
Abstract
Cardiovascular toxicity is a potential complication of cancer chemotherapy (CC) that increases the morbidity and mortality of cancer patients. Cardiac arrhythmias have been reported as an adverse effect of many chemotherapeutic drugs, including novel targeted therapies. The relationship between chemotherapy and arrhythmias has not been well-established and the proarrhythmogenic mechanisms remain uncertain as they can be the result of a direct electrophysiological effect or of changes in cardiac structure and function, including myocardial ischaemia and heart failure, which create an arrhythmogenic substrate. In this review we summarise available evidence of proarrhythmia induced by CC, discuss the possible mechanisms involved in this adverse effect and emphasise the importance of cardiac monitoring for the early diagnosis, intervention and surveillance of those patients more susceptible to develop proarrhythmia in an attempt to reduce the morbidity and mortality. Oncologists should be fully aware of proarrhythmia and the close collaboration between cardiologists and oncologists would result in a better cardiovascular assessment, risk stratification, cardiac monitoring and treatment during CC and during the follow-up. The final objective is to understand the mechanisms of proarrhythmia and evaluate its real incidence and clinical relevance so as to select the safest and most effective treatment for cancer patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Several classes of chemotherapy drugs can induce a wide spectrum of cardiac arrhythmias, but this is a poorly recognised and studied aspect of cancer chemotherapy-induced cardiotoxicity. |
Cardiac arrhythmias can be induced by a direct cardiac effect but they can also be initiated/maintained by an arrhythmogenic substrate created by the co-morbidities present in cancer patients or generated by the cardiotoxicity induced by chemotherapy drugs. |
The diagnosis, monitoring and treatment of cardiac arrhythmias and co-morbidities in cancer patients demand a close collaboration between cardiologists and oncologists. |
1 Introduction
In recent years, progress in the early detection of and success of cancer treatment has led to an impressive reduction in both mortality and morbidity [1, 2]. At the same time, however, cardiac complications resulting from cancer chemotherapy (CC) have been recognised as major contributors to morbidity and, ultimately, mortality in cancer survivors [3–7]. Because cancer is an age-related disease, together with the progressive aging of the population, the common occurrence of cardiovascular risk factors and the increased survival after contemporary treatment (almost 60 % of cancer survivors worldwide are older than 65 years) it is progressively likely that cancer, cardiovascular diseases and cardiotoxicity related to the cancer itself or CC may coexist in many patients [2, 6, 8]. This combination represents an important therapeutic challenge and has a significant impact on the overall outcomes of cancer patients, particularly in the elderly and in those with previous cardiovascular diseases [1–8]. Furthermore, in contrast to initial expectations, the risk of cardiotoxicity persists with the novel drugs acting as specific signalling inhibitors (‘targeted therapy’) [1–7], which confirmed that their effects are not specific to the cancer cell but can also exert off-target effects on the heart. CC can produce electrocardiographic (ECG) changes as well as a wide spectrum of both bradyarrhythmias and supraventricular and ventricular tachyarrhythmias. Unfortunately, proarrhythmia, defined as the provocation of a new arrhythmia or the aggravation of a pre-existing one during therapy with a drug at doses or plasma concentrations below those considered to be toxic, has not received the attention it deserves and is still not considered an important part of CC-induced cardiotoxicity. Thus, a better understanding of proarrhythmia induced by CC would lead to a better clinical management and improved patient outcome.
In this article, we review the chemotherapeutic drugs that can produce cardiac arrhythmias, the potential mechanisms involved in their genesis and the risk factors that can increase their incidence. The main chemotherapy drugs are summarised in Electronic Supplementary Material (ESM) Table 1 and the types of cardiac arrhythmias and the possible mechanisms involved in CC-induced cardiac arrhythmias in Tables 1 and 2. The patient characteristics and types of cardiac arrhythmias described in clinical trials and in case reports are presented in ESM Tables 2 and 3, respectively.
2 Methods
We carried out a search of published reports on chemotherapy-induced arrhythmias in English from January 1971 to December 2013 using the Pubmed/MEDLINE (1966–2013) and EMBASE (1980–2012) databases using the following keywords “cardiac arrhythmia”, ‘bradycardia”, “atrio-ventricular block”, “atrial fibrillation”, “QT prolongation”, “ventricular arrhythmias”, “torsades de pointes” and “sudden cardiac death” combined with “chemotherapy”, “associated with chemotherapy” or “chemotherapy-induced”. We also ran searches on typically used chemotherapeutic agents. We excluded publications where cardiac arrhythmias were not properly identified. Two reviewers (ED and RC) independently assessed the eligibility of the articles and abstracts identified by the search, and discrepancies were resolved by consensus. Studies graded as randomised controlled trials, prospective and retrospective studies are summarised in ESM Table 2, while case reports are summarised in ESM Table 3.
3 Chemotherapy-Induced Proarrhythmia
3.1 Alkylating Agents
ECG changes [peaked P waves, low QRS voltage, non-specific ST/T wave changes, prolongation of the corrected QT (QTc) interval] and cardiac arrhythmias, including bradycardia, supraventricular tachycardia (SVT), atrial fibrillation (AF), atrial flutter, premature ventricular beats (PVBs), ventricular tachycardia (VT) and complete atrioventricular (AV) block (AVB) have been reported (ESM Tables 2, 3). Atrial and ventricular tachyarrhythmias appear in 7.9–10 % of patients 24–72 h after the administration of a high dose of cyclophosphamide (HDC), particularly in the context of perimyocarditis and congestive heart failure, but they disappear spontaneously in 1–7 days [9–14]. However, ECG changes and isolated AF may also occur even in the absence of clinical cardiotoxicity. In patients pretreated with other chemotherapeutic agents [CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) therapy] and whole-body irradiation undergoing bone marrow transplantation (BMT), cyclophosphamide produces QT prolongation and cardiac arrhythmias (SVT, paroxysmal AF, PVBs and VT) in 33 % of patients [9, 10]. In 39 patients with breast cancer receiving HDC with BMT, nine developed SVT, two VT [one ventricular fibrillation (VF)] and four AVB [12].
Isolated reports of sinus tachycardia are reported with carmustine (ESM Tables 2, 3). Cytarabine can produce ECG changes (ST segment elevations consistent with pericarditis, persisting at least for a week) and bradycardia, sometimes requiring the discontinuation of drug infusion and the administration of atropine, although in some patients the ECG returns to baseline within 24 h after cessation of the drug [15]. Melphalan administered before BMT produces AF or flutter in 6.6–11 % of patients, PVCs and SVT, but AF is not observed in patients receiving BMT alone [16–19]. Out of 52 patients treated with high doses of ifosfamide and BMT, two patients developed myocardial depression and malignant ventricular arrhythmias [20]. The risk of malignant arrhythmias requiring antiarrhythmic drugs or cardioversion increases at high doses and in patients who developed cardiomyopathy or with chronic kidney disease [3, 4, 20].
3.1.1 Mechanism of Action
Cardiac arrhythmias probably result from direct damage of the cardiomyocytes and ischaemic damage caused by coronary artery vasospasm and vascular coronary endothelial injury followed by intracapillary microthrombi [10]. Heart block may also result from damage of the conduction system secondary to microangiopathy or transient coronary spasms [11, 12]. Some patients develop complete AVB with uncontrolled vomiting, suggesting that increased vagal tone induced by emesis could contribute to the development of AVB. An immune-mediated process or a type of hypersensitivity reaction has also been proposed for cytarabine [10, 21]. Histopathological studies demonstrated an arrhythmogenic substrate characterised by cardiac hypertrophy, fibrosis, interstitial oedema and haemorrhagic myocardial necrosis.
3.1.2 Risk Factors
The risk of proarrhythmia increases with age (>70 years), in females and in patients treated with single high doses followed by BMT, with hypertension, coronary artery disease (CAD), myocarditis, heart failure or chronic kidney disease, and with previous mediastinic irradiation or anthracycline exposure [3–5, 13, 14].
3.2 Cisplatin
Cisplatin produces ECG changes (ST–T changes, T wave inversion) [3, 4], cardiac arrhythmias (bradycardia, left bundle branch block, SVT, AF) and chest pain with elevated cardiac enzymes indicative of myocardial infarction (ESM Tables 2, 3). Intrapericardial and intrapleural cisplatin produces AF (12–32 %) and non-sustained VT (NSVT; 8 %) [5, 22–25]. These events are not dose-dependent and may occur at any time, from hours after the first cisplatin infusion is completed to several months after completion of a cycle. However, bradyarrhythmias are rarely observed [26].
3.2.1 Mechanism of Action
Cardiac arrhythmias may result from coronary vasospasms, direct pericardial irritation, electrolyte disturbances (hypomagnesaemia due to injury of proximal tubular epithelial cells that can potentiate arterial vasospasm), endothelial damage, and vascular fibrosis and thrombosis [3–5].
3.2.2 Risk Factors
The risk factors with cisplatin are similar to those described for other alkylating agents, increasing in patients with hypertension, CAD, left ventricular (LV) dysfunction or heart failure and renal impairment. Because cardiotoxiticy can be worsened by cisplatin-induced electrolyte abnormalities, serum creatinine, blood urea nitrogen, creatinine clearance and electrolytes (magnesium, sodium, potassium and calcium plasma levels) should be monitored prior to initiating therapy, and prior to each subsequent course.
3.3 Antimetabolites
Chest pain, ischaemic-like ECG changes (decrease in QRS amplitude, ST elevation, Q waves, peaked T waves) and elevation of creatinine kinase-MB levels suggestive of myocardial ischaemia occur in up to 18 % of patients treated with high doses of 5-fluorouracil (5-FU) [5, 27–33]. The risk of cardiac ischaemia appears in up to 68 % of patients treated with high-dose infusions of 5-FU [34, 35]. Cardiac arrhythmias may manifest as sinus bradycardia/tachycardia, AF and ventricular tachyarrhythmias [PVCs, polymorphic VT (Torsades de pointes) and sudden cardiac death (SCD)] have been reported [27–33, 36, 37]. The risk of proarrhythmia is similar with 5-FU and capecitabine, an oral prodrug of 5-FU [29]. QTc prolongation with Torsades de pointes has been described with capecitabine plus 5-FU or oxiplatin [38, 39]. Ischaemic-like ECG changes and ventricular arrhythmias appear within 2–5 days and disappear within a few hours to 5 days after drug discontinuation, but recurrences can be observed in 90 % of patients with drug re-exposure, which confirms the proarrhythmic effect of antimetabolites [28, 32, 40, 41]. However, arrhythmias without ischaemic events are rare.
A review of the literature (1969–2007) has identified 23 unspecified arrhythmias in 377 patients (6 %) [31]. Of 644 patients, three present conduction abnormalities and 11 develop tachyarrhythmias (12 sinus tachycardia, two premature atrial beats, one PSVT, five AF, four VPCs and three VT) [37]. In a prospective study in 367 patients, 5-FU induced cardiotoxic events in 28 (8 %), including seven supraventricular arrhythmias and one SCD [27]. After drug discontinuation, four patients experienced SCD. Five of 910 patients (0.55 %) treated with 5-FU presented ST elevation and ventricular arrhythmias consistent with CAD during the 5-day infusion [32]. Four patients developed an acute myocardial infarction and two cardiac arrest. Another study described 36 instances of bradycardia (12 %) and 11 of VT (3.7 %) in 301 patients treated with 5-FU [21]. In another 72 patients, the combination of 5-FU and cisplatin was associated with AF (4–6.5 %), VF (2.6 %) and SCD [36]. Asymptomatic bradycardia persisting for more than 24 h has been observed in six (2.9 %) of 207 patients [42]. Bradycardia requiring pacemaker implantation has been described with cytarabine (ESM Table 3).
3.3.1 Mechanism of Action
Antimetabolite-induced arrhythmias can been explained by multiple mechanisms, including coronary vasospasm due to a direct toxic effect on vascular endothelium involving endothelial nitric oxide synthase and endothelium-independent vasoconstriction via protein kinase C, arteritis and/or thrombosis (due to alteration in the coagulation system and decreased fibrinolytic activity) leading to myocardial ischaemia [1, 3, 31, 37, 43, 44]. The temporal relationship between chest pain and ECG changes and the recurrence of pain with rechallenge supports the role of myocardial ischaemia. Indeed, circulating levels of endothelin-1, a potent vasoconstrictor, increase particularly in patients treated with 5-FU experiencing cardiac events [45]. Other mechanisms include direct cardiac injury, the release of vasoactive compounds, an immune-mediated cardiac damage, the production of fluoroacetate, a degradation cardiotoxic product of 5-FU that depletes cardiac high-energy phosphates and is a potent inhibitor of the tricarboxylic acid cycle, or cytotoxic metabolites of gemcitabine [37, 44]. Cardiac histopathological findings include diffuse interstitial oedema, intracytoplasmatic vacuolation and necrosis of myocytes, fibrosis, pericardial haemorrhage and inflammatory infiltrates compatible with a myocarditis or cardiomyopathy as an arrhythmogenic substrate [3].
3.3.2 Risk Factors
The risk associated with antimetabolites increases among the elderly, at high doses or continuous infusion schedules, and with previous CAD, renal failure, concomitant cisplatin treatment and prior mediastinal radiation which produces small-vessel thrombosis [27, 29, 31, 37]. Thus, ECG and electrolyte balance should be regularly monitored during treatment, especially in patients with CAD or when antimetabolites are associated with cisplatin, because hypomagnesaemia increases the risk of arrhythmias [27]. If proarrhythmia occurs, 5-FU should be immediately discontinued, since prophylaxis or treatment of CAD with nitrates or calcium channel blockers can be ineffective [31, 36, 43].
3.4 Antimicrotubule Drugs
3.4.1 Taxanes
Chest pain indicative of myocardial ischaemia and serious hypersensitivity reactions due to paclitaxel (and its vehicle Cremophor® EL) were observed in phase I trials. Thus, continuous cardiac monitoring was regularly performed which allowed the determination of the incidence of a wide spectrum of arrhythmias. In isolated coronary perfused hearts taxol induces conduction arrhythmias (bradycardia, AVB and asystole), reduces coronary flow and LV systolic pressure, and blocks the vasodilator effect of bradykinin [46, 47]. In dogs, ECG changes progresses through a widening of the QRS to polymorphic PVCs and eventually to VF and death.
The most common arrhythmias reported in patients treated with paclitaxel are asymptomatic sinus bradycardia (0.1–31 %) and first-degree AVB (25 %) [48, 49], although other arrhythmias occur, including left bundle branch block (3 %), AF, SVT, VPCs and NSVT [4, 48–52] (ESM Table 2). In four phase I and II studies involving 140 patients treated with paclitaxel, asymptomatic bradycardia occurred in up to 29 % of patients, and other cardiac disturbances, including a range of AVB, left bundle branch block, VT and manifestations of cardiac ischaemia, occurred in seven patients (5 %) [51]. In the National Cancer Institute database including more than 4,000 patients without significant cardiac risk factors, sinus bradycardia appears in ~30 % of patients, and the incidence of supraventricular and ventricular arrhythmias and heart block is 0.24, 0.26 and 0.11 % of patients, respectively [49]. Arrhythmias appear within the first 24 h after initiation of paclitaxel infusion (range 2.5 h to 6 days) and resolve spontaneously over the next 48–72 h, although in some patients brief episodes of SVT or PVCs can occur even 10 days after drug discontinuation [49, 51]. The close time–effect relationship and the fact that most patients had no cardiac risk factors supports that paclitaxel is the cause of these arrhythmias [51].
3.4.1.1 Mechanism of Action
Paclitaxel (and Cremophor® EL) can induce cardiac arrhythmias by different mechanisms, including the following: (a) the release of histamine. In animal models, stimulation of histamine H1 and H2 receptors produces bradycardia, conduction delay through the AV node and the Purkinje system and atrial and ventricular arrhythmias [48, 51, 52]. H2 receptor stimulation also increases ventricular ectopic automaticity and can induce faster rhythms via re-entry and/or after depolarisations [53, 54]. Moreover, paclitaxel increases myocardial oxygen demands and reduces coronary blood flow, producing an ischaemic arrhythmogenic substrate. (b) The induction of cardiac muscle damage via effects on subcellular organelles [55]. (c) In rat ventricular cardiomyocytes paclitaxel increases the levels of the neuronal Ca2+ sensor 1 (NCS-1) and the binding of NCS-1 to the inositol 1,4,5-trisphosphate (InsP3) receptor, leading to an increase in the frequency of spontaneous cytosolic Ca2+ oscillations that can manifest themselves as PVBs, VT and VF [56].
3.4.1.2 Risk Factors
The risk of bradyarrhythmias increases in patients with a previous history of bradycardia, AVB (Mobitz types I and II and complete heart block), bundle branch block, myocardial infarction or congestive heart failure, those treated with AV blocking drugs (β-blockers, digoxin or calcium antagonists) or with electrolyte disturbances [3, 48, 50, 51]. The combination of docetaxel with thalidomide in solid tumours produces dose-limiting bradycardia and myocardial infarction [57].
Paclitaxel enhances doxorubicin-induced cardiotoxicity by decreasing its metabolism and increasing the cardiomyocyte formation of doxorubicinol, the major metabolite of doxorubicin [58]. Thus, it is recommended that anthracyclines be administered prior to paclitaxel, and that the infusions be separated by 24 h and/or the cumulative doxorubicin dose is limited to 380 mg/m2. This interaction was not observed for epirubicin, so this drug should be selected preferentially when combined with taxanes [49]. Additionally, Cremophor® EL lowers the renal and hepatic clearance of anthracyclines, resulting in their increased plasma concentrations [55, 59].
Cardiac monitoring is not generally recommended in asymptomatic patients and in those without risk factors [49, 51]. However, in patients with symptomatic bradycardia or progressive conduction disturbances, continuous ECG monitoring is recommended, drug discontinuation should be considered and some patients may require a pacemaker implantation depending on the severity of symptoms, haemodynamic consequences and the need to continue paclitaxel therapy [4, 50]. Pretreatment with antihistamines and corticosteroids prevents the release of histamine and reduces the risk of bradycardia.
3.4.2 Vinca Alkaloids
Vinca alkaloids produce chest pain and ECG changes (T wave inversions, ST changes) associated with myocardial ischaemia, PVCs and AF that rapidly reverses within 10 days [60]. In an uncontrolled open-label study, eribulin mesylate prolongs the QTc using the Fridericia’s formula (QTcF) [61], but no patients had a QTcF interval >450 ms or a change in QTcF >30 ms over baseline values [62].
3.4.2.1 Mechanism of Action
The mechanism of cardiotoxicity is unknown but may include changes in pre-existing atherosclerotic coronary vessels or coronary artery spasm [60, 63], an increase in cardiac sensitivity to hypoxia and an inhibitory effect on the vagal chronotropic control of the heart [64].
3.4.2.2 Risk Factors
The risk of cardiac arrhythmias increases in women, in patients with a previous history of CAD or hypertension, or who have had previous mediastinal radiation therapy [1, 4, 65].
3.5 Anthracyclines
Anthracyclines are effective chemotherapeutic agents, but their usefulness is limited by potentially fatal dose-dependent cardiotoxicity [66, 67]. Anthracycline-induced cardiotoxicity can be developed in an acute, subacute, or chronic manner. Acute cardiotoxicity manifests itself as arrhythmias, a pericarditis–myocarditis syndrome or acute heart failure occurring during or immediately after a single dose or a course of anthracycline therapy. ECG changes (e.g. non-specific ST/T and T wave changes, decreased QRS voltage, T wave flattening and QTc prolongation) are present in 6–38.4 % of the patients, and transient arrhythmias (supraventricular and ventricular tachycardias, AVB) are seen in 0.5–3 % of the patients [5, 55, 66–72] (ESM Tables 2, 3). PVCs are the most common arrhythmias after the administration of doxorubicin (they appear in 3 and 24 % of patients 1 and 24 h post-infusion, respectively) but up to 6 % of the patients can develop NSVT 24 h post-infusion [69, 71]. These arrhythmias appear during and shortly after drug administration, are usually reversible, are not dose dependent and do not preclude further anthracycline use [4, 55, 68, 72]. However, there are reports of QTc prolongation followed by Torsades de pointes and SCD in patients with hypokalaemia and life-threatening arrhythmias (VT/VF) causing cardiac arrest been reported in the first 24 h after drug administration [3, 4, 55, 70, 72–76]. Isolated cases of asymptomatic bradycardia and advanced AVB leading to syncopal episodes and ventricular asystole requiring pacemaker implantation have been described with doxorubicin and epirubicin, while mitoxantrone can produce sinus bradycardia (ESM Tables 2, 3). Nevertheless, AF appears to be a rather common complication of anthracyclines and in one study it was described in 10.3 % of the patients during the first course of doxorubicin-based chemotherapy [69].
Subacute cardiomyopathy clinically resembling a myocarditis generally occurs within a few weeks to months after the last dose of anthracyclines and is accompanied by diastolic dysfunction and severe dysrrhythmias (VT, VF and SCD) [69, 77, 78]. Chronic anthracycline cardiotoxicity associated with serious ventricular arrhythmias, including VT, VF and SCD, can appear many years after chemotherapy has been completed in symptomatic or asymptomatic patients with late cardiomyopathy [69, 77–79]. In one study, nine of 201 patients followed throughout 4–20 years presented late symptoms, including cardiac failure and dysrhythmia, and three patients died suddenly [79]. QT prolongation can be a delayed effect of anthracyclines that may precede development of subacute cardiac failure [80].
3.5.1 Mechanism of Action
Several mechanisms have been proposed to explain the anthracycline-induced cardiac arrhythmias [1, 4, 72, 73, 81–84]. (a) Direct electrophysiological effects. In isolated cardiomyocytes, doxorubicin produces proarrhythmic effects that are prevented with β-blockers [85]. In ventricular myocytes isolated from cardiomyopathic rats treated long-term with the drug, doxorubicin increases the amplitude of L-type Ca2+ currents [86, 87] and in canine Purkinje fibres it decreases the amplitude and prolongs the duration of the cardiac action potential. This latter effect was attributed to the direct inhibition of the transient and the delayed outward currents and the inhibition of Na+–Ca2+ exchange [88, 89]. In rats, doxorubicin produces a widening of the QRS complex and QT prolongation and in mice it induces PVCs, VT and fatal arrhythmias [90]. (b) Generation and accumulation of reactive oxygen species (ROS) (superoxide anions) via the formation of iron–anthracycline complexes, the redox cycling of the quinone and semiquinone moieties of doxorubicin or downstream effects of topoisomerase IIβ inhibition. ROS can activate MAPK (mitogen-activated protein kinase), SAPK (stress-activated protein kinase) and caspase 9 activity that induce myocyte apoptosis [81]. Moreover, anthracyclines increase cardiomyocyte susceptibly to ROS by reducing cardiac production of protective endogenous antioxidants (glutathione peroxidase, superoxide dismutase and catalase) [73]. Furthermore, accumulation of ROS causes severe lipid peroxidation and membrane damage. (c) Doxorubicin and ROS increase the release of Ca2+ from intracellular stores and decrease the expression of sarcoplasmic reticulum calcium–ATPase, resulting in cytosolic Ca2+ overload, contractile dysfunction and myocardial necrosis [72]. (d) Decrease of adenosine monophosphate (AMP)-activated protein kinase (AMPK) expression, which results in mitochondrial dysfunction, adenosine triphosphatase (ATP) depletion, ROS formation, release of cytochrome c from mitochondria into cytoplasm, activation of caspases and myocyte apoptosis [81]. (e) Impairment of prosurvival signalling pathways via the inhibition of the neuroregulin/ErbB signalling pathway. (f) Cardiac accumulation of doxorubicinol, a toxic metabolite of doxorubicin, which inhibits ion-dependent pumps (calcium and sodium ion exchange pumps) in the mitochondria and the sarcolemma, leading to ROS production [91, 92]. Other possible mechanisms include the release of vasoactive mediators [histamine and tumour necrosis factor (TNF)-α from macrophages, interleukin-2 from monocytes], autonomic imbalance secondary to adverse events (i.e. nausea, vomiting) and allergic reactions [31, 69].
Histopathological cardiac changes in endomyocardial biopsies demonstrate sarcoplasmic reticulum dilation, cytoplasmic vacuolisation, fibrosis, mitochondrial swelling or disruption, acute inflammation of the pericardium, myofibrolysis and necrosis [3, 65, 77].
3.5.2 Risk Factors
The risk factors relating to anthracyclines include extremes of age (<15 and >65 years), female sex, total cumulative doses (doxorubicin >400 mg/m2; epirubicin >450–600 mg/m2), pre-existing cardiovascular diseases (CAD, hypertension, diabetes mellitus), hypokalaemia, liver/renal impairment, concomitant use of CC (cyclophosphamide, taxanes, trastuzumab, bleomycin, mytomicin, vincristine, amsacrine, mitoxantrone) and prior mediastinal irradiation [2, 77, 93].
In patients with non-Hodgkin’s lymphoma, genetic factors possibly associated with anthracycline-induced cardiotoxicity include variants in genes encoding doxorubicin transport systems [multidrug resistance proteins (MRP) 1 and 2, human concentrative nucleoside transporter (hCNT3), adenosine triphosphate–binding cassette transporters (ABCB1, ABCB4 and ABCC1), enterohepatic reuptake (SLC10A2)], three subunits (NCF4, p22phox and RAC2) of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase involved in ROS formation and glutathione-S-transferase P (GSTP1), a phase II detoxification enzyme [94–96].
Measures to reduce or prevent cardiac arrhythmias include identification and treatment of cardiovascular risk factors before anthracycline treatment, ECG monitoring before, during and a long time after completion of anthracycline treatment, aggressive treatment of anthracycline-induced cardiomyopathy, avoidance of the coadministration of doxorubicin with other cardiotoxic drugs (i.e. introducing a drug-free interval between the two agents), administration of cardioprotective drugs (dexrazoxane) or administration of less cardiotoxic anthracyclines (epirubicin, idarubicin, mitoxantrone) in high-risk patients, changes of dosage schedules and new doxorubicin formulations [2, 59, 67, 69, 77, 78, 97]. Dexrazoxane is an iron-chelating agent that prevents the generation of anthracycline–iron complexes and ROS, decreasing the risk of heart failure without affecting overall survival; this benefit is not observed with other cardioprotective agents [97]. Whether dexrazoxane interferes with the anti-tumour effects of anthracyclines and its potential for an increase in secondary leukaemias remains controversial.
Because cardiotoxicity depends on peak plasma drug concentrations, while antineoplastic activity depends on total systemic exposure over time or tissue concentration over time, doxorubicin is less cardiotoxic when administered as a prolonged intravenous infusion over 48–96 h or as lower weekly injections instead of a bolus injection every 3 weeks [3, 4, 65–67, 72, 93, 97–99]. Liposome-encapsulated or pegylated doxorubicin reduces cardiotoxicity, while providing comparable anti-tumour efficacy [100, 101]. Finally, since anthracycline-induced arrhythmias can occur many years after the treatment, survivors of malignancies treated with anthracyclines should undergo ambulatory ECG monitoring as part of their follow-up to detect potentially life-threatening arrhythmias even if asymptomatic [78].
3.6 Arsenic Trioxide
In patients with refractory or relapsed acute promyelocytic leukaemia arsenic trioxide (ATO) produces ECG changes (QTc prolongation, QRS widening, ST/T changes, T wave inversion/flattening), bradyarrhythmias (sinus bradycardia, AVB requiring pacemaker insertion) [102–105] and life-threatening ventricular tachyarrythmias, including Torsades de pointes and SCD within the first 24 h after drug administration (ESM Table 2). In two trials, six out of 30 patients died suddenly during the first cycle of treatment [106, 107]. ATO prolongs the QT interval in 26–93 % of patients and life-threatening ventricular tachyarrythmias are reported in up to 30 % of patients, including accelerated idioventricular rhythm and Torsades de pointes [4, 8, 102–105, 108]. QT prolongation is observed 1–5 weeks after drug infusion, and return towards baseline by the end of 8 weeks, i.e. before the second course [104, 105].
In 99 patients enrolled in phase I or II trials, ATO prolonged the QTc interval by a mean of 47 ms in 36.6 % of the treatment courses and by >60 ms from baseline in 35.4 % of the patients; 26 patients presented QTc intervals ≥500 ms [104]. In another 40 patients, ATO produced bradycardia in 24 (55 %), QTc prolongation in 16 (40 %) and one patient presented a Torsades de pointes [105]. In another study, in 13 of 14 patients ATO produced QTc prolongation (13 ms) and arrhythmias (two paroxysmal SVT, eight VPCs, four NSVT) [103]. During the second course of therapy, nine patients developed PVCs or NSVT and four required antiarrhythmic treatment. In six of 18 patients in a further study, ATO prolonged the QTc interval and three of the patients developed VT [108].
3.6.1 Mechanism of Action
ATO increases cardiac Ca2+ currents and inhibits several potassium currents, including the rapid (I Kr) and slow (I Ks) components of the delayed rectifier K+ current and the ATP-sensitive current (I KATP) [5, 109]. Additionally, ATO reduces surface expression of human ether-á-go-go-related potassium channels (hERG, Kv11.1, KCNH2) by inhibiting the formation of channel–chaperone (Hsp70/90) complexes and channel trafficking from the endoplasmic reticulum and Golgi complex to the cell surface [110]. As a consequence, ATO prolongs ventricular action potential duration [111] and the QT interval in the surface ECG [105, 110, 112].
3.6.2 Risk Factors
Possible risk factors for QT prolongation include female sex, old age, bradycardia, electrolyte abnormalities (hypokalaemia), pre-existing heart disease, congenital long QT syndrome, concomitant use of QT-prolonging drugs, high cumulative anthracycline dose, and a history of cardiac ischaemia and myocardial infarction [3, 4].
Because ATO-induced ventricular arrhythmias are often resistant to chemical and electrical cardioversion, the US FDA safety guidelines for QT interval monitoring with ATO administration recommend that all patients must have a baseline QTc <500 ms, close ECG monitoring (1–2 times per week in stable patients; more often if needed) for early recognition of the arrhythmia, and must maintain serum potassium >4 mEq/L and magnesium >1.8 mg/dL during the treatment. Patients with a QTc ≥500 ms or symptoms (palpitations or syncope) should be immediately hospitalised, electrolyte abnormalities corrected and the ECG closely monitored. ATO may be temporarily discontinued until the QT interval regresses to <460 ms, electrolyte abnormalities are corrected and irregular heartbeats cease [103–105, 108].
3.7 Cytokines
3.7.1 Interferons
Reversible arrhythmias, including AVB, atrial flutter, AF, atrial premature beats (APBs), ventricular premature beats (VPBs), NSVT and VF have been reported in up to 20 % of patients treated with interferon-α (ESM Table 2). In a phase I trial that enrolled 15 patients, one died in VF while experiencing chest pain after receiving 5 days of treatment with recombinant interferon-α-2a [113]. In another study, five patients (20 %) treated with recombinant interferon-α-2a presented cardiac arrhythmias that required treatment [114]. Among 18 patients with normal basal ECGs treated with interferon-γ, eight presented complex cardiac ectopy and seven NSVT [115]. In a review of cardiotoxicity in patients treated with all types of interferons, supraventricular and ventricular arrhythmias were reported in 25 patients, most of them having underlying heart disease [116]. Cardiac arrhythmias are usually reversible following cessation of drug therapy.
3.7.1.1 Mechanism of Action
Cardiotoxicity has been related to coronary vasospasm, an autoimmune or inflammatory reaction and/or a decrease in the ATP levels [116]. Cases of cardiomyopathy have been observed on rare occasions in patients treated with interferon-α.
3.7.1.2 Risk Factors
In a review of cardiotoxicity induced with all types of interferons, most cardiac arrhythmias were reported in patients with underlying heart disease [116].
3.7.2 Interleukin-2
Interleukin (IL)-2 induces supraventricular tachyarrhythmias (9.7–17 %), AF (5–8 %) and life-threatening VTs (<1 %) (ESM Table 2). Arrhythmias appear within 2–8 h and reverse rapidly after drug discontinuation [4, 117]. In three phase II trials enrolling patients treated with high-dose IL-2 and lymphokine-activated killer cells, 20 patients (21.5 %) developed SVT, AF, PVCs and one VT requiring cardioversion [118]; additionally, four patients presented transient cardiac ischaemia. In an analysis of 270 patients with metastatic melanoma, SVT appeared in 17 % of patients, but life-threatening VTs occurred in <1 % of patients [119]. In 317 patients treated with 423 courses of IL-2 therapy, 8 % of the courses were associated with AF, 1.7 % with a prolonged atrial arrhythmia and hypotension, and 0.2 % with NSVT; in general, ventricular and supraventricular arrhythmias occur in 14–21 % of the patients [117]. Among 199 patients who received 310 courses of treatment, arrhythmias occurred during 6 % of the courses, including one VT [120].
3.7.2.1 Mechanism of Action
A direct myocardial toxicity possibly related to coronary vasospasm and myocarditis has been proposed as the mechanism of action of IL-2 [3, 4]. IL-2 administration often causes a capillary leak syndrome characterised by enhanced capillary permeability, hypovolaemia, peripheral vasodilation and hypotension, leading to a reflex increase in heart rate [3, 4]. This syndrome is associated to supraventricular and ventricular arrhythmias in up to 10 % of patients [117].
3.7.2.2 Risk Factors
Patients with cardiac or pulmonary disease may be at greater risk during IL-2 administration and should not be selected to undergo this treatment. Myocardial ischaemia can be aggravated by IL-2 [3, 4].
3.8 Histone Deacetylase Inhibitors
ECG changes (marked QTc prolongation, ST/T wave flattening, T wave inversion), SVT, AF and ventricular tachyarrhythmias, including Torsades de pointes and unexpected deaths, have been reported with romidepsin [8, 121–127]. Among 42 patients with T cell lymphoma, romidepsin (depsipeptide) produced SVT in 14 (38 %), VT in five (14 %), and APBs and VPBs were observed in 24 (65 %) and 14 (38 %) patients, respectively; QTc interval values >450 ms were detected in 28 patients and QTc intervals >500 ms in four patients [123]. In 37 patients with refractory solid tumours, romidepsin produced three asymptomatic arrhythmic events, one atrial bigeminy, one 3 s sinus pause and an asymptomatic five-beat run of VT [122]. Both QTc prolongation and arrhythmias usually disappeared before the subsequent cycle [122, 123]. However, in 15 patients with metastatic neuroendocrine tumours mostly pretreated with QT-prolonging drugs (octreotide and ondansetron), romidepsin was suspended due to serious cardiac adverse events, including one SCD possibly related to VF, two episodes of asymptomatic VT, and three instances of QT prolongation (grade 2) [124]. Moreover, among more than 500 individuals treated with romidepsin, six unexpected deaths were reported [121]. QTcF prolongation (>470 ms) was reported in 10 % of the patients treated with dacinostat, 6.3–28 % treated with panobinostat and 3.5–6 % treated with vorinostat [122] (ESM Table 2). In clinical studies in patients with cutaneous T cell lymphoma (CTCL), three of 86 CTCL patients exposed to 400 mg once daily exhibit grade 1 (>450–470 ms) or 2 (>470–500 ms or increase of >60 ms above baseline) QTc prolongation. In a retrospective analysis of three phase I and two phase II studies recruiting 116 patients, four presented grade 2 and one grade 3 QTc prolongation [128] and three in 86 CTCL patients presented a QTc prolongation >450–4,500 ms (ESM Table 2).
3.8.1 Mechanism of Action
Histone deacetylase (HDAC) inhibitors with different chemical structures block hERG channels, suggesting that QT prolongation is a class effect related to their underlying mechanism of action. In dogs and rats, depsipeptide prolongs the QT interval, increases cardiac enzymes, and induces myocardial inflammatory changes and epicardial and endocardial haemorrhages [129]. HDAC inhibitors alter gene expression and modulate cell cycle arrest and apoptosis. Histopathologic findings include chronic inflammatory changes in the myocardium or epicardial and endocardial haemorrhage of the right atrium or left ventricle [125].
3.8.2 Risk Factors
QT prolongation increases with the dose, rate and duration of drug infusion (a 4 h infusion is better tolerated than a 10 min bolus) in patients treated with QT-prolonging drugs or with QT-related risk factors (see Sect. 4.4).
3.9 Monoclonal Antibodies
Rituximab produces AF, PVCs and VT reversible upon drug discontinuation [130, 131]. In 197 patients, the CHOP–rituximab group had a higher incidence of SVT and grade 1 events (24 vs. 13 %) than the CHOP group, although the incidence of grade 3–4 cardiac toxicity was similar in both groups [130]. Ten of 131 patients with mantle-cell lymphoma developed bradycardia, AF, PVCs and VT during or immediately after the rituximab infusion [132]. Interestingly, trastuzumab-related cardiotoxicity is not dose dependent but is reversible upon drug discontinuation. Isolated reports of proarrhythmia have been described with alemtuzumab and trastuzumab [133] (ESM Table 3).
3.9.1 Mechanism of Action
The cardiotoxicity produced by trastuzumab in clinical practice was an unexpected finding since in preclinical safety studies there was no evidence of acute or multiple dose-related cardiotoxicity [134]. Several mechanisms have been proposed, including induction of immune-mediated destruction of cardiomyocytes, inhibition of NRG-1/Erb signalling and a cytokine release syndrome. When the heart is exposed to oxidative stress or anthracyclines, the ErbB2 protein activates a protective intracellular signalling pathway (NRG-1/ErbB2). Consistently, inhibition of NRG-1/ErbB2 leads to cardiomyocyte apoptosis and mice deficient in the erbB2 gene develop dilated cardiomyopathy and present an increased susceptibility to oxidative stress and anthracycline toxicity [135, 136]. Thus, inhibition of the NRG-1/ErbB2 signalling seems to play a central role in the mechanism of trastuzumab-induced cardiotoxicity [133]. Alemtuzumab-induced arrhythmias in T cell malignancies can be attributed to a cytokine release syndrome associated with an increase in the plasma levels of TNF-α, interferon-γ and IL-6, which can lead to coronary vasospasm, myocarditis and myocardium “stunning” [132, 137].
3.9.2 Risk Factors
Advanced age (>60 years), prior hypertension, previous chest radiation or LV dysfunction increase the risk of trastuzumab-associated cardiotoxicity [138]. The concomitant use of trastuzumab with anthracyclines (doxorubicin) markedly increases the risk of cardiotoxicity (from 4–10 % in monotherapy to 27 % in combination); this increased risk is substantially ameliorated by introducing a drug-free interval between the two agents [32, 138].
3.10 Proteasome Inhibitors
Bortezomib is a proteasome inhibitor for the treatment of patients with multiple myeloma. Four out of 69 patients (5.8 %) with multiple myeloma treated with bortezomib developed bradycardia, AF and AVB leading to pacemaker implantation [139]. In other 63 patients, bortezomib produced one SVT and one complete AVB [140]. Isolated case reports of bradycardia and complete AVB are summarised in ESM Table 1. However, in a review of 25 clinical trials involving 5,718 patients bortezomib did not increase the risk of proarrhythmia compared with control patients [141]. Deaths due to cardiac arrest occurred within 1 day of carfilzomib administration [142].
3.10.1 Mechanism of Action
Bortezomib increases the apoptosis of smooth muscle cells, leading to atherosclerotic plaque instability and rupture by a weakening of the fibrous cap and an enlargement of the necrotic pore resulting in ischaemic complications. Furthermore, it inhibits myocardial preconditioning leading to ischaemic complications and it causes mitochondrial abnormalities resulting in decreased ATP synthesis and cardiac contractility [139, 141].
3.10.2 Risk Factors
Patients with risk factors for or existing heart disease should be closely monitored when prescribing bortezomib. The overall incidence of all-grade and high-grade cardiotoxicity with bortezomib among multiple myeloma patients was higher than that of non-multiple myeloma patients. Additionally, the incidence of all-grade and high-grade cardiotoxicity was higher in bortezomib monotherapy than with bortezomib combination therapy, which suggested that concurrent drugs with bortezomib might not increase the incidence of cardiotoxicity [141].
3.11 Thalidomide
In patients with multiple myeloma treated with thalidomide, alone or plus dexamethasone, mild sinus bradycardia occurs in up to 55 % of the patients and severe sinus bradycardia in only 1–3 % (ESM Table 1) [143–148]. However, in a post-marketing surveillance study carried out among 10,450 patients with neoplastic/non-neoplastic diseases, bradycardia appeared in 0.12 % of the patients [149]. Bradycardia recovers by decreasing the dose or after drug discontinuation, suggesting a reversible effect on sinus node function, but it often requires a pacemaker implantation.
Among 668 patients with multiple myeloma pretreated with intensive melphalan-based chemotherapy and autologous haematopoietic stem-cell transplantation, 12 % of the patients randomised to thalidomide developed symptomatic bradycardia and syncopal episodes, so that a cardiac pacemaker needed to be implanted in nearly a third of them [143]. In another study, 52 of 96 patients developed bradycardia; eight patients presented a heart rate of 30 bpm and five required pacemaker implantation [144]. However, no changes in the PR interval, QRS duration, and QTc intervals were observed over the 12 months of follow-up.
3.11.1 Mechanism of Action
The underlying mechanism of thalidomide-induced bradycardia remains unclear. It has been hypothesised that thalidomide inhibits TNF-α expression and activity, which causes a rapid and complete inhibition of the dorsal motor neurons of the nucleus of the vagus nerve, leading to an over-reactivity of the parasympathetic nervous system resulting in bradycardia and conduction disturbances [144]. Bradycardia has also been related to thalidomide-induced hypothyroidism [4, 144, 146].
3.11.2 Risk Factors
The risk of bradycardia increases in the elderly and in patients with co-morbidities or when thalidomide is coadministered with bradycardic agents (β-blockers, calcium channel blockers, digoxin, antiarrhythmic drugs), doxorubicin or cyclophosphamide, or following radiotherapy for breast cancer [146]. Therefore, patients should be monitored closely for signs and symptoms (fatigue, limitations in physically activity, syncope, lightheadedness or dizziness) of bradycardia during the administration of thalidomide and a thyroid-stimulating hormone level should be obtained to rule out hypothyroidism.
3.12 Topoisomerase Inhibitors
Amsacrine prolongs the QTc interval and produces non-specific ST/T wave changes without changes in RR, QRS and PR intervals, bradycardia, atrial tachycardias, AF (when combined with cisplatin), ventricular tachyarrhythmias (VPCs, VF) and SCD [150–155]. ECG changes and arrhythmias occur within minutes to hours after the first dose or with the first course of treatment and disappear within 24 h after drug discontinuation. Sixty-five arrhythmias were described in 5,340 patients treated with amsacrine, including 34 VT/VF (14 of these patients died during or within 4 h after drug infusion) [98]. Interestingly, 37 % of patients with serious cardiac disturbances presented hypokalaemia.
3.12.1 Mechanism of Action
The reports of QT interval prolongation, ventricular arrhythmia and death associated with amsacrine suggest a drug effect on cardiac repolarisation. Indeed, it blocks cardiac hERG potassium channels predominantly in the open and inactivated states [156].
3.12.2 Risk Factors
The risk of proarrhythmia increases in patients with electrolyte abnormalities (hypokalaemia), CAD or pretreated with anthracyclines [98, 151]. Thus, serum potassium should be monitored during amsacrine infusion. Factors that prolong the QT interval (hypokalaemia, ischaemia, QT-prolonging drugs) should be avoided in patients treated with amsacrine.
3.13 Tyrosine Kinase Inhibitors
The introduction of new drugs that inhibit the activity of protein kinases that are mutated and/or overexpressed in cancer has revolutionised the treatment of some cancers and significantly improved survival rates in many others [157, 158]. It was thought that these targeted therapies might exert a high efficacy on cancer cells with minimal adverse effects but tyrosine kinase inhibitors (TKIs) produce important cardiotoxicity, including acute coronary syndromes, systemic hypertension, LV dysfunction, chronic heart failure and cardiac arrhythmias. This can be explained because their effects are not specific to the cancer cells, but can also exert off-target effects on heart cells (myocytes, fibroblasts, endothelial and vascular smooth cells) that share the same signalling pathways. Thus, protein kinase inhibitor (PKI)-induced cardiotoxicity is directly related to non-selective inhibition of tyrosine kinases not intended to be inhibited by a drug [159]. Indeed, only a few PKIs (axitinig, erlotinib, gefitinib, ruxolitinib, vemurafenib) are selective enough for one or two protein kinases, whereas some PKIs present a lack of selectivity (sorafenib and sunitinib can inhibit in vitro between 15 and 90 kinases) [160].
Several PKIs (crizotinib, dasatinib, gefitinib, nilotinib, sorafenib, sunitinib, vandetanib, vemurafenib) inhibit hERG channels and prolong the ventricular action potential duration and the QTc interval [134, 160]. However, some discrepancies were observed with lapatinib and nilotinib, as these drugs prolong the ventricular action potential, but have no effect on the QTc interval in monkeys or dogs [160]. Overall, the effect of the PKIs that prolong the QTc interval is relatively mild (95 % upper bound around their mean effects being ~15 ms), except for sunitinib, lapatinib, nilotinib and vandetanib (95 % upper bound around their mean effects being 22.4, 23.4, 25.8 and 36.4 ms, respectively) [4, 6, 8, 160, 161].
In the FDA database of >5,000 sunitinib-exposed patients with baseline and on-treatment ECG monitoring, a QTc prolongation >500 ms was observed in <2.3 % of patients and Torsades de pointes in <0.1 % of patients; similar results were observed with dasatinib, nilotinib and sunitinib [4, 8]. In 911 patients, only 11 (2 %) presented cardiac arrhythmias [162]. In four studies involving 445 patients with chronic myeloid leukaemia treated with dasatinib, only three patients (0.7 %) presented a QTcF >500 ms, but no Torsades de pointes was reported [163]. Among 519 patients with chronic-phase chronic myeloid leukemia, six patients in the dasatinib group (2 %) and nine in the imatinib group (3.5 %) presented QTc intervals between 450 and 500 ms, but only one patient in each group (0.4 %) presented a QTc interval >500 ms [164]. In a meta-analysis of nine trials with 2,188 patients, the overall incidence of all-grade and high-grade QTc interval prolongation with vandetanib was 16.4 and 3.7 %, respectively, among patients with non-thyroid cancer, and 18.0 and 12.0 %, respectively, among patients with thyroid cancer [165]. Patients with thyroid cancer who have longer treatment duration also have a higher incidence of high-grade events.
In patients with CML and imatinib resistance or intolerance, nilotinib prolongs the QTcF interval by 5–15 ms but only 1–1.2 % presented a QTcF >500 ms and no Torsades de pointes or SCD were reported [166–168]. In another group of 119 patients with CML, nilotinib prolonged the QTcF interval from baseline by >60 ms in five patients [169]. Thirteen of 81 patients (16 %) treated with lapatinib presented either a QTc interval >480 ms or an increase in QTc interval >60 ms from baseline [4], and in 449 patients treated with ponatinib symptomatic bradyarrhythmias requiring a pacemaker implantation occurred in three, SVT in five and AF in 20 patients; in 13 of these patients the arrhythmia led to the hospitalisation of the patient [170]. QTc prolongation (>500 ms) was identified in 2 % (11/558) of patients and Torsades de pointes occurred in <1 % (2/977) of patients treated with pazopanib (ESM Table 2).
Case reports of AF have been described with sorafenib and sunitinib [171–174] and case reports of bradyarrhythmias and AVB have been described with imatinib, ponatinib, soratinib and sunitinib (ESM Table 3) [170, 173–175]. Sorafenib can also induce acute coronary syndromes, including myocardial infarction, in 2.9 % of patients [176]. Mild (grade 1: 50–54 bpm) and moderate (grade 2: 45–49 bpm) sinus bradycardia occurred in 5 % of the patients treated with crizotinib [177], although in a retrospective analysis 69 % of treated patients experienced sinus bradycardia with a mean decrease in heart rate of 26 bpm [178].
3.13.1 Mechanism of Action
Preclinical studies indicate that several TKIs (sunitinib and its active metabolite SU012662) block HERG K+ channels conducting the I Kr and prolong the ventricular action potential and the QT interval [8, 160, 161]. However, in contrast to other drugs that produce a direct blockade of hERG channels, PKIs induce channel closure via the inhibition of serine/tyrosine phosphorylation [179]. Interestingly, the concentration of dasatinib that inhibits the hERG current in vitro is 150 times higher than the human peak plasma concentration of the drug, suggesting that the mechanism underlying QT interval prolongation must be independent of hERG inhibition. Indeed, recent evidence suggests that TKIs can prolong the QTc interval through the inhibition of the phosphoinositide 3-kinase (PI3K) signalling pathway, which affects multiple cardiac ion currents. In fact, PKIs decrease the I Kr and I Ks, the L-type Ca2+ current and the peak Na+ current, while they increase the late Na+ current [180, 181]. These results indicate that down-regulation of PI3K signalling directly or indirectly via tyrosine kinase inhibition prolongs the QT interval by affecting multiple ion channels.
Other possible molecular mechanisms of proarrhythmia include the following [1, 8, 83, 134, 157, 160]: (1) Sunitinib, through the inhibition of ribosomal S6 kinase could lead to the release of the proapoptotic factor bcl2 and of cytochrome c into the cytosol, which can activate the mitochondrial pathway for cell death or apoptosis. Sunitinib also inactivates AMPK, which is crucial in the response to hypoxia and cardiomyocyte survival, leading to mitochondrial dysfunction, ATP depletion, reduced myocardial protection against stress-related injury and activation of the proapoptotic factor Bcl2. Conversely, lapatinib activates AMPK and ATP production. (2) Inhibition of the antiapoptotic Bcr-abl tyrosine kinase. (3) Inhibition of platelet-derived growth factor (PDGF) receptor, which reduces myocardial protection against stress-related injury. (4) Inhibition of hypoxia-inducible factor 1α and vascular endothelial growth factor expression leading to vasoconstriction and microvascular rarefaction, LV hypertrophy and dilatation, and contractile dysfunction. (5) Sunitinib-inhibition of cardiac RAF1 and BRAF kinase activity may disrupt the pro-survival ERK (or MAPK) cascade, which plays an important role in cardiomyocyte survival under stress conditions; in fact, conditional cardiac-specific deletion of RAF-1 results in LV dilatation and decreases cardiac contractility. (6) Imatinib increases the expression of protein kinase Cδ, which has cardiac proapoptotic effects.
Histopathological examination from endomyocardial biopsies from imatinib-, sorafenib- and sunitinib-treated patients show myocardial hypertrophy and mitochondrial abnormalities (swollen mitochondria, membrane whorls, effaced cristae) [172].
3.13.2 Risk Factors
The risk of arrhythmias increases in women, patients with a previous history of CAD, hypertension, LV dysfunction, heart failure or diabetes, and in those pretreated with anthracyclines. Crizotinib, dasatinib, lapatinib, nilotinib, pazopanib, sorafenib, sunitinib, vandetanib and vemurafenib should be administered with caution in patients with pre-existing QT prolongation or QT-related risk factors (see Sect. 4.4) [182].
4 Discussion
There is clinical evidence that an increasing number of chemotherapy drugs can produce electrophysiological changes (QTc prolongation) and a wide spectrum of arrhythmias, including bradyarrhythmias, supraventricular and ventricular arrhythmias that can lead to life-threatening VTs and SCD (Table 1). The risk of proarrhythmia persists even with the novel ‘targeted’ drugs acting as specific signalling inhibitors (monoclonal antibodies, PKIs), which confirm that they do no exert a specific effect on the cancer cells but can also exert off-target effects on the heart. The complex pharmacological profile of chemotherapy drugs frequently associated with cardiac arrhythmias, the identification of patients who are at increased risk of proarrhythmic events and the increasing number of co-morbidities that, in many cases, represent an arrhythmogenic substrate in the elderly population demand a close collaboration between cardiologists and oncologists to allow continuously effective and integrated patient care. The onco-cardio/cardio-oncologist team must perform a judicious balance between risk of proarrhythmia and the benefits of CC, as an excessive concern regarding potential arrhythmias may compromise the administration of highly beneficial anticancer therapies, while under-appreciation of proarrhythmia may worsen the outcome of patients who have been cured of their cancer.
4.1 Incidence of Cardiac Arrhythmias
The real incidence of CC-induced arrhythmias is uncertain and can be underestimated in clinical trials for several reasons. First, for many years oncology trials were designed mainly by non-cardiologists, systematic cardiac monitoring was not part of the protocol, some arrhythmias may not be detected during routine monitoring (12-lead ECGs and 1-min rhythm strips), patients at risk of cardiotoxicicty were sometimes excluded in clinical trials, and there were marked variations in methodology and in the definition of cardiac arrhythmias [1, 2, 6, 7]. Moreover, the criteria for reporting adverse cardiovascular events in oncological trials are markedly different from methods used in cardiovascular drug trials; these discrepancies compromise detection of cardiotoxicity and limit the ability to compare safety data between trials. Second, even after exclusion of confounding factors, including the chemotherapy itself, some cancer patients present basal ECG abnormalities and cardiac arrhythmias [2, 6–8, 104, 183–188]. This is not a surprising as cancer is an age-related disease (~60 % of patients are older than 65 years) and this population sector presents other cardiovascular co-morbidities that can create an arrhythmogenic substrate. Moreover, cancer itself creates an arrhythmogenic substrate (see Sect. 4.2). Unfortunately, in many clinical trials baseline ECGs prior to beginning CC are missing, making it difficult to ascertain whether ECG changes and arrhythmias reflect the baseline state of the patient or represent an adverse effect of CC [36, 50, 104, 124]. Third, the heterogeneity of the enrolled population and the great number of new agents used in the treatment are such that studies on sufficiently large populations are frequently not available, which makes comparisons between and among groups difficult. Finally, many patients are treated simultaneously with complex poly-chemotherapy regimens, so it is difficult to identify which drug causes the arrhythmia. All of these discrepancies compromise detection of cardiac arrhythmias during the clinical development programme and limit the ability to compare safety data between trials. For all of these reasons, the real incidence of CC-induced arrhythmias will only become apparent when the drug is prescribed in patients with co-morbidities [1, 2, 6, 7]. In general, asymptomatic self-limiting arrhythmias are reported in studies performing a systematic monitoring of heart rhythm, while symptomatic recurrent and life-threatening arrhythmias are mainly described in single-centre studies and case reports.
4.2 Mechanisms of Cardiac Arrhythmias
Multiple mechanisms have been proposed to explain CC-induced cardiac arrhythmias, and they are summarised in Table 2. In general, cardiac arrhythmias can arise from direct electrophysiological effects or cardiac indirect effects, as discussed below.
4.2.1 Direct Electrophysiological Effects
Direct electrophysiological effects are derived from their effects on cardiac Na+, Ca2+ and K+ ion channels and/or pumps involved in the genesis of the cardiac action potentials [3–5, 8, 25, 189]. Most drugs that prolong the QTc interval, including chemotherapeutic drugs, produce a blockade of hERG channels. However, there are important risk variations among drugs of the same family group. Indeed, dasatinib, imatinib and nilotinib differentially inhibited the hERG currents in vitro with an IC50 (concentration of drug producing 50 % inhibition) of 14.3, 15.6 and 0.66 µmol/L, respectively, i.e. at concentrations approximately 150-, 3- and 0.1-fold the expected human peak plasma concentration for the three TKIs, respectively. Thus, the mechanism underlying QT interval prolongation for dasatinib may be independent of hERG inhibition [184]. Whether these differences are related to differences in the chemical structure (presence/absence of a hydroxamate group in HDAC inhibitors and of a fluorophenyl or fluoromethyl-phenyl ring in PKIs) is a matter of discussion [160]. Thus, QTc interval prolongation can be produced by many other mechanisms. Very recent evidence suggests that down-regulation of PI3 K signalling directly or indirectly via tyrosine kinase inhibition prolongs the QTc interval by affecting multiple ion channels, including a decrease in delayed rectifier outward K+ currents (I Kr and I Ks), L-type Ca2+ and the peak Na+ inwards currents and an increase in the late Na+ current [180, 181].
Modulation of ions channels/pumps can also be mediated via the release of vasoactive substances (histamine: doxorubicin, paclitaxel; endothelin-1: 5-FU; cytokine-release syndrome: alkylating agent, angiogenesis inhibitors, antimetabolites, some PKIs), changes in cardiac metabolism and ATP production (anthracycline, interferons, proteasome inhibitors, sunitinib) or cardiac autonomic nervous system modulation, including a reflex increase in cardiac sympathetic tone in response to hypotension or modulation of the vagal tone (thalidomide, vinca alkaloids). Some immediate proarrhythmic effects can be related to myocardial ischaemia due to coronary spasm, endothelial dysfunction and/or vascular thrombosis or to a direct irritative effect of CC. The direct irritation of the pericardium can explain, in part, the episodes of AF observed following the intrapericardial administration of cisplatin in patients with lung adenocarcinoma and malignant cardiac tamponade [5, 22–24]. Finally, some drugs, such as anthracyclines, can modify intracellular Ca2+ handling as they decrease the expression of the sarcoplasmic reticulum Ca2+-ATPase leading to an increase in [Ca2+] i and possibly to the appearance of triggered focal activity due to delayed after-depolarisations.
4.2.2 Cardiac Indirect Effects
Many cardiac arrhythmias are initiated/maintained by an arrhythmogenic substrate created by the co-morbidities present in cancer patients or generated by the cardiotoxicity induced by CC (Table 2). Some chemotherapy agents can produce arterial (or pulmonary) hypertension, myocardial ischaemia, myocarditis/pericarditis, LV dysfunction or heart failure. Additionally, many patients treated with CC present cardiovascular co-morbidities (hypertension, CAD, heart failure). As a result, there is a structural remodelling of the heart characterised by myocardial hypertrophy, necrosis/apoptosis, fibrosis, ischaemia, chamber dilatation and inflammation, which create a permanent arrhythmogenic substrate. This arrhythmogenic substrate leads to a decrease in intracardiac conduction and to a heterogeneous dispersion of repolarisation, two effects that facilitate the genesis of cardiac arrhythmias in patients with CC-induced structural heart disease even years after CC has been completed [6, 25, 77, 78, 190]. Additionally, the cardiac fibrosis observed in elderly patients, that is produced by primary cardiac tumours or that occurs following radiation therapy can also affect the cardiac conduction system [186] and facilitate the appearance of bradyarrhythmias [6]. Thus, arterial hypertension produced by some chemotherapy drugs increases pressure load, leading to myocardial hypertrophy and eventually progressing to congestive heart failure, while both arterial hypertension and heart failure markedly increase the incidence of AF, one of the most common cardiac arrhythmias among the elderly population.
4.3 Diagnosis
Because many CC can induce cardiac arrhythmias, the challenge for the cardio-oncologist team is to identify and treat those patients more susceptible to developing cardiac arrhythmias in an attempt to individualise the therapeutic strategy, optimise clinical monitoring, and reduce morbidity and mortality. A careful baseline cardiovascular evaluation should be performed in all patients undergoing chemotherapy, including cardiac family and personal history, physical examination and cardiovascular risk assessment, pre-existing cardiovascular diseases (CAD, hypertension, LV dysfunction or heart failure, cardiomyopathies, thromboembolic events), metabolic (hypo-/hyperthyroidism, especially with neck radiation) and electrolyte abnormalities, and previous treatments with CC and radiation therapy [77]. Cardiac electrophysiological evaluation should include a baseline ECG, chest radiography, exercise testing, and an assessment of LV function using echocardiography and/or magnetic resonance imaging, if necessary. A 12-lead ECG and/or ECG Holter monitoring are readily available, inexpensive and non-invasive methods to detect brady-/tachyarrhythmias, conduction disturbances and repolarisation abnormalities (QTc interval prolongation) as well as signs of myocardial ischaemia, LV hypertrophy and cardiomyopathy before and during the treatment. The final objective of cardiac monitoring is to identify signs of cardiac disease early enough to potentially prevent, reverse or slow down both cardiac electrophysiological and structural damage during cancer treatment. Unfortunately, uniform recommendations regarding cardiac monitoring in patients undergoing CC and guidelines for cancer treatment that take cardiovascular co-morbidities into account are currently lacking and need to be developed as a result of a collaboration between cardiologists and oncologists [80].
Cancer patients with basal ECG abnormalities, impaired exercise capacity or pre-existing cardiovascular diseases, and those receiving treatment regimens known to be associated with significant cardiotoxicity are more susceptible to CC-induced arrhythmias [7, 80, 187, 188]. Furthermore, older age, the dose of the anticancer drug administered during each session, cumulative dose, simultaneous use of other chemotherapeutic agents, sequence of drug administration, and previous exposure to CC or history of radiation therapy are important factors to be considered to reduce the risk of cardiac arrhythmias [4, 14]. Special attention should be paid to patients’ co-morbidities that represent possible arrhythmogenic substrates, especially in patients receiving multitargeted agents as these can have a dramatic effect on long-term survival after cancer therapy [4–6, 187, 188]. Furthermore, co-morbidities can represent a contraindication for some chemotherapeutic agents and sometimes do not allow the optimal cancer therapeutic regimen to be reached in certain patients. Therefore, co-morbidities representing a possible arrhythmogenic substrate should be identified and treated aggressively prior to and during chemotherapy. It has been demonstrated that renin–angiotensin–aldosterone inhibitors, β-blockers or statins (HMG-CoA reductase inhibitors) might improve the arrhythmogenic substrate and exert antiarrhythmic effects in patients with CAD, arterial hypertension, LV dysfunction or heart failure [4, 80, 186]. Other strategies include the selection of drugs with lower risk of cardiotoxicity (i.e. epirubicin and liposomal or pegylated doxorubicin formulations instead of doxorubicin) and the addition of cardioprotective drugs (i.e. dexarozoxane) [4, 80].
A better understanding of the proarrhythmic profile of chemotherapy drugs, and the early identification and aggressive treatment of those patients at higher risk of developing cardiac arrhythmias to prevent and/or reverse the structural cardiac remodelling (i.e. the arrhythmogenic substrate) are the best strategies to improve outcomes in patients with CC-induced arrhythmias. These successive steps require a close collaboration between cardiologists and oncologists.
4.4 QT Prolongation
The prolongation of the QTc interval above the upper limit of normal (460 ms for women and 450 ms for men) or >60 ms over baseline during CC raises concern about the possible risk of potentially malignant cardiac arrhythmias, such as Torsades de pointes, and SCD [8, 189, 190]. However, QTc intervals above the upper limit of normal are present in 10–36 % of cancer patients [8, 124, 186, 191], which suggests that these patients may present intrinsic repolarisation abnormalities or that standard QT reference ranges may not apply to oncology patients. Other explanations might be that cancer is an age-related disease, and age has been associated with QT prolongation, and the high prevalence of co-morbid diseases in cancer patients, which is likely to contribute to distinct baseline ECG characteristics [8, 188, 191].
Risk factors for Torsades de pointes include female sex, old age, bradyarrhythmias, electrolyte abnormalities (hypokalaemia and/or hypomagnesaemia), structural heart diseases (CAD, heart failure, LV hypertrophy), congenital long QT syndrome and coadministration of QT-prolonging drugs (see http://www.qtdrugs.org) [4]. Cancer patients present co-morbidities (bradyarrhythmias, cardiac hypertrophy or ischaemia, renal and hepatic dysfunction) that represent independent risk factors for QT prolongation [8, 188, 189] and some chemotherapy drugs can inhibit hepatic metabolism (via cytochrome P450 3A4: imatinib) or renal clearance (cisplatin) of some QT-prolonging drugs. Furthermore, cancer patients frequently receive QT-prolonging drugs, including 5-hydroxytryptamine receptor 3 antagonists (for prevention and treatment of chemotherapy- and radiation-induced emesis), octreotide, tamoxifen, antipsychotics, antidepressants, antifungals and quinolone antibacterials and often experience diarrhoea- and vomiting-induced electrolyte disturbances (hypokalaemia) that may further prolong the QTc interval, increasing the risk of ventricular arrhythmias [4, 8, 186, 187, 190].
In patients treated with QT-prolonging drugs, ECG (1–2 times per week in stable patients; more often if needed), serum electrolyte and other risk factors should be closely monitored and corrected [4, 5, 8, 189] and coadministration of other QT-prolonging agents should be avoided before and during the treatment. Additionally, patients should be informed of the risk of arrhythmias and asked to report any cardiac symptoms such as palpitations.
Anticancer drugs that induce QT prolongation in preclinical studies should undergo intensive cardiac monitoring in subsequent phase clinical trials using standardised and corrected QT measurements. Baseline ECG characteristics and a complete cardiac history, including the concomitant medications and co-morbidities, should be obtained in all trial patients, drugs that prolong the QT interval should be avoided if possible, electrolytes should be closely monitored and aggressively replaced during the trial, and cardiac data, including post-marketing information, should be centralised and made available to investigators [8].
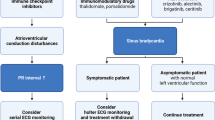
4.5 Treatment of Cardiac Arrhythmias
Asymptomatic cardiac tachyarrhythmias [grade 1 National Cancer Institute (NCI)-Common Terminology Criteria for Adverse Events (CTCAE)] detected during cardiac monitoring are self-terminating; thus, drug discontinuation is not required without cardiac risk factors, although it may be considered if ECG abnormalities persist. Symptomatic tachyarrhythmias (grade 2) require dose reduction or permanent drug discontinuation and patients should be referred to the cardiologist for evaluation and treatment. Recurrent symptomatic life-threatening tachyarrhythmias (grade 3–4) with haemodynamic consequences require urgent intervention including defibrillation [4]. In patients with symptomatic bradycardia (fatigue, lightheadedness, dizziness, syncope) or progressive AV conduction disturbances, drug (paclitaxel, thalidomide) discontinuation may be required and a permanent pacemaker implantation should be considered, particularly when recovery of sinus rhythm does not occur after drug discontinuation or an alternative chemotherapy treatment cannot be administered [4, 143, 146, 186]. Thus, in multiple myeloma patients responsive to thalidomide therapy where no other therapeutic alternatives are available, patients have had pacemakers implanted to be able to continue thalidomide [143, 144, 146]. In these patients, concomitant bradycardic agents (β-blockers, calcium channel blockers, digoxin and antiarrhythmic drugs) should be avoided and a hypothyroidism should be ruled out.
Treatment of patients with QTc prolongation begins with the recognition and immediate withdrawal of any offending drug and correction of any known risk factors [192, 193] (Table 3). Drug administration should be temporarily withheld in patients who develop a QTc interval >500 ms or which has increased >60 ms from pre-treatment values; upon recovery to a QTc interval <460 ms (grade 2), treatment should be restarted at a reduced dose. If the QTc interval is >500 ms and signs and symptoms (palpitations, heart failure, hypotension or syncope) or Torsades de pointes occur, the patient should be hospitalised and drug administration should be permanently discontinued. Treatment for Torsades de pointes includes immediate withdrawal of any potentially offending drug (see http://www.torsades.org), correction of risk factors (hypokalaemia, hypomagnesaemia and bradycardia) and administration of magnesium sulphate (1–2 g intravenous in 30–60 s, which then can be repeated in 5–15 min, or a continuous intravenous infusion at a rate of 3–10 mg/min) regardless of the serum magnesium level. Cardiac pacing or isoproterenol titrated to ≥90 beats/min shorten the QTc interval and are highly effective in terminating and preventing recurrences when Torsades de pointes are precipitated by bradycardia or are pause dependent and when refractory to magnesium [8]. If the patient is unresponsive an electrical cardioversion can be attempted.
4.6 Preclinical and Clinical Detection of Cardiotoxicity
In recent years, the prolongation of the QTc interval by non-cardiac drugs has become the most common cause of delays in drug development, non-approvals and post-marketing withdrawals. This has led to scrutiny of regulations for preclinical and early phase examination of the impact of novel therapeutics on cardiac repolarisation as reflected by changes in the duration of the QTc interval (‘QT liability’) of the surface ECG [194, 195]. Therefore, much of the preclinical screening and early phase clinical investigation into the impact of novel chemotherapeutic drugs has been focused on hERG-transfected expression systems and action potential duration and in vivo studies (usually in dogs) to identify drugs that prolong the QT interval [194]. However, it is clear that CC produce different types of bradyarrhythmias and supraventricular arrhythmias that are unrelated to a prolongation of the QT interval. Thus, the risk of proarrhythmias goes far beyond QT prolongation.
Interestingly, we recently learnt that some serious clinical cardiovascular complications produced by new targeted therapies (trastuzumab, imatinib, lapatinib) were not predicted in preclinical safety studies, but were reported at the post-approval stage [134]. This finding has several explanations, including [134, 157, 158] (1) the lack of translation of specific toxicological mechanisms from animal species to humans; (2) the preclinical cardiac dysfunction is subtle and/or asymptomatic with no overt pathology; (3) the specific issue was not assessed preclinically or was not deemed biologically meaningful; (4) some adverse events cannot be detected in preclinical studies in young, healthy and drug-naive animals, but can be observed in cancer patients, who are older and present several cardiovascular co-morbidities; and (5) the models are not predictive of cardiotoxicity, as cell cultures have been questioned as a reliable predictor of organ toxicity in vivo and studies have revealed that different cell lines can exhibit dramatically different responses to a given drug (i.e. to PKIs).
Thus, we have an unmet need to develop preclinical models more predictive of cardiotoxicity. New models have been proposed, including zebrafish [196], human embryonic stem cell-derived cardiomyocytes [197, 198] and engineered heart tissue, which allow us to monitor effects on cardiac membrane potential, Ca2+ handling, myofilament function, gene expression and cell survival [118]. However, these models are also far from the elderly patient with co-morbidities. It seems much more interesting that safety studies were carried out in adult animal models with “preexisting cardiovascular diseases” to more accurately reproduce the impact of primary disease and co-morbidities on the cardiac response to new developmental drugs [134].
The detection of potentially cardiac arrhythmias needs to be an integral part of the development programs of novel anticancer agents. Oncology drugs can be cytotoxic or genotoxic or not otherwise tolerated in healthy volunteers. In this case, the primary approach is to carry out intense and robust ECG monitoring and evaluation during early phase clinical trials, together with characterisation of the concentration–QTc interval relationship, and to follow this up with an appropriate intensity of ECG monitoring in the later phases of development [199]. A rigorous cardiac monitoring, assessment and reporting of cardiac arrhythmias is needed to develop registries/databases that may allow for a better understanding of the incidence of cardiac arrhythmias and their impact on the outcomes in cancer population. Finally, after drug approval, when ‘real-world’ patients with co-morbidities are treated, it is especially important to monitor the actual incidence and clinical significance of proarrhythmia. This is the reason why the post-marketing surveillance studies are so important for potentially problematic agents in order to confirm that the efficacy and safety of novel chemotherapy drugs is similar to that previously reported in clinical trials [7].
In the last decade, several targeted therapies (mainly PKIs) were granted accelerated approval despite the fact that serious adverse cardiac events (LV dysfunction, heart failure, acute coronary syndromes, thromboembolisms) were reported during clinical trials because these agents emerged as first-line therapy in cancers where treatment options were very limited. The clinical benefits of PKIs are considered sufficiently valuable as, apart from some of them (bosutinib, gefitinib, lapatinib, nilotinib, sorafenib and vantetanib), contraindications applied to these drugs are modest and none is absolutely contraindicated in the USA despite exerting clinical effects that would otherwise have required certain contraindications [160]. Conversely, in the EU, all TKIs are contraindicated in patients with hypersensitivity to the drug and there are some specific contraindications for several agents (e.g. vandetanib in patients with QT-related risk factors). Thus, in oncology drug development, the challenges of achieving anticancer efficacy are so great that cardiotoxicity risk is considered on balance and is not necessarily a regulatory barrier. However, patients with risk factors for PKIs-induced cardiotoxicty were excluded from clinical trials which, in general, recruited a small number of patients, and this may aggravate the lack of adequate risk/benefit assessment in routine clinical practice. Therefore, the post-marketing efficacy and safety of PKIs requires ongoing reassessment through diligent pharmacovigilance studies [160].
4.7 Gaps in Knowledge
CC-induced arrhythmias are an important but still poorly understood adverse effect and, therefore, there are multiple gaps in knowledge and areas for future research (Table 4). We need a better understanding of the mechanisms involved in the genesis/maintenance of CC-induced cardiac arrhythmias. Few prospective studies have analysed the incidence of proarrhythmia in the general population or whether there are differences in the spectrum of cardiac arrhythmias according to the therapeutic indications of the drugs. Furthermore, because the incidence of life-threatening arrhythmias is small, further studies are needed to reach definitive conclusions on the role of proarrhythmia on the long-term outcomes in the general population. A better understanding of these mechanisms could ultimately provide biologic insights in order to exploit tissue-specific differences between cancerous tissues and the healthy myocardium and to develop safer and more effective targeted chemotherapy drugs that spare the cardiovascular system from damage while offering maximum anticancer benefits. Furthermore, because many major chemotherapeutic agents are associated with strong adverse effects on the cardiovascular system, a key goal will be the identification of new cardioprotective drugs. There is also a need to improve early diagnosis, risk stratification, cardiac monitoring, selection and optimisation of therapy, and screening for pre-clinical disease. To date, no guidelines have been developed specifically for the definition, detection, monitoring or therapy of patients undergoing anticancer treatment taking into account cardiologic conditions. Thus, it is imperative to develop joint guidelines between cardiologists and oncologists.
The identification of genetic polymorphisms associated with genetic predisposition for chemotherapy-induced cardiotoxicity [94–96] raised the possibility of identifying valuable surrogate markers that might allow a more personalised oncologic therapy in the near future. Finally, the clinical development of safer and more effective chemotherapy drugs should be the result of teamwork between cardiologists and oncologists to allow a continuous effective and integrated patient care and to ensure that cardiotoxicity does not inappropriately impede progress in chemotherapy drug development [2].
5 Conclusions
Clinicians should be aware of the fact that a progressively increasing number of chemotherapy agents can produce proarrhythmic effects. A better understanding of the arrhythmogenic mechanisms associated with CC is needed as this would result in the development of optimal evidence-based cardiac monitoring guidelines to assess the long-term impact of CC, in the development of newer more effective targeted drugs with a lower risk of proarrhythmia and in the development of cardioprotective regimens [54]. Additionally, as clinicians continue learning about CC-induced proarrhythmia (and cardiotoxicity in general), the importance of primary prevention, early detection and cardiac-specific treatment will become clearer. The progressive aging of cancer patients and the presence of co-morbidities that, in many cases, represent an arrhythmogenic substrate, demand a team approach between cardiologists and oncologists to allow a continued effective and integrated patient care. The challenge for this cardio-oncologist (or onco-cardiologist) team is to balance the risk of proarrhythmia against the expected benefit of CC and to ensure that proarrhythmia does not inappropriately impede progress in chemotherapy drug development.
References
Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300–11.
Suter TM, Ewer MS. Cancer drugs and the heart: importance and management. Eur Heart J. 2013;34(15):1102–11.
Floyd JD, Nguyen DT, Lobins RL, Bashir Q, Doll DC, Perry MC. Cardiotoxicity of cancer therapy. J Clin Oncol. 2005;23(30):7685–96.
Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53(24):2231–47.
Guglin M, Aljayeh M, Saiyad S, Ali R, Curtis AB. Introducing a new entity: chemotherapy-induced arrhythmia. Europace. 2009;11(12):1579–86.
Albini A, Pennesi G, Donatelli F, Cammarota R, De FS, Noonan DM. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardiooncological prevention. J Natl Cancer Inst. 2010;102(1):14–25.
Eschenhagen T, Force T, Ewer MS, de Keulenaer GW, Suter TM, Anker SD, et al. Cardiovascular side effects of cancer therapies: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2011;13(1):1–10.
Strevel EL, Ing DJ, Siu LL. Molecularly targeted oncology therapeutics and prolongation of the QT interval. J Clin Oncol. 2007;25(22):3362–71.
Cazin B, Gorin NC, Laporte JP, Gallet B, Douay L, Lopez M, et al. Cardiac complications after bone marrow transplantation. A report on a series of 63 consecutive transplantations. Cancer. 1986;57(10):2061–9.
Kupari M, Volin L, Suokas A, Timonen T, Hekali P, Ruutu T, et al. Cardiac involvement in bone marrow transplantation: electrocardiographic changes, arrhythmias, heart failure and autopsy findings. Bone Marrow Transplant. 1990;5(2):91–8.
Gottdiener JS, Appelbaum FR, Ferrans VJ, Deisseroth A, Ziegler J. Cardiotoxicity associated with high dose cyclophosphamide therapy. Arch Intern Med. 1981;141(6):758–63.
Ando M, Yokozawa T, Sawada J, Takaue Y, Togitani K, Kawahigashi N, et al. Cardiac conduction abnormalities in patients with breast cancer undergoing high-dose chemotherapy and stem cell transplantation. Bone Marrow Transplant. 2000;25(2):185–9.
Curigliano G, Mayer EL, Burstein HJ, Winer DP. Golodhirsch. Cardiac toxicity from systemic cancer therapy: a comprehensive review. Prog Cardiovasc Dis. 2010;53(1):94–104.
Morandi P, Ruffini PA, Benvenuto GM, Raimondi R, Fosser V. Cardiac toxicity of high-dose chemotherapy. Bone Marrow Transplant. 2005;35(4):323–34.
Cil T, Kaplan MA, Altintas A, Pasa S, Isikdogan A. Cytosine-arabinoside induced bradycardia in patient with non-Hodgkin lymphoma: a case report. Leuk Lymphoma. 2007;48(6):1247–9.
Olivieri A, Corvatta L, Montanari M, Brunori M, Offidani M, Ferretti GF, et al. Paroxysmal atrial fibrillation after high-dose melphalan in five patients autotransplanted with blood progenitor cells. Bone Marrow Transplant. 1998;21(10):1049–53.
Moreau P, Milpied N, Mahé B, Juge-Morineau N, Rapp MJ, Bataille R, et al. Melphalan 220 mg/m2 followed by peripheral blood stem cell transplantation in 27 patients with advanced multiple myeloma. Bone Marrow Transplant. 1999;23(10):1003–6.
Phillips GL, Meisenberg B, Reece DE, Adams VR, Badros A, Brunner J, et al. Amifostine and autologous hematopoietic stem cell support of escalating-dose melphalan: a phase I study. Biol Blood Marrow Transplant. 2004;10(7):473–83.
Feliz V, Saiyad S, Ramarao SM, Khan H, Leonelli F, Guglin M. Melphalan-induced supraventricular tachycardia: incidence and risk factors. Clin Cardiol. 2011;34(6):356–9.
Quezado ZM, Wilson WH, Cunnion RE, Parker MM, Reda D, Bryant G, et al. High-dose ifosfamide is associated with severe, reversible cardiac dysfunction. Ann Intern Med. 1993;118(1):31–6.
Williams SF, Larson RA. Hypersensitivity reaction to high-dose cytarabine. Br J Haematol. 1989;73(2):274–5.
Tomkowski WZ, Wiśniewska J, Szturmowicz M, Kuca P, Burakowski J, Kober J, et al. Evaluation of intrapericardial cisplatin administration in cases with recurrent malignant pericardial effusion and cardiac tamponade. Support Care Cancer. 2004;12(1):53–7.
Bischiniotis TS, Lafaras CT, Platogiannis DN, Moldovan L, Barbetakis NG, Katseas GP. Intrapericardial cisplatin administration after pericardiocentesis in patients with lung adenocarcinoma and malignant cardiac tamponade. Hellenic J Cardiol. 2005;46(5):324–9.
Richards WG, Zellos L, Bueno R, Jaklitsch MT, Jänne PA, Chirieac LR, et al. Phase I to II study of pleurectomy/decortication and intraoperative intracavitary hyperthermic cisplatin lavage for mesothelioma. J Clin Oncol. 2006;24(10):1561–7.
Tamargo J, Caballero R, Delpón E. Drug-induced atrial fibrillation. Expert Opin Drug Saf. 2012;11(4):615–34.
Yavaş Ö, Aytemır K, Çelık I. The prevalence of silent arrhythmia in patients receiving cisplatin-based chemotherapy. Turkish J Cancer. 2008;38(1):12–5.
de Forni M, Malet-Martino MC, Jaillais P, Shubinski RE, Bachaud JM, Lemaire L, et al. Cardiotoxicity of high dose continuous infusion fluorouracil: a prospective clinical study. J Clin Oncol. 1992;10(11):1795–801.
Hrovatin E, Viel E, Lestuzzi C, Tartuferi L, Zardo F, Brieda M, et al. Severe ventricular dysrhythmias and silent ischemia during infusion of the antimetabolite 5-fluorouracil and cis-platin. J Cardiovasc Med (Hagerstown). 2006;7(8):637–40.
Polk A, Vaage-Nilsen M, Vistisen K, Nielsen DL. Cardiotoxicity in cancer patients treated with 5-fluorouracil or capecitabine: a systematic review of incidence, manifestations and predisposing factors. Cancer Treat Rev. 2013;39(8):974–84.
Khan MA, Masood N, Husain N, Ahmad B, Aziz T, Naeem A. A retrospective study of cardiotoxicities induced by 5-fluouracil (5-FU) and 5-FU based chemotherapy regimens in Pakistani adult cancer patients at Shaukat Khanum Memorial Cancer Hospital and Research Center. J Pak Med Assoc. 2012;62(5):430–4.
Saif MW, Shah MM, Shah AR. Fluoropyrimidine-associated cardiotoxicity: revisited. Expert Opin Drug Saf. 2009;8(2):191–202.
Keefe DL, Roistacher N, Pierri MK. Clinical cardiotoxicity of 5-fluorouracil. J Clin Pharmacol. 1993;33(11):1060–70.
Yilmaz U, Oztop I, Ciloglu A, Okan T, Tekin U, Yaren A, et al. 5-fluorouracil increases the number and complexity of premature complexes in the heart: a prospective study using ambulatory ECG monitoring. Int J Clin Pract. 2007;61(5):795–801.
Rezkalla S, Kloner RA, Ensley J, Al-Sarraf M, Revels S, Olivenstein A, et al. Continuous ambulatory ECG monitoring during fluorouracil therapy: a prospective study. J Clin Oncol. 1989;7(4):509–14.
Tsibiribi P, Descotes J, Lombard-Bohas C, Barel C, Bui-Xuan B, Belkhiria M, et al. Cardiotoxicity of 5-fluorouracil in 1350 patients with no prior history of heart disease. Bull Cancer. 2006;93(3):E27–30.
Eskilsson J, Albertsson M. Failure of preventing 5-fluorouracil cardiotoxicity by prophylactic treatment with verapamil. Acta Oncol. 1990;29(8):1001–3.
Kosmas C, Kallistratos MS, Kopterides P, Syrios J, Skopelitis H, Mylonakis N, et al. Cardiotoxicity of fluoropyrimidines in different schedules of administration: a prospective study. J Cancer Res Clin Oncol. 2008;134(1):75–82.
Stewart T, Pavlakis N, Ward M. Cardiotoxicity with 5-fluorouracil and capecitabine: more than just vasospastic angina. Intern Med J. 2010;40(4):303–7.
Ng M, Cunningham D, Norman AR. The frequency and pattern of cardiotoxicity observed with capecitabine used in conjunction with oxaliplatin in patients treated for advanced colorectal cancer (CRC). Eur J Cancer. 2005;41(11):1542–6.
Robben NC, Pippas AW, Moore JO. The syndrome of 5-fluorouracil cardiotoxicity. Cancer. 1993;71(2):493–509.
Santini D, Tonini G, Abbate A, Di Cosimo S, Gravante G, Vincenzi B, et al. Gemcitabine-induced atrial fibrillation: a hitherto unreported manifestation of drug toxicity. Ann Oncol. 2000;11(4):479–81.
Talapatra K, Rajesh I, Rajesh B, Selvamani B, Subhashini J. Transient asymptomatic bradycardia in patients on infusional 5-fluorouracil. J Cancer Res Ther. 2007;3(3):169–71.
Alter P, Herzum M, Soufi M, Schaefer JR, Maisch B. Cardiotoxicity of 5-fluorouracil. Cardiovasc Hematol Agents Med Chem. 2006;4(1):1–5.
Becker K, Erckenbrecht JF, Häussinger D, Frieling T. Cardiotoxicity of the antiproliferative compound fluorouracil. Drugs. 1999;57(4):475–84.
Porta C, Moroni M, Ferrari S, Nastasi G. Endothelin-1 and 5-fluorouracil-induced cardiotoxicity. Neoplasma. 1998;45(2):81–2.
Alloatti G, Penna C, Gallo MP, Levi RC, Bombardelli E, Appendino G. Differential effects of paclitaxel and derivatives on Guinea pig isolated heart and papillary muscle. J Pharmacol Exp Ther. 1998;284(2):561–7.
Bryant J, Picot J, Levitt G, Sullivan I, Baxter L, Clegg A. Cardioprotection against the toxic effects of anthracyclines given to children with cancer: a systematic review. Health Technol Assess. 2007;11(27):iii, ix–x, 1–84.
McGuire WP, Rowinsky EK, Rosenshein NB, Grumbine FC, Ettinger DS, Armstrong DK, et al. Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann Intern Med. 1989;111(4):273–9.
Arbuck SG, Strauss H, Rowinsky E, Christian M, Suffness M, Adams J, et al. A reassessment of cardiac toxicity associated with taxol. J Natl Cancer Inst Mono. 1993;15:117–30.
Kietpeerakool C, Tiyayon J, Suprasert P, Kanjanavanit R, Srisomboon J. Benefit of electrocardiography during front-line combination paclitaxel and carboplatin chemotherapy for epithelial ovarian cancer. J Med Assoc Thai. 2006;89(11):1805–10.
Rowinsky EK, Eisenhauer EA, Chaudhry V, Arbuck SG, Donehower RC. Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol. 1993;20(4 Suppl 3):1–15.
Bristow MR, Sageman WS, Scott RH, Billingham ME, Bowden RE, Kernoff RS, et al. Acute and chronic cardiovascular effects of doxorubicin in the dog: the cardiovascular pharmacology of drug-induced histamine release. J Cardiovasc Pharmacol. 1980;2(5):487–515.
Levi R, Zavecz JH. Acceleration of idioventricular rhythms by histamine in the guinea pig heart: mediation by H2 receptors. Circ Res. 1979;44(6):847–55.
Hageman GR, Urthaler F, Isobe JH, James TN. Chronotropic and dromotropic effects of histamine on the canine heart. Chest. 1979;75(5):597–604.
Schimmel KJ, Richel DJ, Van den Brink RB, Guchelaar HJ. Cardiotoxicity of cytotoxic drugs. Cancer Treat Rev. 2004;30(2):181–91.
Zhang K, Heidrich FM, DeGray B, Boehmerle W, Ehrlich BE. Paclitaxel accelerates spontaneous calcium oscillations in cardiomyocytes by interacting with NCS-1 and the InsP3R. J Mol Cell Cardiol. 2010;49(5):829–35.
Sanborn SL, Cooney MM, Dowlati A, Brell JM, Krishnamurthi S, Gibbons J, et al. Phase I trial of docetaxel and thalidomide: a regimen based on metronomic therapeutic principles. Invest New Drugs. 2008;26(4):355–62.
Salvatorelli E, Menna P, Cascegna S, Liberi G, Calafiore AM, Gianni L, et al. Paclitaxel and docetaxel stimulation of doxorubicinol formation in the human heart: implications for cardiotoxicity of doxorubicin–taxane chemotherapies. J Pharmacol Exp Ther. 2006;318(1):424–33.
Gianni L, Herman EH, Lipshultz SE, Minotti G, Sarvazyan N, Sawyer DB. Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol. 2008;26(22):3777–84.
Yancy RS, Talpaz M. Vindesine associated angina and ECG changes. Cancer Treat Rep. 1982;66(3):587–9.
Lesimple T, Edeline J, Carrothers TJ, Cvitkovic F, Darpo B, Delord JP, et al. A phase I, open-label, single-arm study for QT assessment of eribulin mesylate in patients with advanced solid tumors. Invest New Drugs. 2013;31(4):900–9.
Full prescribing information for HALAVEN® (eribulin mesylate) for intravenous use. http://www.halaven.com/sites/default/files/HALAVEN_full_Prescribing_Information.pdf. Accessed 20 Nov 2014.
Subar M, Muggia FM. Apparent myocardial ischemia associated with vinblastine administration. Cancer Treat Rep. 1986;70(5):690–1.
Roca E, Bruera E, Politi PM, Barugel M, Cedaro L, Carraro S, et al. Vinca alkaloid-induced cardiovascular autonomic neuropathy. Cancer Treat Rep. 1985;69(2):149–51.
Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869–79.
Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, et al. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer. 2010;10:337.
Dindogru A, Barcos M, Henderson ES, Wallace HJ Jr. Electrocardiographic changes following adriamycin treatment. Med Pediatr Oncol. 1978;5(1):65–71.
Kilickap S, Barista I, Akgul E, Aytemir K, Aksoy S, Tekuzman G. Early and late arrhythmogenic effects of doxorubicin. South Med J. 2007;100(3):262–5.
O’Bryan RM, Luce JK, Talley RW, Gottlieb JA, Baker LH, Bonadonna G. Phase II evaluation of adriamycin in human neoplasia. Cancer. 1973;32(1):1–8.
Steinberg JS, Cohen AJ, Wasserman AG, Cohen P, Ross AM. Acute arrhythmogenicity of doxorubicin administration. Cancer. 1987;60(6):1213–8.
Outomuro D, Grana DR, Azzato F, Milei J. Adriamycin-induced myocardial toxicity: new solutions for an old problem? Int J Cardiol. 2007;117(1):6–15.
Hayek ER, Speakman E, Rehmus E. Acute doxorubicin cardiotoxicity. N Engl J Med. 2005;352(23):2456–7.
Wortman JE, Lucas VS Jr, Schuster E, Thiele D, Logue GL. Sudden death during doxorubicin administration. Cancer. 1979;44(5):1588–91.
Couch RD, Loh KK, Sugino J. Sudden cardiac death following adriamycin therapy. Cancer. 1981;48(1):38–9.
Rudzinski T, Ciesielczyk M, Religa W, Bednarkiewicz Z, Krzeminska-Pakula M. Doxorubicin-induced ventricular arrhythmia treated by implantation of an automatic cardioverter–defibrillator. Europace. 2007;9(5):278–80.
Lipshultz SE, Adams MJ, Colan SD, Constine LS, Herman EH, Hsu DT, et al. Long-term cardiovascular toxicity in children, adolescents, and young adults who receive cancer therapy: pathophysiology, course, monitoring, management, prevention, and research directions a scientific statement from the American Heart Association. Circulation. 2013;128(17):1927–95.
Steinherz LJ, Steinherz PG, Tan C. Cardiac failure and dysrhythmias 6–19 years after anthracycline therapy: a series of 15 patients. Med Pediatr Oncol. 1995;24(6):352–61.
Larsen RL, Jakacki RI, Vetter VL, Meadows AT, Silber JH, Barber G. Electrocardiographic changes and arrhythmias after cancer therapy in children and young adults. Am J Cardiol. 1992;70(1):73–7.
Altena R, Perik PJ, van Veldhuisen DJ, de Vries EG, Gietema JA. Cardiovascular toxicity caused by cancer treatment: strategies for early detection. Lancet Oncol. 2009;10(4):391–9.
Chen B, Peng X, Pentassuglia L, Lim CC, Sawyer DB. Molecular and cellular mechanisms of anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):114–21.
Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. 2014;3(2):e000665.
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229.
Geisber G, Pentassuglia L, Sawyer DB. Cardiac side effects of anticancer treatments: new mechanistic insights. Curr Heart Fail Rep. 2012;9(3):211–8.
Gorelik J, Vodyanoy I, Shevchuk AI, Diakonov IA, Lab MJ, Korchev YE. Esmolol is antiarrhythmic in doxorubicin-induced arrhythmia in cultured cardiomyocytes-determination by novel rapid cardiomyocyte assay. FEBS Lett. 2003;548(1–3):74–8.
Keung EC, Toll L, Ellis M, Jensen RA. L-type cardiac calcium channels in doxorubicin cardiomyopathy in rats: morphological, biochemical, and functional correlations. J Clin Invest. 1991;87(6):2108–13.
Earm YE, Ho WK, So I. Effects of adriamycin on ionic currents in single cardiac myocytes of the rabbit. J Mol Cell Cardiol. 1994;26(2):163–72.
Binah O, Cohen IS, Rosen MR. The effects of adriamycin on normal and ouabaintoxic canine Purkinje and ventricular muscle fibers. Circ Res. 1983;53(5):655–62.
Wang YX, Korth M. Effects of doxorubicin on excitation-contraction coupling in guinea pig ventricular myocardium. Circ Res. 1995;76(4):645–53.
Aversano RC, Boor PJ. Acute doxorubicin-induced cardiac arrhythmias during ether anesthesia. Res Commun Chem Pathol Pharmacol. 1983;41(2):345–8.
Olson RD, Mushlin PS, Brenner DE, Fleischer S, Cusack BJ, Chang BK, et al. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci USA. 1988;85(10):3585–9.
Boucek RJ Jr, Olson RD, Brenner DE, Ogunbunmi EM, Inui M, Fleischer S. The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J Biol Chem. 1987;262(33):15851–6.
Pai VB, Nahata M. Cardiotoxicity of chemotherapeutic agents. Drug Saf. 2000;22(4):263–302.
Swain SM, Vici P. The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: expert panel review. J Cancer Res Clin Oncol. 2004;130(1):1–7.
Wojnowski L, Kulle B, Schirmer M, Schluter G, Schmidt A, Rosenberger A, et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation. 2005;112(24):3754–62.
Deng S, Wojnowski L. Genotyping the risk of anthracycline-induced cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):129–34.
Visscher H, Ross CJ, Rassekh SR, Barhdadi A, Dubé MP, Al-Saloos H, Canadian Pharmacogenomics Network for Drug Safety Consortium, et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol. 2012;30(13):1422–8.
van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev. 2011;6:CD003917.
Weiss AJ, Manthel RW. Experience with the use of Adriamycin in combination with other anticancer agents using a weekly schedule with particular reference to lack of cardiac toxicity. Cancer. 1977;40(5):2046–52.
Torti FM, Bristow MR, Howes AE, Aston D, Stockdale FE, Carter SK, et al. Reduced cardiotoxicity of doxorubicin delivered on a weekly schedule. Assessment by endomyocardial biopsy. Ann Intern Med. 1983;99(6):745–9.
Harris L, Batist G, Belt R, Rovira D, Navari R, Azarnia N, TLC D-99 Study Group, et al. Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer. 2002;94(1):25–36.
O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYXTM/Doxil(R)) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15(3):440–9.
Huang SY, Chang CS, Tang JL, Tien HF, Kuo TL, Huang SF, et al. Acute and chronic arsenic poisoning associated with treatment of acute promyelocytic leukaemia. Br J Haematol. 1998;103(4):1092–5.
Ohnishi K, Yoshida H, Shigeno K, Nakamura S, Fujisawa S, Naito K, et al. Arsenic trioxide therapy for relapsed or refractory Japanese patients with acute promyelocytic leukemia: need for careful electrocardiogram monitoring. Leukemia. 2002;16(4):617–22.
Barbey JT, Pezzullo JC, Soignet SL. Effect of arsenic trioxide on QT interval in patients with advanced malignancies. J Clin Oncol. 2003;21(19):3609–15.
Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, et al. United states multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol. 2001;19(18):3852–60.
Beer TM, Tangen CM, Nichols CR, Margolin KA, Dreicer R, Stephenson WT, et al. Southwest Oncology Group phase II study of arsenic trioxide in patients with refractory germ cell malignancies. Cancer. 2006;106(12):2624–9.
Westervelt P, Brown RA, Adkins DR, Khoury H, Curtin P, Hurd D, et al. Sudden death among patients with acute promyelocytic leukemia treated with arsenic trioxide. Blood. 2001;98(2):266–71.
Unnikrishnan D, Dutcher JP, Garl S, Varshneya N, Lucariello R, Wiernik PH. Cardiac monitoring of patients receiving arsenic trioxide therapy. Br J Haematol. 2004;124(5):610–7.
Drolet B, Simard C, Roden DM. Unusual effects of a QT-prolonging drug, arsenic trioxide, on cardiac potassium currents. Circulation. 2004;109(1):26–9.
Ficker E, Kuryshev Y, Dennis AT, Obejero-Paz C, Wang L, Hawryluk P, et al. Mechanisms of arsenic-induced Prolongation of Cardiac Repolarization. Mol Pharmacol. 2004;66(1):33–44.
Chiang CE, Luk HN, Wang TM, Ding PY. Prolongation of cardiac repolarization by arsenic trioxide. Blood. 2002;100:2249–52.
Wu MH, Lin CJ, Chen CL, Su MJ, Sun SSM, Cheng AL. Direct cardiac effects of As2O3 in rabbits: evidence of reversible chronic toxicity and tissue accumulation of arsenicals after parenteral administration. Toxicol Appl Pharmacol. 2003;189(3):214–20.
Budd GT, Bukowski RM, Miketo L, Yen-Lieberman B, Proffitt MR. Phase I trial of ultrapure human leukocyte interferon in human malignancy. Cancer Chemother Pharmacol. 1984;12(1):39–42.
Martino S, Ratanatharathorn V, Karanes C, Samal BA, Sohn YH, Rudnick SA. Reversible arrhythmias observed in patients treated with recombinant alpha 2 interferon. J Cancer Res Clin Oncol. 1987;113(4):376–8.
Fries GG, Brown TD, Wrenn RC. Cardiovascular rhythm effects of gamma recombinant DNA interferon. Invest New Drugs. 1989;7(2–3):275–80.
Sonnenblick M, Rosin A. Cardiotoxicity of interferon: a review of 44 cases. Chest. 1991;99(3):557–61.
Lee RE, Lotze MT, Skibber JM, Tucker E, Bonow RO, Ognibene FP, et al. Cardiorespiratory effects of immunotherapy with interleukin-2. J Clin Oncol. 1989;7(1):7–20.
Margolin KA, Rayner AA, Hawkins MJ, Atkins MB, Dutcher JP, Fisher RI, et al. Interleukin-2 and lymphokine-activated killer cell therapy of solid tumors: analysis of toxicity and management guidelines. J Clin Oncol. 1989;7(4):486–98.
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16.
White RL Jr, Schwartzentruber DJ, Guleria A, MacFarlane MP, White DE, Tucker E, et al. Cardiopulmonary toxicity of treatment with high dose interleukin-2 in 199 consecutive patients with metastatic melanoma or renal cell carcinoma. Cancer. 1994;74(12):3212–22.
Bates SE, Rosing DR, Fojo T, Piekarz RL. Challenges of evaluating the cardiac effects of anticancer agents. Clin Cancer Res. 2006;12(13):3871–4.
Full prescribing information for ZOLINZA® (vorinostat) [package insert]. https://www.merck.com/product/usa/pi_circulars/z/zolinza/zolinza_pi.pdf. Accessed 1 Sep 2014.
Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, et al. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res. 2002;8(3):718–28.
Piekarz RL, Frye AR, Wright JJ, Steinberg SM, Liewehr DJ, Rosing DR, et al. Cardiac studies in patients treated with depsipeptide, FK229, in a phase II trial for T-cell lymphoma. Clin Cancer Res. 2006;12(12):3762–73.
Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997–4003.
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25(21):3109–15.
Rowinsky E, Bono J, Deangelo D, van Oosteron A, Morganroth J, Laird GH, et al. Cardiac monitoring in phase I trials of a novel histone deacetylase (HDAC) inhibitor LAQ825 in patients with advanced solid tumor and hematologic malignancies. J Clin Oncol 2005;23:3131 (Abstract).
Lynch DR Jr, Washam JB, Newby LK. QT interval prolongation and torsades de pointes in a patient undergoing treatment with vorinostat: a case report and review of the literature. Cardiol J. 2012;19(4):434–8.
Page JG, Rodman LE, Heath JE, et al. Effect of infusion rate on the toxicity of depsipeptide (NSC630176) [abstract no. 2193]. Proc Am Assoc Cancer Res. 1995;36:368.
Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–42.
Arai Y, Tadokoro J, Mitani K. Ventricular tachycardia associated with infusion of rituximab in mantle cell lymphoma. Am J Hematol. 2005;78(4):317–8.
Foran JM, Rohatiner AZ, Cunningham D, Popescu RA, Solal-Celigny P, Ghielmini M, et al. European phase II study of rituximab (chimeric anti-CD20 monoclonal antibody) for patients with newly diagnosed mantle-cell lymphoma and previously treated mantle-cell lymphoma, immunocytoma, and small B-cell lymphocytic lymphoma. J Clin Oncol. 2000;18(9):317–24.
Perez EA, Rodeheffer R. Clinical cardiac tolerability of trastuzumab. J Clin Oncol. 2004;22(2):322–9.
Mellor HR, Bell AR, Valentin JP, Roberts RRA. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol Sci. 2011;120(1):14–32.
Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8(5):459–65.
Grazette LP, Boecker W, Matsui T, Semigran M, Force TL, Hajjar RJ, et al. Inhibition of ErbB2 causes mitochondrial dysfunction in cardiomyocytes: implications for herceptin-induced cardiomyopathy. J Am Coll Cardiol. 2004;44(11):2231–8.
Lenihan DJ, Alencar AJ, Yang D, Kurzrock R, Keating MJ, Duvic M. Cardiac toxicity of alemtuzumab in patients with mycosis fungoides/Sézary syndrome. Blood. 2004;104(3):655–8.
Suter TM, Procter M, van Veldhuisen DJ, Muscholl M, Bergh J, Carlomagno C, et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol. 2007;25(25):3859–65.
Orcioulo E, Buda G, Cecconi N, Galimberi S, Versari D, Cervetti G, et al. Unexpected cardiotoxicity in haematological bertozomib treated patients. Br J Haematol. 2007;138(3):396–403.
Berenson JR, Jagannath S, Barlogie B, Siegel DT, Alexanian R, Richardson PG, et al. Safety of prolonged therapy with bortezomib in relapsed or refractory multiple myeloma. Cancer. 2005;104(10):2141–8.
Xiao Y, Yin J, Wei J, Shang Z. Incidence and risk of cardiotoxicity associated with bortezomib in the treatment of cancer: a systematic review and meta-analysis. PLoS One. 2014;9(1):e87671.
Full prescribing information for KYPROLIS™ (carfilzomib) for intravenous use. http://www.kyprolis.com/prescribing-information. Accessed 1 Sep 2014.
Barlogie B, Tricot G, Anaissie E, Shaughnessy J, Rasmussen E, van Rhee F, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354(10):1021–30.
Fahdi IE, Gaddam V, Saucedo JF, Kishan CV, Vyas K, Deneke MG, et al. Bradycardia during therapy for multiple myeloma with thalidomide. Am J Cardiol. 2004;93(8):1052–5.
Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24(3):431–6.
Kaur A, Yu SS, Lee AJ, Chiao TB. Thalidomide-induced sinus bradycardia. Ann Pharmacother. 2003;37:1040–3.
Palumbo A, Facon T, Sonneveld P, Bladè J, Offidani M, Gay F, et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood. 2008;111(8):3968–77.
Palladini G, Perfetti V, Perlini S, Obici L, Lavatelli F, Caccialanza R, Invernizzi R, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood. 2005;105(7):2949–51.
Clark TE, Edom N, Larson J, Lindsey LJ. Thalomid (Thalidomide) capsules: a review of the first 18 months of spontaneous postmarketing adverse event surveillance, including off-label prescribing. Drug Saf. 2001;24(2):87–117.
Von Hoff DD, Elson D, Polk G, Coltman C Jr. Acute ventricular fibrillation and death during infusion of 4′-(9-acridinylamino) methanesulfon-m-ansidide (AMSA). Cancer Treat Rep. 1980;64(2–3):356–8.
Louie AC, Issell BF. Amsacrine (AMSA)—a clinical review. J Clin Oncol. 1985;3(4):562–92.
Falkson G. Multiple ventricular extrasystoles following administration of 4′-(9-aciridinylamino) methanesulfon-m-anisidide (AMSA). Cancer Treat Rep. 1980;64(2–3):358.
Steinherz LJ, Steinherz PG, Mangiacasale D, Tan C, Miller DR. Cardiac abnormalities after AMSA administration. Cancer Treat Rep. 1982;66(3):483–8.
Shinar E, Hasin Y. Acute electrocardiographic changes induced by amsacrine. Cancer Treat Rep. 1984;68(9):1169–72.
Fenaux P, Tertian G, Castaigne S, Tilly H, Leverger G, Guy H, et al. A randomized trial of amsacrine and rubidazone in 39 patients with acute promyelocytic leukemia. J Clin Oncol. 1991;9(9):1556–61.
Thomas D, Hammerling BC, Wu K, Wimmer AB, Ficker EK, Kirsch GE, et al. Inhibition of cardiac HERG currents by the DNA topoisomerase II inhibitor amsacrine: mode of action. Br J Pharmacol. 2004;142(3):485–94.
Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10(2):111–26.
Lal H, Kolaja KL, Force T. Cancer genetics and the cardiotoxicity of the therapeutics. J Am Coll Cardiol. 2013;61(3):267–74.
Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol. 2010;244(2):190–5.
Shah RR, Morganroth J, Shah DR. Cardiovascular safety of tyrosine kinase inhibitors: with a special focus on cardiac repolarisation (QT Interval). Drug Saf. 2013;36(5):295–316.
Lenihan DJ, Kowey PR. Overview and management of cardiac adverse events associated with tyrosine kinase inhibitors. Oncologist. 2013;18(8):900–8.
Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Ther. 2007;29(11):2289–308.
Brave M, Goodman V, Kaminskas E, Farrell A, Timmer W, Pope S, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14(2):352–9.
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260–70.
Zang J, Wu S, Tang L, Xu X, Bai J, Ding C, et al. Incidence and risk of QTc interval prolongation among cancer patients treated with vandetanib: a systematic review and meta-analysis. PLoS One. 2012;7(2):e30353.
Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–51.
Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR–ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110(10):3540–6.
Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz JA, Larson RA, Gattermann N, et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase following imatinib resistance or intolerance: 24-month follow-up results. Blood. 2011;117(4):1141–5.
le Coutre P, Ottmann OG, Giles F, Kim DW, Cortes J, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–9.
Prescribing information for ICLUSIG® (ponatinib) tablets for oral use. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203469lbl.pdf. Accessed 1 Sep 2014.
Petrini I, Lencioni M, Ricasoli M, Iannopollo M, Orlandini C, Oliveri F, et al. Phase II trial of sorafenib in combination with 5-fluorouracil infusion in advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2012;69(3):773–80.
Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370(9604):2011–9.
Bello CL, Mulay M, Huang X, Patyna S, Dinolfo M, Levine S, et al. Electrocardiographic characterization of the QTc interval in patients with advanced solid tumors: pharmacokinetic-pharmacodynamic evaluation of sunitinib. Clin Cancer Res. 2009;15(22):7045–52.
Mego M, Reckova M, Obertova J, Sycova-Mila Z, Brozmanova K, Mardiak J. Increased cardiotoxicity of sorafenib in sunitinib-pretreated patients with metastatic renal cell carcinoma. Ann Oncol. 2007;18(11):1906–7.
Ribeiro AL, Marcolino MS, Bittencourt HN, Barbosa MM, Nunes Mdo C, Xavier VF, et al. An evaluation of the cardiotoxicity of imatinib mesylate. Leuk Res. 2008;32(12):1809–14.
Prescribing information for NEXAVAR® (sorafenib) tablets, oral use. http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/021923s008s009lbl.pdf. Accessed 1 Sep 2014.
Prescribing information for XALKORI® (crizotinib) capsules. http://labeling.pfizer.com/showlabeling.aspx?id=676. Accessed 20 Nov 2014.
Ou SH, Azada M, Dy J, Stiber JA. Asymptomatic profound sinus bradycardia (heart rate ≤45) in non-small cell lung cancer patients treated with crizotinib. J Thorac Oncol. 2011;6(12):2135–7.
Zhang DY, Wang Y, Lau CP, Tse HF, Li GR. Both EGFR kinase and Src-related tyrosine kinases regulate human ether-a-go-go related gene potassium channels. Cell Signal. 2008;20(10):1815–21.
Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS, Lin RZ. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med. 2012;4(131):131ra50.
Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C, Roden DM. Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation. 2014;130(3):224–34.
Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26(32):5204–12.
Kim TD, le Coutre P, Schwarz M, Grille P, Levitin M, Fateh-Moghadam S, et al. Clinical cardiac safety profile of nilotinib. Haematologica. 2012;97(6):883–9.
Freebern WJ, Fang SH, Slade MD, Wells S, Canale J, Mehgill J, et al. In vitro cardiotoxicity potential comparative assessments of chronic myelogenous leukemia tyrosine kinase inhibitor therapies: dasatinib, imatinib and nilotinib. Blood. 2007;110 (ASH Annual Meeting Abstracts):Abstract 4582.
Postma A, Elzenga NJ, Haaksma J, Schasfoort-Van Leeuwen MJ, Kamps WA, et al. Cardiac status in bone tumor survivors up to nearly 19 years after treatment with doxorubicin: a longitudinal study. Med Pediatr Oncol. 2002;39(2):86–92.
Yusuf SW, Razeghi P, Yeh ET. The diagnosis and management of cardiovascular disease in cancer patients. Curr Probl Cardiol. 2008;33(4):163–96.
Doyle JJ, Neugut AI, Jacobson JS, Grann VR, Hershman DL. Chemotherapy and cardiotoxicity in older breast cancer patients: a population-based study. J Clin Oncol. 2005;23(34):8597–605.
Aksoy S, Aksoy H, Harputluoglu H. Treatment of comorbidities besides the treatment of primary tumor may further increase effective management of cancer. Med Hypotheses. 2006;67(4):744–6.
Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62(4):760–81.
Gupta A, Lawrence AT, Krishnan K, Kavinsky CJ, Trohman RG. Current concepts in the mechanisms and management of drug induced QT prolongation and torsade de pointes. Am Heart J. 2007;153(6):891–9.
Varterasian M, Fingert H, Agin M, Meyer M. Consideration of QT/QTc interval data in a phase I study in patients with advanced cancer. Clin Cancer Res. 2004;10(17):5967–8.
National Cancer Institute. Cancer therapy evaluation program, common terminology for adverse events, version 3.0, DCTD, NCI, NIH, DHHS, 2006. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 30 Nov 2014.
Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(10):e385–484.
Committee for Medicinal Products for Human Use. ICH note for guidance: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs (ICH E14) (CHMP/ICH/2/04). London: EMA; 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002879.pdf. Accessed 20 Nov 2014.
Food and Drug Administration. Product reviews and labels. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. Accessed 20 Nov 2014.
Cheng H, Kari G, Dicker AP, Rodeck U, Koch WJ, Force T. A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ Res. 2011;109(12):1401–9.
Pekkanen-Mattila M, Chapman H, Kerkelä E, Suuronen R, Skottman H, Koivisto AP, et al. Human embryonic stem cell-derived cardiomyocytes: demonstration of a portion of cardiac cells with fairly mature electrical phenotype. Exp Biol Med (Maywood). 2010;235(4):522–30.
Cohen JD, Babiarz JE, Abrams RM, Guo L, Kameoka S, Chiao E, et al. Use of human stem cell derived cardiomyocytes to examine sunitinib mediated cardiotoxicity and electrophysiological alterations. Toxicol Appl Pharmacol. 2011;257(1):74–83.
Morganroth J, Shah RR, Scott JW. Evaluation and management of cardiac safety using the electrocardiogram in oncology clinical trials: focus on cardiac repolarization (QTc interval). Clin Pharmacol Ther. 2010;87(2):166–74.
Acknowledgments
We thank Paloma Vaquero for her invaluable technical assistance.
Conflicts of interest
Juan Tamargo, Ricardo Caballero and Eva Delpón have no conflicts of interest that are directly relevant to the content of this review.
Funding
This work was supported by the Instituto de Salud Carlos III (PI11/01030, SAF-2011/30088, SAF-2011/30112, Red HERACLES RD06/0009 and Red Española de Investigación Cardiovascular RD12/0042/0011) and Comunidad Autónoma de Madrid (S2010/BMD-2374).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tamargo, J., Caballero, R. & Delpón, E. Cancer Chemotherapy and Cardiac Arrhythmias: A Review. Drug Saf 38, 129–152 (2015). https://doi.org/10.1007/s40264-014-0258-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-014-0258-4