with most (88%) resolving within 7 days (median, 3 days). A higher incidence of ISRs was reported at week 4 after the loading dose, decreasing over time to week 48 (FLAIR: 69–11%; ATLAS: 71–20%). Similar ISR outcomes were seen in ATLAS-2M, despite the larger dosing volume in the 8-week dosing arm, as illustrated by the proportion with ISR at weeks 4 and 48, respectively (4-week arm: 53% and 19%; 8-week arm: 70% and 20%) [123]. Reassuringly, low discontinuation rates due to ISRs (range: < 1–2%) were observed [24, 25, 123]. Aside from ISRs, the most common drug-related adverse events were headache and pyrexia (Table 3) [24, 25].
6.1 Neuropsychiatric Adverse Events
NPAEs have commonly been reported in clinical trials and post-marketing studies of INSTIs. The reported NPAEs primarily include dizziness, depression, anxiety, headache, insomnia, and other psychiatric disorders [132]. Higher rates of psychiatric disorders were observed when comparing dolutegravir with darunavir/ritonavir-based ART (6/242 [2.5%] vs. 1/242 [0.4%]) [104]. Yet, most NPAEs secondary to INSTI-based ART have been compared with NNRTI-based regimens and between INSTI-based regimens.
In comparative clinical trials, dolutegravir- and elvitegravir-based ART were associated with fewer NPAEs compared with efavirenz-based ART. In STARTMRK, NPAEs were higher for participants receiving efavirenz compared with those receiving raltegravir at weeks 8 (18% vs. 10%, p = 0.0149) and 48 (23% vs. 14%, p = 0.0044) [77]. Dizziness (24% vs. 7%), abnormal dreams (27% vs. 15%), and insomnia (14% vs. 9%) were significantly more common (all p < 0.01) with efavirenz in comparison with elvitegravir [86]. Further, treatment-naïve participants receiving dolutegravir-based ART experienced less NPAEs (dizziness, abnormal dreams, anxiety, somnolence) compared with efavirenz-based ART in the SINGLE trial; however, insomnia was more common in the dolutegravir arm (15% vs. 10%) [102]. Discontinuations across these trials were low and rarely associated with NPAEs.
Similar rates of NPAEs (range, 4–6%) were seen within the SPRING trials comparing dolutegravir and raltegravir [106]. This was consistent with randomized clinical trials investigating bictegravir- versus dolutegravir-based ART, which found similar, but rare, rates of NPAE or discontinuation between treatment arms [116, 120, 142]. Further, NPAEs were rarely observed in the randomized clinical trials of long-acting cabotegravir/rilpivirine in both treatment-naïve and -experienced persons [25, 123][24]. In contrast, the SWORD studies found NPAEs more common (5% vs. < 1%) in the dolutegravir/rilpivirine treatment arm compared with the standard-of-care arm (INSTI-, PI-, or NNRTI-based ART) and more participants in the dolutegravir/rilpivirine arm discontinued due to NPAE (n = 5 vs. 1) [111].
Numerous observational cohort studies have evaluated the rates of NPAEs and associated ART discontinuation among patients initiated on, or switched to, INSTI-based regimens. In a cohort study of 323 participants, higher rates of NPAEs (depression, vertigo, and sleep disturbances) were observed for dolutegravir compared to elvitegravir or raltegravir (10% vs. 1–2%, respectively), while a larger cohort of 1344 participants on the same regimens found a similar trend for higher, but not statistically different, rate of NPAEs in those receiving dolutegravir (3.5%) compared to raltegravir (2.8%) or elvitegravir (1.6%) [139, 145]. Several cohort studies found higher rates of discontinuation associated with NPAEs in patients receiving dolutegravir compared to either elvitegravir or raltegravir [11, 146,147,148]. Female sex at birth, elderly persons (> 60 years), lower CD4 + cell counts, and use of dolutegravir with or without abacavir have all been identified as predictive factors for therapy discontinuation related to NPAEs in European cohorts [11, 147,148,149]. However, these predictive factors have not been consistently identified across studies [126, 146]. Few cohort data are available with regard to NPAE incidence among patients treated with bictegravir or cabotegravir; however, similar rates of NPAE were observed between bictegravir/emtricitabine/tenofovir AF and dolutegravir/abacavir/lamivudine in one switch study [120].
Overall, though NPAEs are associated with INSTIs, the occurrence of INSTI discontinuation appears to be low based on the evidence to date. Therefore, providers should educate and monitor patients for NPAEs, and modify therapy as needed. Some NPAEs, such as sleep disturbances, may be managed by revising the dose time from evening to morning, while severe NPAEs may require therapy modification.
6.2 Fetal Toxicity
Raltegravir use among pregnant women has demonstrated an acceptable safety profile and low rates of fetal toxicity [49, 50]. The prevalence of birth defects among infants exposed to raltegravir during the first trimester of pregnancy was 3.09% (95% CI 1.42, 5.79), which is comparable with the US population average rate of 2.72% [150, 151]. One retrospective cohort of pregnant individuals (n = 497) receiving raltegravir-based ART found similar rates of birth defects among infants exposed to raltegravir during the first versus the second/third trimesters (5.7% vs. 3.5%, respectively; p = 0.29) [152].
Zash et el. reported higher rates of birth defects among infants exposed to dolutegravir at the time of conception, which raised concern around the use of dolutegravir at the time of conception or during early pregnancy [153, 154]. In the initial report, rates of birth defects for those who started dolutegravir at conception, started on dolutegravir during pregnancy, started on a non-dolutegravir regimen at conception, or were HIV-negative were 0.94% (4/426), 0.00% (0/2812), 0.12% (14/11,300), and 0.09% (61/66,057), respectively [153]. After further observations of the same study cohort, the prevalence of neural tube defects was 0.30% (5/1683) among infants exposed to dolutegravir regimens at conception compared with 0.10% (15/14,792) among infants exposed to non-dolutegravir regimens at conception (difference, 0.20%; 95% CI 0.01, 0.59) [154]. According to the Antiretroviral Pregnancy Registry, the rate of birth defects was 3.6% (11/302) for infants exposed to dolutegravir at any time during pregnancy, while the rate of neural tube defects was 0.004% (1/248) for infants with dolutegravir exposure at periconception [150].
No increased risk of birth defects have been observed in infants exposed to elvitegravir-based ART during pregnancy [150]. The prevalence of birth defects in infants with elvitegravir exposure during the first trimester was 2.50% (6/240) compared with the US overall prevalence of 2.72% [150, 151]. However, as described in Sect. 4.1.1, elvitegravir exposure is decreased during pregnancy, suggesting it may not be the optimal regimen for pregnant women [43].
The DHHS Perinatal HIV Treatment Guidelines currently recommend raltegravir- and dolutegravir-based regimens for pregnant PLWH, while dolutegravir-based regimens are an alternative regimen during periconception [43]. The World Health Organization recommends dolutegravir as preferred therapy for all PLWH, irrespective of pregnancy or childbearing potential [9]. There are no data to support the use of bictegravir or cabotegravir among pregnant women, as the prevalence of birth defects among infants exposed to either is unknown.
6.3 Weight Gain and Metabolic Concerns
Recent reports of weight gain associated with the use of INSTI-based ART is a rising concern [155, 156]. A pooled analysis of eight randomized controlled trials among ART-naïve persons with 96 weeks of follow-up found INSTIs, specifically dolutegravir and bictegravir, associated with more weight gain compared with NNRTIs and PIs [157]. The use of tenofovir AF in combination with the INSTI was associated with more weight gain than other NRTI partner drugs. Predictive clinical factors associated with weight gain were lower CD4 + cell count, higher HIV-RNA, no injection drug use, female sex at birth, and black race.
The NAMSAL trial was a randomized, non-inferiority trial in Africa evaluating three once-daily treatments consisting of dolutegravir in combination with emtricitabine/tenofovir AF, dolutegravir plus emtricitabine/tenofovir DF, or efavirenz 400 mg plus emtricitabine/tenofovir DF in treatment-naïve participants; a majority of participants (65.9%) were female [158]. Analogous with Sax et al. [157], larger weight gain and obesity incidence were observed at 48 weeks in participants in the dolutegravir compared with efavirenz arms (median weight gain, 5.0 kg vs. 3.0 kg; incident obesity, 12.3% vs. 5.4%). The ADVANCE trial was a randomized trial in Africa among treatment-naïve participants with the same treatment arms as NAMSAL, except efavirenz was dosed as 600 mg daily [159]. The population was nearly all black race and 59% were female. At 96 weeks, weight gain was most common in the dolutegravir groups, particularly those receiving tenofovir AF, and higher among females. Incidence in new obesity was higher overall for females, but a similar trend was observed among treatment arms and sex (incident obesity male vs. female: tenofovir AF group, 20% vs. 7%; tenofovir DF group, 11% vs. 3%; efavirenz group, 9% vs. 3%). Further, increases in mean changes of limb and truncal lean and fat mass were observed.
A retrospective, observational cohort study evaluated differences in weight gain among treatment-naïve persons 18 months after starting ART in the US. Of the 1152 patients, significantly more weight gain was observed with dolutegravir compared with NNRTIs or elvitegravir (6.0 kg vs. 2.6 kg vs. 0.5 kg, respectively; p < 0.05) [160]. Similarly, another observational cohort study of virologically suppressed persons found larger increases in weight over an 18-month period after switching from efavirenz to either an INSTI- or a PI-based regimen compared with continuing efavirenz-based ART [161]. Weight gain was greatest with the INSTI-based regimen (efavirenz-based regimen: 0.9 kg, vs. INSTI: 2.9 kg, p = 0.003; or vs. PI: 0.7 kg, p = 0.81).
Minimal data exist from clinical trials to inform our understanding of weight gain among persons treated with bictegravir or cabotegravir. Wohl et al. found a median weight gain of 3.6 kg (interquartile range (IQR), 0.0–8.5) in persons treated with bictegravir/emtricitabine/tenofovir AF compared with 2.5 kg (IQR − 0.4 to 5.8) over 96 weeks in those randomized to dolutegravir/abacavir/lamivudine [117]. Small weight increases were observed in persons receiving injectable cabotegravir/rilpivirine in FLAIR (injectable: 1.80 kg [IQR − 0.30 to 4.90]; oral: 0.30 kg (IQR − 1.60 to 2.50]) and ATLAS trials (injectable: 1.30 kg [IQR − 1.0 to 5.0]; oral: 1.50 kg (IQR − 1.0 to 3.9]) [24, 25]. No changes in weight or fasting metabolic parameters were observed in the phase 2 study evaluating cabotegravir versus placebo in HIV-negative participants using cabotegravir for HIV prevention over 41 weeks [162]. However, the phase 3 trial observed a median 1.3 kg (95% CI 0.99, 1.6) increase per year in HIV-negative men receiving cabotegravir compared to emtricitabine/tenofovir DF [+ 0.31 (95% CI − 0.12, − 0.49) kg per year] [163].
Treatment-naïve persons often experience weight gain after ART initiation, which has been described as a return to health [164]. Yet, several observational cohorts have associated the risk of onset diabetes mellitus and cardiovascular risk with weight gain after ART initiation [165, 166]. At week 96, the ADVANCE trial found the dolutegravir/emtricitabine/tenofovir AF arm had significantly higher risk of metabolic syndrome (International Diabetes Foundation; p = 0.031), myocardial infarction or coronary death (QRISK; p = 0.027), and 10-year risk of emergent diabetes (QDIABETES, change from baseline; p = 0.004) [167]. However, no significant differences in cardiovascular risk (Framingham Risk Equation) were observed among arms. Several case reports highlight incident diabetes mellitus and hyperglycemia in patients taking dolutegravir [168, 169]. A single-center, retrospective cohort study among Ugandan patients identified a higher rate of hyperglycemia after initiation of dolutegravir-based ART compared to ART without dolutegravir (0.47% [16/3417] vs. 0.03% [1/3230]; p = 0.0004) [170]. The median time to hyperglycemia after initiation of dolutegravir-based ART initiation was 4 months (IQR 2.5–4.5) and weight loss preceding hyperglycemia was observed in 80% (12/15).
Weight gain is a clinical challenge with INSTI-based regimens, particularly second-generation INSTI regimens in combination with tenofovir AF [117, 119, 157, 159, 160, 171]. Further, data suggest a higher incidence of weight gain among specific populations including women, those of African descent, and Hispanic ethnicity [157, 159, 172]. Understanding the intersection of weight gain and other resultant metabolic and cardiovascular co-morbidities is critical, and further data are needed to support clinical decisions on the selection of ART regimens to minimize weight gain and any associated metabolic comorbidities.
7 INSTIs in the Management of HIV-1 Infection
Clinical trials have demonstrated that INSTIs produce a rapid decline in, and maintain durable suppression of, plasma HIV-RNA, and are generally safe and well tolerated. The rapid decline in plasma HIV-RNA associated with INSTIs is a unique characteristic compared with older antiretroviral classes, and is particularly relevant to prevent HIV transmission through rapid virologic suppression [8, 43, 74,75,76]. All INSTIs, except cabotegravir, are indicated for antiretroviral-naïve persons [12, 13, 16, 18, 114, 173]. With the exception of raltegravir, all INSTIs are also indicated for individuals currently suppressed on an ART regimen, who do not have INSTI-related resistance mutations, and desire treatment switch [13, 16, 18, 114, 173]. Intramuscular cabotegravir plus rilpivirine is currently approved in Canada for a non-oral ART option for virologically suppressed adults [14]. Dolutegravir and raltegravir may be used in treatment-experienced persons with a detectable viral load in the presence of viral resistance [12, 17]. For pediatric patients, the agents have different indications for use, with raltegravir approved for use immediately after birth, dolutegravir for children at least 4 weeks of age, and bictegravir and elvitegravir for adolescents weighing at least 25 kg [40].
Within the INSTI class, raltegravir has the longest period of use and has minimal drug-drug interactions that cannot be managed by dose adjustment. Based on these characteristics, both the twice-daily and once-daily formulations remain a recommended antiretroviral for most individuals initiating ART in the US DHHS Adult and Adolescent Treatment Guidelines [8] but not the International AIDS Society guidelines [10]. In addition, the twice-daily formulation is a preferred INSTI for use during pregnancy [43]. Disadvantages related to raltegravir include a higher pill burden compared with other INSTI-based regimens, and a lower barrier to resistance compared with the second-generation INSTIs dolutegravir and bictegravir [22, 23]. It offers flexibility for combination with preferred NRTIs based on an individual’s characteristics and may be combined with other antiretrovirals to form an ART regimen for persons with drug-resistant virus.
Elvitegravir requires pharmaco-enhancement to achieve adequate concentrations, therefore, its co-formulation with cobicistat results in a significantly higher risk of drug-drug interactions. In addition, a higher rate of gastrointestinal adverse events, fatigue, and malaise are reported with elvitegravir FDCs [139]. These disadvantages, along with a requirement that the medication be co-administered with food and a lower barrier to antiretroviral resistance, highlight why elvitegravir is no longer preferred for most PLWH [8, 10]. Elvitegravir is not recommended during pregnancy due to suboptimal concentrations and associated risk for virologic failure [43]. One benefit of elvitegravir is a lower risk of weight gain compared with dolutegravir in retrospective evaluations [160]. Elvitegravir is co-formulated with either tenofovir DF or tenofovir AF plus emtricitabine, allowing use of the elvitegravir FDCs in patients with chronic hepatitis B coinfection.
Dolutegravir and bictegravir are recommended options for most antiretroviral-naïve individuals due to their minimal drug-drug interactions, ease of use as a FDC, and higher barrier to antiretroviral resistance compared with other INSTIs and some other antiretroviral classes [8, 10]. These characteristics also support the use of both for ART rapid start programs after diagnosis of HIV, and before resistance testing is available [174]. Dolutegravir is also the World Health Organization’s only preferred option for ART in all adults, including pregnant women [56]. Dolutegravir is a preferred regimen for pregnant women according to the US DHHS Perinatal HIV Treatment Guidelines, while bictegravir does not yet have data to support its use during pregnancy [43]. Dolutegravir was non-inferior to raltegravir in ART-naïve persons [107], and superior to raltegravir in those with some drug resistance [108]. Though bictegravir has not been evaluated in a phase 3 trial compared with raltegravir, dolutegravir and bictegravir were non-inferior initial therapies for ART-naïve individuals [118, 142]. The choice between dolutegravir and bictegravir as part of triple therapy in adults or adolescents is often based upon the desired NRTI companion drugs present in the FDC. Though a complete discussion regarding the benefits of emtricitabine/tenofovir AF versus abacavir/lamivudine is outside the scope of this review, there are concerns related to cardiovascular risks associated with abacavir use and with weight gain related to tenofovir AF, while the emtricitabine/tenofovir AF combination is preferable for individuals with chronic hepatis B virus co-infection [8]. Concern regarding the contribution of dolutegravir to weight gain after ART initiation is growing; however, it is not known if there is a difference in the associated risk between the two agents due to a shorter duration of clinical use of bictegravir. Finally, the bictegravir FDC is more susceptible to drug-drug interactions that cannot be overcome with dose adjustment and it is not indicated in combination with any other antiretroviral [16]. In contrast, dolutegravir may be given twice daily, to overcome drug-drug interactions and as a treatment option for individuals with drug resistance [17].
The two-drug regimens of dolutegravir/lamivudine or dolutegravir/rilpivirine may be useful for individuals at risk for adverse events related to the companion drugs in INSTI FDCs, such as abacavir or the tenofovir prodrugs. The dolutegravir/lamivudine FDC is the only two-drug combination recommended as an initial regimen for most PLWH (with an HIV-RNA level ≤ 500,000 copies/mL) according to the DHHS Adult and Adolescent Treatment Guidelines [8], while either two-drug regimen may be useful for treatment switch in those already suppressed on another ART regimen [175]. The dolutegravir/rilpivirine combination has added food restrictions and drug-drug interaction considerations, due to the rilpivirine component. Neither dual-therapy agent has been evaluated in adolescents, nor can they be used for patients co-infected with chronic hepatitis B virus.
Cabotegravir is not currently included in HIV treatment guidelines, but this long-acting agent, in combination with rilpivirine, offers an alternative to PLWH who do not want to take daily oral medications. Although clinical trials have been conducted in treatment-naïve individuals initiating therapy and virologically suppressed individuals desiring treatment simplification [24, 25], the recent Canadian approval was only for treatment simplification [14]. In clinical trials, participants preferred long-acting injectable ART over oral therapy and 8-week dosing over every-4-week dosing [24, 25, 123]. These results, however, may be skewed given all subjects volunteered to participate in long-acting therapy trials. The acceptability and demand for intramuscular injections outside of a clinical trial setting is unknown. Intramuscular injection will avoid some absorption-related drug-drug interactions with divalent cations and food requirements for rilpivirine, but it is not yet clear how co-administration of UGT1A1 inducers or inhibitors will be managed in combination with long-acting cabotegravir.
INSTIs are part of the standard of care for most PLWH. Future ART strategies will need to demonstrate non-inferiority to the second-generation INSTIs to have a role in first-line ART therapy. As long-acting ART emerges as a treatment option, clinicians will need to confront challenges managing the prolonged pharmacokinetic tail after long-acting ART discontinuation [37], how monthly or bimonthly injections will be administered in clinical settings, and non-adherence. Virologic failure has occurred in clinical trial participants receiving long-acting therapy who had perfect adherence (i.e., did not miss any injections) [24, 25]. These findings indicate the need for dosing-regimen optimization, particularly as this strategy moves to settings of less than perfect adherence, and investigations of individual characteristics, such as sex and high body mass index that may be associated with an increased risk for virologic failure. The contribution of individual INSTIs to weight gain remains an area of clinical investigation and may influence the choice of ART. INSTIs now represent an essential component of most ART regimens and are likely to remain an important class of antiretrovirals for the foreseeable future due to their anti-HIV potency, efficacy, safety, and tolerability.
References
UNAIDS. Global HIV & AIDS statistics—2019 fact sheets. https://www.unaids.org/en/resources/fact-sheet Accessed 2 Apr 2020.
Samji H, Cescon A, Hogg RS, Modur SP, Althoff KN, Buchacz K, et al. Closing the gap: increases in life expectancy among treated HIV-positive individuals in the United States and Canada. PLoS One. 2013;8(12):e81355. https://doi.org/10.1371/journal.pone.0081355.
INSIGHT START Study Group, Lundgren JD, Babiker AG, Gordin F, Emery S, Grund B et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373(9):795–807. https://doi.org/10.1056/nejmoa1506816.
TEMPRANO ANRS 12136 Study Group, Danel C, Moh R, Gabillard D, Badje A, Le Carrou J et al. A trial of early antiretrovirals and isoniazid preventive therapy in Africa. N Engl J Med. 2015;373(9):808–22. https://doi.org/10.1056/nejmoa1507198.
Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Antiretroviral therapy for the prevention of HIV-1 transmission. N Engl J Med. 2016;375(9):830–9. https://doi.org/10.1056/NEJMoa1600693.
Rodger AJ, Cambiano V, Bruun T, Vernazza P, Collins S, van Lunzen J, et al. Sexual activity without condoms and risk of HIV transmission in serodifferent couples when the HIV-positive partner is using suppressive antiretroviral therapy. JAMA. 2016;316(2):171–81. https://doi.org/10.1001/jama.2016.5148.
Townsend CL, Cortina-Borja M, Peckham CS, de Ruiter A, Lyall H, Tookey PA. Low rates of mother-to-child transmission of HIV following effective pregnancy interventions in the United Kingdom and Ireland, 2000-2006. AIDS. 2008;22(8):973–81. https://doi.org/10.1097/QAD.0b013e3282f9b67a.
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. Accessed 19 Aug 2020.
Update of recommendations on first- and second-line antiretroviral regimens. Geneva, Switzerland: World Health Organization; 2019 (WHO/CDS/HIV/19.15). Licence: CC BY-NC-SA 3.0 IGO.
Saag MS, Benson CA, Gandhi RT, Hoy JF, Landovitz RJ, Mugavero MJ, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the international antiviral society-USA panel. JAMA. 2018;320(4):379–96. https://doi.org/10.1001/jama.2018.8431.
Peñafiel J, De LE, Padilla M, Rojas J, Gonzalez-Cordon A, Blanco JL, et al. Tolerability of integrase inhibitors in a real-life setting. J Antimicrob Chemother. 2017;72(6):1752–9. https://doi.org/10.1093/jac/dkx053.
Isentress® [package insert]. Whitehouse Station, NJ. Merck & Co., Inc. January 2019.
Stribild® [package insert]. Foster City, CA. Gilead Sciences, Inc. January 2019.
Cabenuva® and Vocabria® [Product Monograph]. Laval, Quebec. Viiv Healthcare ULC. March 2020.
Dehority W, Abadi J, Wiznia A, Viani RM. Use of integrase inhibitors in HIV-infected children and adolescents. Drugs. 2015;75(13):1483–97. https://doi.org/10.1007/s40265-015-0446-2.
Biktarvy® [package insert]. Foster City, CA. Gilead Sciences, Inc. February 2018.
Tivicay® [package insert]. Research Triangle Park, NC. Viiv Healthcare. June 2020.
Genvoya® [package insert]. Foster City, CA. Gilead Sciences, Inc. February 2019.
Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, et al. In vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55(2):813–21. https://doi.org/10.1128/AAC.01209-10.
Podany AT, Scarsi KK, Pham MM, Fletcher CV. Comparative clinical pharmacokinetics and pharmacodynamics of HIV-1 integrase strand transfer inhibitors: an updated review. Clin Pharmacokinet. 2020. https://doi.org/10.1007/s40262-020-00898-8.
Wainberg MA, Zaharatos GJ, Brenner BG. Development of antiretroviral drug resistance. N Engl J Med. 2011;365(7):637–46. https://doi.org/10.1056/NEJMra1004180.
Anstett K, Brenner B, Mesplede T, Wainberg MA. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology. 2017;14(1):36. https://doi.org/10.1186/s12977-017-0360-7.
Tsiang M, Jones GS, Goldsmith J, Mulato A, Hansen D, Kan E, et al. Antiviral activity of bictegravir (GS-9883), a novel potent HIV-1 integrase strand transfer inhibitor with an improved resistance profile. Antimicrob Agents Chemother. 2016;60(12):7086–97. https://doi.org/10.1128/AAC.01474-16.
Orkin C, Arasteh K, Gorgolas Hernandez-Mora M, Pokrovsky V, Overton ET, Girard PM, et al. Long-acting cabotegravir and rilpivirine after oral induction for HIV-1 infection. N Engl J Med. 2020;382(12):1124–35. https://doi.org/10.1056/NEJMoa1909512.
Swindells S, Andrade-Villanueva JF, Richmond GJ, Rizzardini G, Baumgarten A, Masia M, et al. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. N Engl J Med. 2020;382(12):1112–23. https://doi.org/10.1056/NEJMoa1904398.
Boffito M, Waters L, Cahn P, Paredes R, Koteff J, van WJ et al. Perspectives on the barrier to resistance for dolutegravir + lamivudine, a 2-drug antiretroviral therapy for HIV-1 infection. AIDS Res Hum Retrovirus. 2019. https://doi.org/10.1089/aid.2019.0171.
Spreen W, Min S, Ford S, Chen S, Lou Y, Bomar M, et al. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin Trials. 2013;14(5):192–203. https://doi.org/10.1310/hct1405-192.
Min S, Sloan L, Dejesus E, Hawkins T, McCurdy L, Song I, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS. 2011;25(14):1737–45. https://doi.org/10.1097/QAD.0b013e32834a1dd9.
DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, et al. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr. 2006;43(1):1–5. https://doi.org/10.1097/01.qai.0000233308.82860.2f.
Gallant JE, Thompson M, DeJesus E, Voskuhl GW, Wei X, Zhang H, et al. Antiviral activity, safety, and pharmacokinetics of bictegravir as 10-day monotherapy in HIV-1-infected adults. J Acquir Immune Defic Syndr. 2017;75(1):61–6. https://doi.org/10.1097/QAI.0000000000001306.
Rizk ML, Hang Y, Luo WL, Su J, Zhao J, Campbell H, et al. Pharmacokinetics and pharmacodynamics of once-daily versus twice-daily raltegravir in treatment-naïve HIV-infected patients. Antimicrob Agents Chemother. 2012;56(6):3101–6. https://doi.org/10.1128/AAC.06417-11.
Food and Drug Administration (FDA). Guidance for industry, antiviral product development, conducting and submitting virology studies to the agency. 2006. https://www.fda.gov/media/71223/download Accessed 8 Mar 2020.
Cattaneo D, Gervasoni C. Pharmacokinetics and pharmacodynamics of cabotegravir, a long-acting HIV integrase strand transfer inhibitor. Eur J Drug Metab Pharmacokinet. 2019;44(3):319–27. https://doi.org/10.1007/s13318-018-0526-2.
Bowers GD, Culp A, Reese MJ, Tabolt G, Moss L, Piscitelli S, et al. Disposition and metabolism of cabotegravir: a comparison of biotransformation and excretion between different species and routes of administration in humans. 2016;46(2):147–62. https://doi.org/10.3109/00498254.2015.1060372.
Ford S, Crauwels H, Han K, Rossenu S, Zhang F, Huang JO, et al. Cabotegravir and rilpivirine PK following long-acting HIV treatment discontinuation. In: Conference on Retroviruses and Opportunistic Infections. March 8–11, 2020. Boston, MA. Abstract #466.
Spreen W, Ford SL, Chen S, Wilfret D, Margolis D, Gould E, et al. GSK1265744 pharmacokinetics in plasma and tissue following single-dose long-acting (la) injectable administration in healthy subjects. J Acquir Immune Defic Syndr. 2014. https://doi.org/10.1097/QAI.0000000000000301.
Landovitz RJ, Li S, Eron JJ Jr, Grinsztejn B, Dawood H, Liu AY, et al. Tail-phase safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in HIV-uninfected adults: a secondary analysis of the HPTN 077 trial. Lancet HIV. 2020;7(7):e472–81. https://doi.org/10.1016/S2352-3018(20)30106-5.
Vitekta® [package insert]. Foster City, CA. Gilead Sciences, Inc. September 2014.
Wohl DA, Dumond JB, Blevins S, Pittard D, Ragan D, Wang R, et al. Raltegravir pharmacokinetics in treatment-naive patients is not influenced by race: results from the raltegravir early therapy in african-americans living with HIV (REAL) Study. Antimicrob Agents Chemother. 2013;57(2):784–8. https://doi.org/10.1128/AAC.01826-12.
Panel on Antiretroviral Therapy and Medical Management of Children Living with HIV. Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection. http://aidsinfo.nih.gov/contentfiles/lvguidelines/pediatricguidelines.pdf. Accessed 19 Aug 2020.
Lu H, Rosenbaum S. Developmental pharmacokinetics in pediatric populations. J Pediatr Pharmacol Ther. 2014;19(4):262–76. https://doi.org/10.5863/1551-6776-19.4.262.
Elliot ER, Wang X, Singh S, Simmons B, Vera JH, Miller RF, et al. Increased dolutegravir peak concentrations in people living with human immunodeficiency virus aged 60 and over, and analysis of sleep quality and cognition. Clin Infect Dis. 2019;68(1):87–95. https://doi.org/10.1093/cid/ciy426.
Panel on Treatment of Pregnant Women with HIV Infection and Prevention of Perinatal Transmission. Recommendations for the use of antiretroviral drugs in pregnant women with HIV infection and interventions to reduce perinatal HIV transmission in the United States. http://aidsinfo.nih.gov/contentfiles/lvguidelines/PerinatalGL.pdf. Accessed 19 Aug 2020.
Colbers A, De HM, Van CR, Kruijssen M, Duisenberg-Van EM, Abbink E et al. Pharmacokinetics of crushed elvitegravir combination tablet given with drip feed. Top Antiviral Med. 2016;24(-1):166.
Jeong H, Choi S, Song JW, Chen H, Fischer JH. Regulation of UDP-glucuronosyltransferase (UGT) 1A1 by progesterone and its impact on labetalol elimination. Xenobiotica. 2008;38(1):62–75. https://doi.org/10.1080/00498250701744633.
van der Galien R, Ter Heine R, Greupink R, Schalkwijk SJ, van Herwaarden AE, Colbers A, et al. Pharmacokinetics of HIV-integrase inhibitors during pregnancy: mechanisms, clinical implications and knowledge gaps. Clin Pharmacokinet. 2019;58(3):309–23. https://doi.org/10.1007/s40262-018-0684-z.
Mulligan N, Best BM, Wang J, Capparelli EV, Stek A, Barr E, et al. Dolutegravir pharmacokinetics in pregnant and postpartum women living with HIV. AIDS. 2018;32(6):729–37. https://doi.org/10.1097/QAD.0000000000001755.
Waitt C, Orrell C, Walimbwa S, Singh Y, Kintu K, Simmons B, et al. Safety and pharmacokinetics of dolutegravir in pregnant mothers with HIV infection and their neonates: a randomised trial (DolPHIN-1 study). PLoS Med. 2019;16(9):e1002895. https://doi.org/10.1371/journal.pmed.1002895.
Blonk MI, Colbers APH, Hidalgo-Tenorio C, Kabeya K, Weizsäcker K, Haberl AE, et al. Raltegravir in HIV-1-infected pregnant women: pharmacokinetics, safety, and efficacy. Clin Infect Dis. 2015;61(5):809–16. https://doi.org/10.1093/cid/civ366.
Watts DH, Stek A, Best BM, Wang J, Capparelli EV, Cressey TR, et al. Raltegravir pharmacokinetics during pregnancy. J Acquir Immune Defic Syndr. 2014. https://doi.org/10.1097/QAI.0000000000000318.
Momper JD, Best BM, Wang J, Capparelli EV, Stek A, Barr E, et al. Elvitegravir/cobicistat pharmacokinetics in pregnant and postpartum women with HIV. AIDS. 2018;32(17):2507–16. https://doi.org/10.1097/QAD.0000000000001992.
Patel P, Thiagarajah S, Ford S, Margolis DA, Romach BH, Baker M, Sutton K, Harrinton CM, Shaefer MS, Spreen W, Smith K, Vannappagari V. Cabotegravir pharmacokinetic tail in pregnancy and neonatal outcomes. In: Conference on retroviruses and opportunistic infections. March 8–11, 2020. Boston, MA. Abstract #775.
Schalkwijk S, Greupink R, Colbers AP, Wouterse AC, Verweij VG, van Drongelen J, et al. Placental transfer of the HIV integrase inhibitor dolutegravir in an ex vivo human cotyledon perfusion model. J Antimicrob Chemother. 2016;71(2):480–3. https://doi.org/10.1093/jac/dkv358.
Schalkwijk S, Colbers A, Konopnicki D, Greupink R, Russel FGM, Burger D. First reported use of elvitegravir and cobicistat during pregnancy. AIDS. 2016;30(5):807–8. https://doi.org/10.1097/QAD.0000000000000976.
McCormack SA, Best BM. Protecting the fetus against HIV infection: a systematic review of placental transfer of antiretrovirals. Clin Pharmacokinet. 2014;53(11):989–1004. https://doi.org/10.1007/s40262-014-0185-7.
World Health Organization, United Nations Children’s Fund. Guideline: updates on HIV and infant feeding: the duration of breastfeeding, and support from health services to improve feeding practices among mothers living with HIV. Geneva: World Health Organization; 2016.
Iwamoto M, Hanley WD, Petry AS, Friedman EJ, Kost JT, Breidinger SA, et al. Lack of a clinically important effect of moderate hepatic insufficiency and severe renal insufficiency on raltegravir pharmacokinetics; Merck and Co(United States). Antimicrob Agents Chemother. 2009;53(5):1747–52. https://doi.org/10.1128/AAC.01194-08.
Weller S, Borland J, Chen S, Johnson M, Savina P, Wynne B, et al. Pharmacokinetics of dolutegravir in HIV-seronegative subjects with severe renal impairment. Eur J Clin Pharmacol. 2014;70(1):29–35. https://doi.org/10.1007/s00228-013-1590-9.
Debinski HS, Lee CS, Danks JA, Mackenzie PI, Desmond PV. Localization of uridine 5′-diphosphate-glucuronosyltransferase in human liver injury. Gastroenterology. 1995;108(5):1464–9. https://doi.org/10.1016/0016-5085(95)90695-9.
Furlan V, Demirdjian S, Bourdon O, Magdalou J, Taburet AM. Glucuronidation of drugs by hepatic microsomes derived from healthy and cirrhotic human livers. J Pharmacol Exp Ther. 1999;289(2):1169–75.
George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21(1):120–8.
Calza L, Danese I, Colangeli V, Manfredi R, Magistrelli E, Verucchi G, et al. Plasma concentrations of efavirenz, darunavir/ritonavir and raltegravir in HIV-HCV-coinfected patients without liver cirrhosis in comparison with HIV-monoinfected patients. Infect Dis. 2015;47(9):625–36. https://doi.org/10.3109/23744235.2015.1034169.
Custodio JM, Rhee M, Shen G, Ling KHJ, Kearney BP, Ramanathan S. Pharmacokinetics and safety of boosted elvitegravir in subjects with hepatic impairment. Antimicrob Agents Chemother. 2014;58(5):2564–9. https://doi.org/10.1128/AAC.02180-13.
Hernández-Novoa B, Moreno A, Pérez-Elías MJ, Quereda C, Dronda F, Casado JL, et al. Raltegravir pharmacokinetics in HIV/HCV-coinfected patients with advanced liver cirrhosis (Child-Pugh C); Merck Sharp and Dohme(United States). J Antimicrob Chemother. 2014;69(2):471–5. https://doi.org/10.1093/jac/dkt386.
Song IH, Borland J, Savina PM, Chen S, Patel P, Wajima T, et al. Pharmacokinetics of single-dose dolutegravir in HIV-seronegative subjects with moderate hepatic impairment compared to healthy matched controls. Clin Pharmacol Drug Dev. 2013;2(4):342–8. https://doi.org/10.1002/cpdd.55.
Shaik JSB, Ford SL, Lou Y, Zhang Z, Bakshi KK, Tenorio AR, et al. A phase 1 study to evaluate the pharmacokinetics and safety of cabotegravir in patients with hepatic impairment and healthy matched controls. Clin Pharmacol Drug Dev. 2019;8(5):664–73. https://doi.org/10.1002/cpdd.655.
Dooley KE, Sayre P, Borland J, Purdy E, Chen S, Song I, et al. Safety, tolerability, and pharmacokinetics of the HIV integrase inhibitor dolutegravir given twice daily with rifampin or once daily with rifabutin: results of a phase 1 study among healthy subjects. J Acquir Immune Defic Syndr. 2013;62(1):21–7. https://doi.org/10.1097/QAI.0b013e318276cda9.
Dooley KE, Kaplan R, Mwelase N, Grinsztejn B, Ticona E, Lacerda M, et al. Dolutegravir-based antiretroviral therapy for patients co-infected with tuberculosis and hiv: a multicenter, noncomparative, open-label, randomized trial. Clin Infect Dis. 2019. https://doi.org/10.1093/cid/ciz256.
Grinsztejn B, De CN, Arnold V, Veloso VG, Morgado M, Pilotto JH, et al. Raltegravir for the treatment of patients co-infected with HIV and tuberculosis (ANRS 12 180 Reflate TB): a multicentre, phase 2, non-comparative, open-label, randomised trial. Lancet Infect Dis. 2014;14(6):459–67. https://doi.org/10.1016/S1473-3099(14)70711-X.
Ford SL, Sutton K, Lou Y, Zhang Z, Tenorio A, Trezza C et al. Effect of rifampin on the single-dose pharmacokinetics of oral cabotegravir in healthy subjects. Antimicrob Agents Chemother. 2017. https://doi.org/10.1128/aac.00487-17.
Rajoli RKR, Curley P, Chiong J, Back D, Flexner C, Owen A, et al. Predicting drug-drug interactions between rifampicin and long-acting cabotegravir and rilpivirine using physiologically based pharmacokinetic modeling. J Infect Dis. 2019;219(11):1735–42. https://doi.org/10.1093/infdis/jiy726.
Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E, et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a hiv integrase inhibitor. Drug Metab Dispos. 2013;41(2):353–61. https://doi.org/10.1124/dmd.112.048918.
Cattaneo D, Resnati C, Rizzardini G, Gervasoni C. Dolutegravir and metformin: a clinically relevant or just a pharmacokinetic interaction? AIDS. 2018;32(4):532–3. https://doi.org/10.1097/QAD.0000000000001720.
Acosta RK, Willkom M, Martin R, Chang S, Wei X, Garner W et al. Resistance analysis of bictegravir-emtricitabine-tenofovir alafenamide in HIV-1 treatment-naive patients through 48 weeks. Antimicrob Agents Chemother. 2019. https://doi.org/10.1128/aac.02533-18.
Kintu K, Malaba TR, Nakibuka J, Papamichael C, Colbers A, Byrne K, et al. Dolutegravir versus efavirenz in women starting HIV therapy in late pregnancy (DolPHIN-2): an open-label, randomised controlled trial. Lancet HIV. 2020;7(5):e332–9. https://doi.org/10.1016/S2352-3018(20)30050-3.
Jacobson K, Ogbuagu O. Integrase inhibitor-based regimens result in more rapid virologic suppression rates among treatment-naive human immunodeficiency virus-infected patients compared to non-nucleoside and protease inhibitor-based regimens in a real-world clinical setting: a retrospective cohort study. Medicine (Baltimore). 2018;97(43):e13016. https://doi.org/10.1097/MD.0000000000013016.
Lennox JL, DeJesus E, Lazzarin A, Pollard RB, Madruga JVR, Berger DS, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet. 2009;374(9692):796–806. https://doi.org/10.1016/S0140-6736(09)60918-1.
Rockstroh JK, DeJesus E, Lennox JL, Yazdanpanah Y, Saag MS, Wan H, et al. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63(1):77–85. https://doi.org/10.1097/QAI.0b013e31828ace69.
Eron JJ, Young B, Cooper DA, Youle M, DeJesus E, Andrade-Villanueva J, et al. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials. Lancet. 2010;375(9712):396–407. https://doi.org/10.1016/S0140-6736(09)62041-9.
Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, et al. Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359(4):339–54. https://doi.org/10.1056/NEJMoa0708975.
Steigbigel RT, Cooper DA, Teppler H, Eron JJ, Gatell JM, Kumar PN, et al. Long-term efficacy and safety of raltegravir combined with optimized background therapy in treatmentexperienced patients with drugresistant hiv infection: week 96 results of the benchmrk 1 and 2 phase III trials. Clin Infect Dis. 2010;50(4):605–12. https://doi.org/10.1086/650002.
Eron JJ, Cooper DA, Steigbigel RT, Clotet B, Gatell JM, Kumar PN, et al. Efficacy and safety of raltegravir for treatment of HIV for 5 years in the BENCHMRK studies: final results of two randomised, placebo-controlled trials. Lancet Infect Dis. 2013;13(7):587–96. https://doi.org/10.1016/S1473-3099(13)70093-8.
Eron JJ, Rockstroh JK, Reynes J, Andrade-Villanueva J, Ramalho-Madruga JV, Bekker LG, et al. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis. 2011;11(12):907–15. https://doi.org/10.1016/S1473-3099(11)70196-7.
Cahn P, Kaplan R, Sax PE, Squires K, Molina JM, Avihingsanon A, et al. Raltegravir 1200 mg once daily versus raltegravir 400 mg twice daily, with tenofovir disoproxil fumarate and emtricitabine, for previously untreated HIV-1 infection: a randomised, double-blind, parallel-group, phase 3, non-inferiority trial. Lancet HIV. 2017;4(11):e486–94. https://doi.org/10.1016/S2352-3018(17)30128-5.
Cahn P, Sax PE, Squires K, Molina JM, Ratanasuwan W, Rassool M, et al. Raltegravir 1200 mg once daily vs 400 mg twice daily, with emtricitabine and tenofovir disoproxil fumarate, for previously untreated HIV-1 infection: week 96 results from ONCEMRK, a randomized, double-blind, noninferiority trial. J Acquir Immune Defic Syndr. 2018;78(5):589–98. https://doi.org/10.1097/QAI.0000000000001723.
Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379(9835):2439–48. https://doi.org/10.1016/S0140-6736(12)60917-9.
Wohl DA, Cohen C, Gallant JE, Mills A, Sax PE, DeJesus E, et al. A randomized, double-blind comparison of single tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF versus single tablet regimen efavirenz/emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2013. https://doi.org/10.1097/QAI.0000000000000057.
Zolopa A, Sax PE, Dejesus E, Mills A, Cohen C, Wohl D, et al. A randomized double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;63(1):96–100. https://doi.org/10.1097/QAI.0b013e318289545c.
DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379(9835):2429–38. https://doi.org/10.1016/S0140-6736(12)60918-0.
Rockstroh JK, DeJesus E, Henry K, Molina JM, Gathe J, Ramanathan S, et al. A randomized, double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus coformulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;62(5):483–6. https://doi.org/10.1097/QAI.0b013e318286415c.
Clumeck N, Molina JM, Henry K, Gathe J, Rockstroh JK, DeJesus E, et al. A randomized, double-blind comparison of single-tablet regimen elvitegravir/cobicistat/emtricitabine/tenofovir DF vs ritonavir-boosted atazanavir plus emtricitabine/tenofovir DF for initial treatment of HIV-1 infection: analysis of week 144 results. J Acquir Immune Defic Syndr. 2014;65(3):e121–4. https://doi.org/10.1097/QAI.0000000000000089.
Pozniak A, Markowitz M, Mills A, Stellbrink HJ, Antela A, Domingo P, et al. Switching to coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus continuation of non-nucleoside reverse transcriptase inhibitor with emtricitabine and tenofovir in virologically suppressed adults with HIV (STRATEGY-NNRTI): 48 week results of a randomised, open-label, phase 3b non-inferiority trial. Lancet Infect Dis. 2014;14(7):590–9. https://doi.org/10.1016/S1473-3099(14)70796-0.
Pozniak A, Flamm J, Antinori A, Bloch M, Ward D, Berenguer J, et al. Switching to the single-tablet regimen of elvitegravir, cobicistat, emtricitabine, and tenofovir DF from non-nucleoside reverse transcriptase inhibitor plus coformulated emtricitabine and tenofovir DF regimens: week 96 results of STRATEGY-NNRTI. HIV Clin Trials. 2017;18(4):141–8. https://doi.org/10.1080/15284336.2017.1338844.
Arribas JR, DeJesus E, van Lunzen J, Zurawski C, Doroana M, Towner W, et al. Simplification to single-tablet regimen of elvitegravir, cobicistat, emtricitabine, tenofovir DF from multi-tablet ritonavir-boosted protease inhibitor plus coformulated emtricitabine and tenofovir DF regimens: week 96 results of STRATEGY-PI. HIV Clin Trials. 2017;18(3):118–25. https://doi.org/10.1080/15284336.2017.1330440.
Arribas JR, Pialoux G, Gathe J, Di Perri G, Reynes J, Tebas P, et al. Simplification to coformulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus continuation of ritonavir-boosted protease inhibitor with emtricitabine and tenofovir in adults with virologically suppressed HIV (STRATEGY-PI): 48 week results of a randomised, open-label, phase 3b, non-inferiority trial. Lancet Infect Dis. 2014;14(7):581–9. https://doi.org/10.1016/S1473-3099(14)70782-0.
Mills A, Crofoot G, Ortiz R, Rashbaum B, Towner W, Ward D, et al. Switching from twice-daily raltegravir plus tenofovir disoproxil Fumarate/Emtricitabine to once-daily Elvitegravir/Cobicistat/Emtricitabine/Tenofovir disoproxil fumarate in virologically suppressed, HIV-1-infected subjects: 48 weeks data. HIV Clinical Trials. 2014;15(2):51–6. https://doi.org/10.1310/hct1502-51.
Sax PE, Wohl D, Yin MT, Post F, DeJesus E, Saag M, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. 2015;385(9987):2606–15. https://doi.org/10.1016/S0140-6736(15)60616-X.
Wohl D, Oka S, Clumeck N, Clarke A, Brinson C, Stephens J, et al. A randomized, double-blind comparison of tenofovir alafenamide versus tenofovir disoproxil fumarate, each coformulated with elvitegravir, cobicistat, and emtricitabine for initial HIV-1 treatment: week 96 results. J Acquir Immune Defic Syndr. 2016;72(1):58–64. https://doi.org/10.1097/QAI.0000000000000940.
Arribas JR, Thompson M, Sax PE, Haas B, McDonald C, Wohl DA, et al. Brief report: randomized, double-blind comparison of tenofovir alafenamide (TAF) vs tenofovir disoproxil fumarate (TDF), each coformulated with elvitegravir, cobicistat, and emtricitabine (E/C/F) for initial HIV-1 treatment: week 144 results. J Acquir Immune Defic Syndr. 2017;75(2):211–8. https://doi.org/10.1097/QAI.0000000000001350.
Mills A, Arribas JR, Andrade-Villanueva J, DiPerri G, Van Lunzen J, Koenig E, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in antiretroviral regimens for virologically suppressed adults with HIV-1 infection: a randomised, active-controlled, multicentre, open-label, phase 3, non-inferiority study. Lancet Infect Dis. 2016;16(1):43–52. https://doi.org/10.1016/S1473-3099(15)00348-5.
DeJesus E, Haas B, Segal-Maurer S, Ramgopal MN, Mills A, Margot N, et al. Superior efficacy and improved renal and bone safety after switching from a tenofovir disoproxil fumarate- to a tenofovir alafenamide-based regimen through 96 weeks of treatment. AIDS Res Hum Retroviruses. 2018;34(4):337–42. https://doi.org/10.1089/AID.2017.0203.
Walmsley SL, Antela A, Clumeck N, Duiculescu D, Eberhard A, Gutieŕrez F, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369(19):1807–18. https://doi.org/10.1056/NEJMoa1215541.
Walmsley S, Baumgarten A, Berenguer J, Felizarta F, Florence E, Khuong-Josses MA, et al. Dolutegravir plus abacavir/lamivudine for the treatment of HIV-1 infection in antiretroviral therapy-naive patients: week 96 and week 144 results from the SINGLE randomized clinical trial. J Acquir Immune Defic Syndr. 2015;70(5):515–9. https://doi.org/10.1097/QAI.0000000000000790.
Clotet B, Feinberg J, Van LJ, Khuong-Josses MA, Antinori A, Dumitru I, et al. Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naive adults with HIV-1 infection (FLAMINGO): 4 8 week results from the randomised open-label phase 3b study. Lancet. 2014;383(9936):2222–31. https://doi.org/10.1016/S0140-6736(14)60084-2.
Molina JM, Clotet B, van Lunzen J, Lazzarin A, Cavassini M, Henry K, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomised, open-label, phase 3b study. Lancet HIV. 2015;2(4):e127–36. https://doi.org/10.1016/S2352-3018(15)00027-2.
Raffi F, Rachlis A, Stellbrink HJ, Hardy WD, Torti C, Orkin C, et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet. 2013;381(9868):735–43. https://doi.org/10.1016/S0140-6736(12)61853-4.
Raffi F, Jaeger H, Quiros-Roldan E, Albrecht H, Belonosova E, Gatell JM, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2013;13(11):927–35. https://doi.org/10.1016/S1473-3099(13)70257-3.
Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet. 2013;382(9893):700–8. https://doi.org/10.1016/S0140-6736(13)61221-0.
Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study. J Infect Dis. 2013;207(5):740–8. https://doi.org/10.1093/infdis/jis750.
Castagna A, Maggiolo F, Penco G, Wright D, Mills A, Grossberg R, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis. 2014;210(3):354–62. https://doi.org/10.1093/infdis/jiu051.
Llibre JM, Hung CC, Brinson C, Castelli F, Girard PM, Kahl LP, et al. Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies. Lancet. 2018;391(10123):839–49. https://doi.org/10.1016/S0140-6736(17)33095-7.
Aboud M, Orkin C, Podzamczer D, Bogner JR, Baker D, Khuong-Josses MA, et al. Efficacy and safety of dolutegravir-rilpivirine for maintenance of virological suppression in adults with HIV-1: 100-week data from the randomised, open-label, phase 3 SWORD-1 and SWORD-2 studies. Lancet HIV. 2019;6(9):e576–87. https://doi.org/10.1016/S2352-3018(19)30149-3.
Cahn P, Madero JS, Arribas JR, Antinori A, Ortiz R, Clarke AE, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet. 2019;393(10167):143–55. https://doi.org/10.1016/S0140-6736(18)32462-0.
Dovato® [package insert]. Research Triangle Park, NC. Viiv Healthcare. March 2020.
Cahn P, Madero JS, Arribas JR, Antinori A, Ortiz R, Clarke AE, et al. Durable efficacy of dolutegravir plus lamivudine in antiretroviral treatment-naive adults with HIV-1 infection: 96-week results from the GEMINI-1 and GEMINI-2 randomized clinical trials. J Acquir Immune Defic Syndr. 2020;83(3):310–8. https://doi.org/10.1097/QAI.0000000000002275.
Gallant J, Lazzarin A, Mills A, Orkin C, Podzamczer D, Tebas P, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS-US-380-1489): a double-blind, multicentre, phase 3, randomised controlled non-inferiority trial. Lancet. 2017;390(10107):2063–72. https://doi.org/10.1016/S0140-6736(17)32299-7.
Wohl DA, Yazdanpanah Y, Baumgarten A, Clarke A, Thompson MA, Brinson C, et al. Bictegravir combined with emtricitabine and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection: week 96 results from a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2019;6(6):e355–63. https://doi.org/10.1016/S2352-3018(19)30077-3.
Sax PE, Pozniak A, Montes ML, Koenig E, DeJesus E, Stellbrink HJ, et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380–1490): a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet. 2017;390(10107):2073–82. https://doi.org/10.1016/S0140-6736(17)32340-1.
Stellbrink HJ, Arribas JR, Stephens JL, Albrecht H, Sax PE, Maggiolo F, et al. Co-formulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide for initial treatment of HIV-1 infection: week 96 results from a randomised, double-blind, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2019;6(6):e364–72. https://doi.org/10.1016/S2352-3018(19)30080-3.
Molina JM, Ward D, Brar I, Mills A, Stellbrink HJ, López-Cortés L, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from dolutegravir plus abacavir and lamivudine in virologically suppressed adults with HIV-1: 48 week results of a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet HIV. 2018;5(7):e357–65. https://doi.org/10.1016/S2352-3018(18)30092-4.
Daar ES, DeJesus E, Ruane P, Crofoot G, Oguchi G, Creticos C, et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48 week results of a randomised, open-label, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2018;5(7):e347–56. https://doi.org/10.1016/S2352-3018(18)30091-2.
Sax PE, Rockstroh JK, Luetkemeyer AF, Yazdanpanah Y, Ward D, Trottier B, et al. Switching to bictegravir, emtricitabine, and tenofovir alafenamide in virologically suppressed adults with HIV. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa988.
Overton ET, Richmond GJ, Rizzardini G, Jaeger H, Orrell C, Nagimova F et al. Cabotegravir + rilpivirine every 2 months is noninferior to monthly: ATLAS-2 M study. Conference on Retroviruses and Opportunistic Infections (CROI). March 8-11, 2020. Boston. Abstract 34.
Landovitz RJ, Li S, Grinsztejn B, Dawood H, Liu AY, Magnus M et al. Safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in low-risk HIV-uninfected individuals: HPTN 077, a phase 2a randomized controlled trial. PLoS Medicine. 2018. https://doi.org/10.1371/journal.pmed.1002690.
Markowitz M, Frank I, Grant RM, Mayer KH, Elion R, Goldstein D, et al. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): a multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV. 2017;4(8):e331–40. https://doi.org/10.1016/S2352-3018(17)30068-1.
Llibre JM, Montoliu A, Miró JM, Domingo P, Riera M, Tiraboschi J, et al. Discontinuation of dolutegravir, elvitegravir/cobicistat and raltegravir because of toxicity in a prospective cohort. HIV Med. 2019;20(3):237–47. https://doi.org/10.1111/hiv.12710.
Lennox JL, Dejesus E, Berger DS, Lazzarin A, Pollard RB, Ramalho Madruga JV, et al. Raltegravir versus Efavirenz regimens in treatment-naive HIV-1-infected patients: 96-week efficacy, durability, subgroup, safety, and metabolic analyses. J Acquir Immune Defic Syndr. 2010;55(1):39–48. https://doi.org/10.1097/QAI.0b013e3181da1287.
Teppler H, Brown DD, Leavitt RY, Sklar P, Wan H, Xu X, et al. Long-term safety from the raltegravir clinical development program. Curr HIV Res. 2011;9(1):40–53. https://doi.org/10.2174/157016211794582650.
Nguyen A, Calmy A, Delhumeau C, Mercier I, Cavassini M, Mello AF, et al. A randomized cross-over study to compare raltegravir and efavirenz (SWITCH-ER study). AIDS. 2011;25(12):1481–7. https://doi.org/10.1097/QAD.0b013e328348dab0.
Lennox JL, Landovitz RJ, Ribaudo HJ, Ofotokun I, Na LH, Godfrey C, et al. Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naïve volunteers infected with HIV-1: a randomized, controlled equivalence trial. Ann Internal Med. 2014;161(7):461–71. https://doi.org/10.7326/M14-1084.
Martinez E, Larrousse M, Llibre JM, Gutierrez F, Saumoy M, Antela A, et al. Substitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL study. AIDS. 2010;24(11):1697–707. https://doi.org/10.1097/QAD.0b013e32833a608a.
Curtis L, Nichols G, Stainsby C, Lim J, Aylott A, Wynne B, et al. Dolutegravir: clinical and laboratory safety in integrase inhibitor-naive patients. HIV Clin Trials. 2014;15(5):199–208. https://doi.org/10.1310/hct1505-199.
Lee FJ, Amin J, Bloch M, Pett SL, Marriott D, Carr A. Skeletal muscle toxicity associated with raltegravir-based combination antiretroviral therapy in HIV-infected adults. J Acquir Immune Defic Syndr. 2013;62(5):525–33. https://doi.org/10.1097/QAI.0b013e3182832578.
Tsai WJ, Lee SS, Tsai HC, Sy CL, Chen JK, Wu KS, et al. Rapid onset of rhabdomyolysis after switching to a raltegravir-based antiretroviral regimen. J Microbiol Immunol Infect. 2016;49(2):286–8. https://doi.org/10.1016/j.jmii.2013.02.008.
Masia M, Enriquez R, Sirvent A, Gutierrez F. Severe acute renal failure associated with rhabdomyolysis during treatment with raltegravir. A call for caution. J Infect. 2010;61(2):189–90. https://doi.org/10.1016/j.jinf.2010.04.011.
Zembower TR, Gerzenshtein L, Coleman K, Palella FJ Jr. Severe rhabdomyolysis associated with raltegravir use. AIDS. 2008;22(11):1382–4. https://doi.org/10.1097/QAD.0b013e328303be40.
Croce F, Vitello P, Dalla Pria A, Riva A, Galli M, Antinori S. Severe raltegravir-associated rhabdomyolysis: a case report and review of the literature. Int J STD AIDS. 2010;21(11):783–5. https://doi.org/10.1258/ijsa.2010.010246.
Dori L, Buonomini AR, Viscione M, Sarmati L, Andreoni M. A case of rhabdomiolysis associated with raltegravir use. AIDS. 2010;24(3):473–5. https://doi.org/10.1097/QAD.0b013e328334cc4a.
Lepik KJ, Yip B, Ulloa AC, Wang L, Toy J, Akagi L, et al. Adverse drug reactions to integrase strand transfer inhibitors. AIDS. 2018;32(7):903–12. https://doi.org/10.1097/QAD.0000000000001781.
Lepist EI, Zhang X, Hao J, Huang J, Kosaka A, Birkus G, et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014;86(2):350–7. https://doi.org/10.1038/ki.2014.66.
Eron J, Kalayjian R, Wurapa A, Stephens J, McDonald C, Wilkin A, et al. Safety and efficacy of elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide (E/C/F/TAF) in HIV-infected adults on chronic haemodialysis. HIV Med. 2018;19:S16.
Sax PE, DeJesus E, Crofoot G, Ward D, Benson P, Dretler R, et al. Bictegravir versus dolutegravir, each with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection: a randomised, double-blind, phase 2 trial. Lancet HIV. 2017;4(4):e154–60. https://doi.org/10.1016/S2352-3018(17)30016-4.
Hill AM, Mitchell N, Hughes S, Pozniak AL. Risks of cardiovascular or central nervous system adverse events and immune reconstitution inflammatory syndrome, for dolutegravir versus other antiretrovirals: meta-analysis of randomized trials. Curr Opin HIV AIDS. 2018;13(2):102–11. https://doi.org/10.1097/COH.0000000000000445.
Hayes E, Derrick C, Smalls D, Smith H, Kremer N, Weissman S. Adverse events with Biktarvy: post-marketing study. ID Week. October 2–6, 2019. Washington, DC. Abstract #2489.
Brehm TT, Franz M, Hufner A, Hertling S, Schmiedel S, Degen O, et al. Safety and efficacy of elvitegravir, dolutegravir, and raltegravir in a real-world cohort of treatment-naive and -experienced patients. Medicine (Baltimore). 2019;98(32):e16721. https://doi.org/10.1097/MD.0000000000016721.
Cuzin L, Pugliese P, Katlama C, Bani-Sadr F, Ferry T, Rey D, et al. Integrase strand transfer inhibitors and neuropsychiatric adverse events in a large prospective cohort. J Antimicrob Chemother. 2019;74(3):754–60. https://doi.org/10.1093/jac/dky497.
Hoffmann C, Llibre JM. Neuropsychiatric adverse events with dolutegravir and other integrase strand transfer inhibitors. AIDS Rev. 2019;21(1):4–10. https://doi.org/10.24875/AIDSRev.19000023.
Hoffmann C, Welz T, Sabranski M, Kolb M, Wolf E, Stellbrink HJ, et al. Higher rates of neuropsychiatric adverse events leading to dolutegravir discontinuation in women and older patients. HIV Med. 2017;18(1):56–63. https://doi.org/10.1111/hiv.12468.
Cid-Silva P, Llibre JM, Fernández-Bargiela N, Margusino-Framiñán L, Balboa-Barreiro V, Pernas-Souto B, et al. Clinical experience with the integrase inhibitors dolutegravir and elvitegravir in HIV-infected patients: efficacy, safety and tolerance. Basic Clin Pharmacol Toxicol. 2017;121(5):442–6. https://doi.org/10.1111/bcpt.12828.
Antiretroviral Pregnancy Registry Steering Committee. Antiretroviral Pregnancy Registry interim report for 1 January 1989–31 January 2019. Wilmington, NC: Registry Coordinating Center. 2019. Available at http://apregistry.com/forms/exec-summary.pdf. Accessed 19 Aug 2020.
Centers for Disease Control and Prevention. Update on overall prevalence of major birth defects–Atlanta, Georgia, 1978-2005. MMWR Morb Mortal Wkly Rep. 2008;57(1):1–5.
Sibiude J, Warszawski J, Blanche S, Dialla O, Faye A, Dollfus C, et al. Evaluation of the risk of birth defects among children exposed to raltegravir in utero in the ANRS-French perinatal cohort EPF. Presented at: 9th IAS Conference on HIV Science; July 23-26, 2017; Paris, France. Abstract MOAB0204.
Zash R, Makhema J, Shapiro RL. Neural-tube defects with dolutegravir treatment from the time of conception. N Engl J Med. 2018;379(10):979–81. https://doi.org/10.1056/NEJMc1807653.
Zash R, Holmes L, Diseko M, Jacobson DL, Brummel S, Mayondi G, et al. Neural-tube defects and antiretroviral treatment regimens in Botswana. N Engl J Med. 2019;381(9):827–40. https://doi.org/10.1056/NEJMoa1905230.
Bakal DR, Coelho LE, Luz PM, Clark JL, De BR, Cardoso SW, et al. Obesity following ART initiation is common and influenced by both traditional and HIV-/ART-specific risk factors. J Antimicrob Chemother. 2018;73(8):2177–85. https://doi.org/10.1093/jac/dky145.
Menard A, Meddeb L, Tissot-Dupont H, Ravaux I, Dhiver C, Mokhtari S, et al. Dolutegravir and weight gain: an unexpected bothering side effect? AIDS. 2017;31(10):1499–500. https://doi.org/10.1097/QAD.0000000000001495.
Sax PE, Erlandson KM, Lake JE, McComsey GA, Orkin C, Esser S, et al. Weight gain following initiation of antiretroviral therapy: risk factors in randomized comparative clinical trials. Clin Infect Dis. 2019. https://doi.org/10.1093/cid/ciz999.
Kouanfack C, Mpoudi-Etame M, Bassega PO, Eymard-Duvernay S, Leroy S, Boyer S, et al. Dolutegravir-based or low-dose efavirenz-based regimen for the treatment of HIV-1. N Engl J Med. 2019;381(9):816–26. https://doi.org/10.1056/NEJMoa1904340.
Venter WDF, Moorhouse M, Sokhela S, Fairlie L, Mashabane N, Masenya M, et al. Dolutegravir plus two different prodrugs of tenofovir to treat HIV; Gilead; Macleods; Mylan. N Engl J Med. 2019;381(9):803–15. https://doi.org/10.1056/NEJMoa1902824.
Bourgi K, Rebeiro PF, Turner M, Castilho JL, Hulgan T, Raffanti SP, et al. Greater weight gain in treatment naive persons starting dolutegravir-based antiretroviral therapy. Clin Infect Dis. 2019. https://doi.org/10.1093/cid/ciz407.
Norwood J, Turner M, Bofill C, Rebeiro P, Shepherd B, Bebawy S, et al. Brief report: weight gain in persons with HIV switched from efavirenz-based to integrase strand transfer inhibitor-based regimens. J Acquir Immune Defic Syndr. 2017;76(5):527–31. https://doi.org/10.1097/QAI.0000000000001525.
Landovitz RJ, Zangeneh SZ, Chau G, Grinsztejn B, Eron JJ, Dawood H, et al. Cabotegravir is not associated with weight gain in human immunodeficiency virus-uninfected individuals in HPTN 077. Clin Infect Dis. 2020;70(2):319–22. https://doi.org/10.1093/cid/ciz439.
Landovitz RJ, Donnell D, Clement M, Hanscom B, Cottle L, Coelho L et al. HPTN083 interim results: Pre-exposure prophylaxis (PrEP) containing long-acting injectable cabotegravir (CAB-LA) is safe and highly effective for cisgender men and transgender women who have sex with men (MSM,TGW). 23rd International HIV Conference (AIDS 2020: Virtual), abstract OAXLB0101, 2020.
Yuh B, Tate J, Butt AA, Crothers K, Freiberg M, Leaf D, et al. Weight change after antiretroviral therapy and mortality. Clin Infect Dis. 2015;60(12):1852–9. https://doi.org/10.1093/cid/civ192.
Achhra AC, Mocroft A, Reiss P, Sabin C, Ryom L, de Wit S, et al. Short-term weight gain after antiretroviral therapy initiation and subsequent risk of cardiovascular disease and diabetes: the D:A: D study. HIV Med. 2016;17(4):255–68. https://doi.org/10.1111/hiv.12294.
Herrin M, Tate JP, Akgun KM, Butt AA, Crothers K, Freiberg MS, et al. Weight gain and incident diabetes among HIV-infected veterans initiating antiretroviral therapy compared with uninfected individuals. J Acquir Immune Defic Syndr. 2016;73(2):228–36. https://doi.org/10.1097/QAI.0000000000001071.
Hill A, McCann KM, Pilkington V, Moorhouse MA, Sokhela S, Serenata CM, et al. Risks of metabolic syndrome, diabetes, and cardiovascular disease in ADVANCE Trial. Conference on Retroviruses and Opportunistic Infections (CROI). March 8–11, 2020. Boston. Abstract 81.
Fong PS, Flynn DM, Evans CD, Korthuis PT. Integrase strand transfer inhibitor-associated diabetes mellitus: a case report. Int J STD AIDS. 2017;28(6):626–8. https://doi.org/10.1177/0956462416675107.
McLaughlin M, Walsh S, Galvin S. Dolutegravir-induced hyperglycaemia in a patient living with HIV. J Antimicrob Chemother. 2018;73(1):258–60. https://doi.org/10.1093/jac/dkx365.
Lamorde M, Atwiine M, Owarwo NC, Ddungu A, Laker EO, Mubiru F, et al. Dolutegravir-associated hyperglycaemia in patients with HIV. Lancet HIV. 2020. https://doi.org/10.1016/S2352-3018(20)30042-4.
Bhagwat P, Ofotokun I, McComsey GA, Brown TT, Moser C, Sugar CA et al. Changes in waist circumference in HIV-infected individuals initiating a raltegravir or protease inhibitor regimen: effects of sex and race. Open Forum Infect Dis. 2018;5(11):ofy201. https://doi.org/10.1093/ofid/ofy201.
Bernardino JI, Mocroft A, Wallet C, de Wit S, Katlama C, Reiss P, et al. Body composition and adipokines changes after initial treatment with darunavir-ritonavir plus either raltegravir or tenofovir disoproxil fumarate-emtricitabine: a substudy of the NEAT001/ANRS143 randomised trial. PLoS One. 2019;14(1):e0209911. https://doi.org/10.1371/journal.pone.0209911.
Triumeq® [package insert]. Research Triangle Park, NC. Viiv Healthcare. March 2020.
New York Department of Health AIDS Insitite. Clinical Guidelines Program. When to initiate ART, with protocol for rapid initiation. Available at https://www.hivguidelines.org/antiretroviral-therapy/when-to-start-plus-rapid-start/ Accessed 5 Apr 2020.
Juluca® [package insert]. Research Triangle Park, NC. Viiv Healthcare. October 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
We acknowledge support from the following grants from the National Institutes of Health: 1R01HD085887-01A1 (to KS), 1K23AI134307 (to ATP), RO1 AI124965-01 and UM1AI06701 (to CVF). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interest
KKS, ATP, SNA, CVF declare no conflict of interest related to this article. JPH reports research grants paid to his institution from Gilead Sciences.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Rights and permissions
About this article

Cite this article
Scarsi, K.K., Havens, J.P., Podany, A.T. et al. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 80, 1649–1676 (2020). https://doi.org/10.1007/s40265-020-01379-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01379-9