
Abstract
Niemann-Pick disease type C (NPC) is a lysosomal storage disorder that presents with a spectrum of clinical manifestations from infancy and childhood or in early or mid-adulthood. Progressive neurological symptoms including ataxia, dystonia and vertical gaze palsy are a hallmark of the disease, and psychiatric symptoms such as psychosis and mood disorders are common. These latter symptoms often present early in the course of NPC and thus these patients are often diagnosed with a major psychotic or affective disorder before neurological and cognitive signs present and the diagnosis is revised. The commonalities and characteristics of psychotic symptoms in both NPC and schizophrenia may share neuronal pathways and mechanisms and provide potential targets for research in both disorders. The neurobiology of NPC and its relationship to the pattern of neuropsychiatric and cognitive symptoms is described in this review. A number of neurobiological models are proposed as mechanisms by which NPC causes psychiatric and cognitive symptoms, informed from models proposed in schizophrenia and other metabolic disorders. There are a number of symptomatic and illness-modifying treatments for NPC currently available. The current evidence is discussed; focussing on two medications which have shown promise, miglustat and hydroxypropyl-β-cyclodextrin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Niemann-Pick disease type C is a rare but important differential diagnosis in the setting of major psychiatric illness with concomitant neurological and cognitive impairments. |
Psychosis is the most common presentation particularly in adolescence and early adulthood. |
Early diagnosis is important particularly with the emerging availability of illness-modifying treatments. |
1 Introduction
Niemann-Pick disease type C (NPC) is an autosomal recessive inborn error of lipid metabolism caused by a mutation in the NPC1 (GenBank accession no. NM_000271.4) or NPC2 (GenBank accession no. NC_000014.9) genes [1], with a combined estimated prevalence of approximately 1:89,000 live births [2]. Recent genetic studies have determined that a late-onset NPC phenotype has a significantly higher incidence of 1/20,000–39,000 [2]. Alterations to the function of NPC1, a large transmembrane protein, and NPC2, a much smaller cytosolic protein, result in disruption of intracellular cholesterol trafficking, particularly between the late endosome and early lysosome [3]. This disruption results in accumulation of cholesterol and glycosphingolipids in lysosomes, particularly in the brain, spleen and liver [4].
The severity of illness, and its onset, is determined by the degree of functional disruption of cholesterol trafficking in NPC. More severe disruptions result in early-life onset, but less severe disruption results in later onset, in adolescence through to mid-life, with a slower illness course. Early-onset forms tend to present with a preponderance of visceral symptoms, with adult presentations presenting predominantly with neurological and neuropsychiatric symptoms [1, 5, 6].
Affected patients display marked clinical heterogeneity [7], including a range of neurological symptoms (such as ataxia, dystonia, vertical gaze palsy, and gelastic cataplexy and seizures) and psychiatric illness, particularly psychosis [8]. NPC can occur across the lifespan, despite previously being mainly recognised as a childhood disease [1, 9, 10], although the proportion of newly diagnosed cases with onset in adolescence or adulthood has increased [10, 11].
As with other treatable inborn errors of metabolism, it is important for the diagnosis of NPC to be made in a timely fashion, as delayed treatment may critically impact early life neurodevelopment [12]. Although treating for a primary psychiatric illness is not harmful in itself, a delay in commencing treatment for NPC—shown to be effective for stabilising neurological symptoms and improving outcomes [13, 14]—may result in irreversible illness progression. Diagnosis is aided by clinical tools such as the NPC Suspicion Index [15], plasma biomarkers such as oxysterols, lyso-SM-509 and bile acids, and genetic testing via gene panels and next-generation sequencing [16].
Given that NPC often presents with psychiatric symptoms prior to the onset of more frank neurological and cognitive symptoms (beyond those associated with ‘typical’ major mental illness), it is critical for clinicians to have a framework for detecting the neuropsychiatric symptoms that may herald the onset of an ‘organic’ psychotic illness such as NPC [17].
The importance of making a timely diagnosis of NPC is highlighted by a recent systematic review of psychiatric symptoms in NPC by Bonnot et al. [7], which examined the temporal course of symptoms in NPC, and the relationship between psychiatric and neurological symptoms. They found that psychiatric manifestations were present in the majority of patients, with psychosis present in 62% and mood symptoms in 38%. Of those with psychosis, half had delusions and approximately a third had visual and/or auditory hallucinations. Strikingly, 39% of this patient group had been misdiagnosed with schizophrenia at some point, and diagnostic delay for some patients has been measured not just in years, but in decades. This diagnostic delay inevitably results in a delay in instigating treatment, such as miglustat (see Sect. 6). As NPC is one of the few neurodegenerative disorders with demonstrated illness-modifying treatment available, minimising any diagnostic delay is crucial to prevent irreversible damage [18,19,20].
2 The Neurobiology of Niemann-Pick Type C (NPC)
The disrupted lipid homeostasis that occurs as a result of impaired cholesterol trafficking in NPC affects a number of intracellular pathways, including glycosphingolipid synthesis (with the accumulation of toxic levels of GM2 and GM3 gangliosides), oxidative stress, lysosomal calcium homeostasis, Rab-mediated vesicle trafficking, fusion of lysosomes and autophagy [21]. At an ultrastructural level, NPC presents with changes commonly seen with other lysosomal storage disorders (LSDs), including neuronal distension, the formation of meganeurites, axonal swelling and formation of axonal spheroids, and ectopic dendritogenesis [22].
Interestingly, there are pathological similarities between NPC and other neurodegenerative disorders such as Alzheimer’s disease (AD). Neurofibrillary tangles (NFTs), tau hyperphosphorylation, altered transition metal homeostasis and β-amyloid accumulation occur in both NPC and AD [21, 23,24,25]. Amyloid precursor protein function may be modulated by cholesterol levels, and apolipoprotein E (APOE), in particular the E4 isoform, influences disease progression in both NPC and AD [26, 27]. Recent studies have also suggested mutations in NPC genes as risk factors for AD, further suggesting bidirectional links between AD and NPC [28]. Recent studies, however, have demonstrated some differences between NPC and AD. For example, NFTs in NPC—otherwise ultrastructurally identical to those seen in AD [23, 24, 29]—tend to have a different distribution, primarily involving subcortical structures such as the thalamus, hippocampus and striatum with sparing of the cerebellum, compared to a more cortical distribution in AD [24, 30, 31]. Furthermore, it has been suggested that, although NFTs are a common feature of NPC, they may be a non-specific reaction to abnormal storage material rather than having a clearer relation to amyloid pathology, as is the case in AD. Finally, although there is β-amyloid accumulation, amyloid plaques, one of the hallmarks of AD, are not characteristic of NPC [21].
Animal studies have also shown a strong neuroinflammatory axis to NPC, with microglial activation and reactive astrocytosis shown to be significant and widespread [32, 33]. Non-steroidal anti-inflammatory medication has demonstrated neuroprotective effects in addition to current standard treatments [34, 35]. Microglial changes may be a primary pathological event in neurodegeneration in NPC, as has been suggested for other neurodegenerative diseases. Additionally, the formation and maintenance of central nervous system (CNS) myelin by oligodendrocytes is heavily NPC1-dependent, and impaired lipid uptake and intraneuronal transport results in impaired oligodendrocyte maturation [36] and significant and marked hypomyelination of white matter [37, 38]. The links between these seemingly parallel and distinct pathways of neurodegeneration are far from elucidated, but some intriguing relationships have emerged, such as increased glial activation resulting in the production of interleukins and APOE and the activation of p38–mitogen-activated protein kinase (MAPK). This leads to hyperphosphorylated tau, destabilisation of the microtubules to which tau binds, and thus ultrastructural change to neurons [39] and altered induction of cellular autophagy [40].
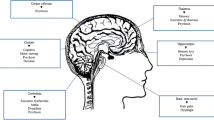
As in most neurodegenerative diseases, some neuronal types, and hence brain regions, are more affected than others. In animal and human models, γ-aminobutyric acid (GABA)-ergic neuronal populations in the cerebellum, brainstem, hippocampus and basal ganglia are particularly affected [41] by neuroaxonal dystrophy, and it is in these regions that the greatest intracellular storage of gangliosides occurs [33, 42], with the most fulminant loss in the cerebellum and thalamus [33, 43, 44]. NFTs also appear to accumulate in these same regions (other than the cerebellum) [23, 24], and neuroinflammation also appears to be most pronounced in cerebellum, thalamus and basal ganglia [45]. This suggests that these regions are where significant cellular pathological ‘overlap’ occurs, with toxic gangliosidosis, tauopathy and ultrastructural morphological change to the neuron all converging. An illustration of this convergence, and the cellular changes driving this, can be seen in Fig. 1.

The progression of neurobiological through to morphological changes in Niemann-Pick disease type C, and the resulting clinical syndromes. a Cellular structural changes; and b key affected brain regions and their relationship to symptom production
These findings have largely been confirmed by human neuroimaging studies, which have revealed widespread grey and white matter changes in NPC. Diffusion tensor imaging (DTI) analyses have demonstrated altered white matter integrity and significant axonal and myelination changes, for example decreased fractional anisotropy in major white matter tracts such as the corona radiata, cingulate gyrus and, in particular, the corpus callosum [46,47,48,49]. Slower rates of white matter changes have been demonstrated in patients treated with miglustat [20, 49]. At a volumetric level, brain magnetic resonance imaging (MRI) and voxel-based morphometry studies have demonstrated widespread changes such as atrophy in the cerebellum, thalamus, hippocampi, caudate nuclei and cortex, an increased pontine-to-midbrain ratio and, once again, slowed progression of volume loss in patients treated with miglustat [18, 20, 49,50,51,52,53,54].
3 Psychiatric Illness in NPC
The delay in diagnosis of NPC is representative of one of the key challenges in metabolic disorders, as the key symptoms of the disease are non-specific, heterogeneous (particularly in adulthood) and may be overshadowed by the presentation of psychiatric illness [55]. The visceral symptoms seen in childhood are often mild, fluctuating and rarely clinically significant in adults, and are likely to be overlooked [8]. The motor signs, particularly dystonia, may be attributed to the extrapyramidal side effects (EPSE) of antipsychotic medications, ataxia may be difficult to assess, and vertical gaze palsy is missed more often than not [7]. The temporal sequence of events is often crucial. The psychiatric presentation of NPC may co-occur alongside subtle neurological changes, but commonly precedes more frank cognitive or motor symptoms by several years, which can lead to an established ‘primary’ psychiatric diagnosis, with new-onset motor and cognitive symptoms often attributed to a psychiatric illness and/or its treatment.
3.1 How Common Are Psychiatric Presentations of NPC?
The first reports of psychosis as the presenting symptom of NPC in adolescents and adults were published more than two decades ago. These reports highlighted that NPC often presents with adolescent-onset psychosis [3, 56,57,58], with cognitive and neurological symptoms often presenting to a degree greater than expected for ‘typical’ schizophrenia. The first series of patients published by Walterfang et al. [3] focused on the likelihood of subtle disconnection being an early feature of neurodegeneration in adolescence and early adulthood in NPC, which may have underpinned the initial presentation being psychiatric, prior to the obvious motor and cognitive symptoms that occur with more frank neurodegeneration. Since this time, numerous similar cases have been published [59,60,61,62,63,64,65,66,67,68], suggesting that the presentation of significant neuropsychiatric illness is common in later-onset NPC patients.
A number of reviews and larger series of adult NPC patients have confirmed an over-representation of psychiatric illness, particularly psychosis, in this age group. Sévin et al. [1] described 13 published cases with onset after the age of 15 years, with up to 40% of patients presenting with a psychotic illness during young adulthood. Maubert et al. [69] reviewed 22 adult patients in France by interviewing patients, relatives and their doctors, finding that psychiatric illness occurred in 86% of patients, 55% with psychotic illness and 36% with depression. When psychiatric diagnoses were made, the chief diagnoses were schizophrenia and major depression, and the median age at onset of psychiatric symptoms was 20 years. Half of these patients had been hospitalised psychiatrically, and most had received antipsychotics. In a review of patients entered into an international NPC registry, Bonnot et al. [70] found that of 386 registry patients, one-third had psychiatric symptomatology, with the most common diagnoses being of a psychotic illness (43%) and mood disorder (39%). The mean age at onset of psychiatric symptoms was 18 years of age, preceding the confirmed diagnosis of NPC by almost 6 years [70] and confirming that diagnostic delay after a psychiatric diagnosis is very common. Psychiatric manifestations present before or at the onset of neurological symptoms in up to 76% of cases [7], suggesting that clinical psychiatrists in particular need to be vigilant for atypicality of symptoms that occur in psychiatric illness that may point to a secondary, rather than primary, illness (see Table 1).
3.2 Psychosis
Psychosis is a common manifestation in adolescent/adult NPC, occurring in up to half of patients, and less commonly can be the sole presenting feature [7, 52]. In cases presenting with schizophrenia-like psychosis alone, these patients can undergo treatment with neuroleptic medications for 5–10 years prior to the onset of defining neurological and/or cognitive symptoms resulting in a diagnosis of NPC being made [1, 3, 71].
Distinguishing psychotic symptoms in NPC as compared to a primary schizophrenic illness can be extremely difficult based on phenomenology alone, given the overlap of commonly reported symptoms between the disorders, such as auditory hallucinations, paranoid delusions and ideas of reference [7, 52]. However there may be an elevated rate of visual hallucinations in NPC, with international registry data finding visual hallucinations reported in 36% of psychotic patients [7], which would be otherwise uncommon in schizophrenia. Treatment resistance, usually defined as insufficient response to two trials of therapy with a second-generation antipsychotic, is common in NPC, occurring in approximately half of patients, and should be considered a red flag for diagnostic revision [7]. Neuroleptic sensitivity has also been described [3], and clearly the co-presentation of ataxia, vertical gaze palsy and greater-than-expected cognitive impairment are concomitant symptoms that are highly suggestive of NPC or another secondary psychotic illness.
3.3 Mood Disorders
Mood disorders are common in NPC, though less frequent than schizophrenia-like psychosis. They are, however, poorly quantified in the literature, due to categorisation and reporting where the term ‘mood disorder/symptom’ may include depression, emotional lability or irritability, or hypomania. A recent review of psychiatric symptoms in the international registry [70] identified 17 patients with a mood disorder out of 94 patients identified as having psychiatric symptomatology. The overall number may have been higher, as only 56 patients had further information provided other than ‘abnormal’, with 44 patients having a specific psychiatric symptom/disorder.
Whilst a number of studies refer to bipolar affective disorder as a possible psychiatric manifestation of NPC [3, 5], there is only one clearly described case. Sullivan et al. [72] published the first case of bipolar affective disorder type I in a patient with confirmed infantile-onset NPC in 2005. This patient presented at age 25 years with a 2-year history of mania that would last 2–3 weeks and occurred every 1–2 months. Remission was maintained at 18 months’ follow-up after commencement of sodium valproate.
3.4 Other Psychiatric Disorders
A multitude of other neuropsychiatric phenomena are reported in NPC, including autism spectrum disorder and attention deficit disorder in children, obsessive–compulsive disorder, and behavioural problems in childhood and adulthood. The recent review by Bonnot et al. [73] found eight of 44 patients with documented psychiatric symptoms, predominately in those over 18 years of age, had an impairment in impulse control.
4 Cognitive Impairment in NPC
Although it may not be part of the initial presentation, progressive cognitive decline is characteristic of the phenotype of NPC [1, 74]. In a third of cases, cognitive dysfunction is a presenting symptom of NPC and it is reported in 60–70% of cases across all age groups in multiple reviews, including those of international registry data [1, 8, 30]. As a late feature, most if not all NPC patients will experience progressive cognitive decline, meeting criteria for dementia. Early recognition of the cognitive phenotype of adolescent/adult NPC is an important aspect of assessment to avoid significant diagnostic delay and/or misdiagnosis.
Despite the prevalence of cognitive impairment and its negative impacts on functioning and quality of life, detailed characterisation of the specific cognitive profile of NPC and its progression is scarce and/or inconsistently reported in the literature. The literature consists predominantly of case reports/series with limited psychometric testing, single-timepoint assessment and various published methodologies resulting in inconclusive findings. For example, many studies report only global cognitive functioning index scores and/or full-scale IQ scores [57]. The Mini-Mental State Examination (MMSE) is often used as a rudimentary cognitive screening tool due to its brevity, including in medication trials [13]. However, the MMSE is not sensitive to identifying the types of early cognitive impairments found in NPC, including changes in higher-order attention and executive function. In a cross-sectional study by Walterfang et al. [47] in 2010, cognitive impairment was measured by the MMSE score and full-scale IQ, with scores ranging from 13 to 30 and 55 to 76, respectively, as compared to the expected range for normal controls of 28+ for the MMSE and 90–110 for full-scale IQ. These cognitive measures were also utilised in a later study by the same group examining ocular–motor function and its relationship to cognition [75]. Two paediatric studies have utilised the MMSE to characterise cognitive improvements in patients treated with miglustat [76, 77], and the tool has also been shown to demonstrate differences between NPC and early-onset AD patients [78]. In a further study in 2013, Walterfang et al. [79] used the global score of the Neuropsychiatry Unit Cognitive Assessment Tool (NUCOG) for nine adult NPC patients as an illness variable for imaging analyses [54], demonstrating a range of 49–94 out of a maximum score of 100 in comparison to the cut-off score for normal function of 80 or above. To date, only four publications have reported a comprehensive neuropsychological battery assessing all major cognitive domains on cross-sectional adolescent/adult NPC cohorts, as shown in Table 2 [78, 80,81,82].
4.1 Is There a Cognitive Profile in NPC?
Careful characterisation of the typical cognitive profile of NPC is critical, as impairments in NPC can be mistaken for other disorders, including primary psychiatric disorders [83] and other neurodegenerative disorders [78]. However, the degree and pattern of neuropsychological impairments varies considerably in adolescent/adult patients with NPC [1, 80, 81]. Neuropsychological testing in adolescent/adult-onset case reports often reveals a steady decline throughout adulthood, with significant deficits in executive function and memory [56, 57, 84, 85]. A systematic review of case reports based on 23 patients reported executive dysfunction as the most frequent deficit in adult NPC patients [30]. In qualitative descriptions of patients’ cognitive profiles, executive dysfunction was the most frequent feature (13/23) followed by memory disturbance (12/23), visuospatial deficits (6/23) and aphasia (4/23). Unfortunately, in these cases, cognitive profiles were inconsistently reported, with very few studies reporting actual test results.
Hinton et al. [80] reported data on 14 patients with severe impairments in verbal fluency, planning and reasoning, and memory, as well as impairments in naming, non-verbal reasoning and drawing skills. Word knowledge (vocabulary) was preserved. All patients exhibited a lack of insight into the degree of deficits. Supporting these findings was Klarner et al. [81] 2007 pilot study of ten adolescent/adult patients, where impairments in verbal fluency, information processing speed, divided attention, fine-motor coordination, visuospatial organisation and executive functions were reported in the early stages of disease. In most patients, there was an early decline in executive functioning, with verbal memory impairment becoming apparent in the moderate stages of illness. Visuospatial working memory was less affected than verbal working memory, and not until later stages of disease. They also noted a frontal lobe behavioural syndrome, with evidence of perseveration and loss of interpersonal distance that manifested as excessive familiarity. More recently, Heitz et al. [82] conducted a retrospective study of 21 adult patients and found that executive functions (85% of the sample had impairment at baseline) and attention (92% of the sample had impairment at baseline) were the most impaired cognitive functions. Visual and verbal attention span scores on the Frontal Assessment Battery were impaired in the majority of patients. The most preserved skill was episodic memory (preserved in 61.5% of cases).
4.2 Cognitive Profile: Differential Diagnoses
Adult-onset NPC is often under-diagnosed, and should be considered in young adults with symptoms that include psychosis with cognitive impairment and/or neurological symptoms such as ataxia and gaze palsy [83]. Although NPC and AD both result in progressive cognitive decline and share pathophysiological features, their neuropsychological profiles differ. Notably, the cognitive impairments in NPC typically occur at a younger age than in early-onset AD. The cognitive profile in AD is typically characterised by predominant and early episodic memory dysfunction (largely amnestic or rapid forgetting, where recall is not aided by cues), followed by impairments in executive, language and visuospatial domains (for a review, see Salmon [86]). In contrast, the cognitive profile in NPC reflects a more subcortical profile reflecting the neuropathology preferentially affecting subcortical grey matter nuclei (thalamus, basal ganglia, hippocampus) and cerebellum, and later the neocortex, with the resulting disconnection of fronto-striatal circuits. This fronto-striatal disconnection leads to psychomotor slowing and prominent executive dysfunction, prior to memory and more global impairments developing. This was demonstrated in Johnen et al. [78] paper, which directly compared the neuropsychological profile of adult NPC patients with a reference group of adults with confirmed early-onset AD. The key neuropsychological difference between NPC and early-onset AD was that verbal memory performance [as measured by the Rey Auditory Verbal Learning Test (RAVLT), a list-learning test] was worse in the AD cohort [78], and early-onset AD patients showed wider, more generalised and severe impairments. Executive functions (as measured using Trail Making Test A/B) and word fluency (measured by a lexical and semantic fluency task) were the most frequently impaired in their NPC cohort.
An additional differential diagnosis worthy of consideration in older adult patients is frontotemporal dementia (FTD), which, like NPC, can present with neuropsychiatric illness, often associated with early executive impairments, before more frank cognitive impairments ensue. Notably, accumulation of tau and transactive response DNA binding protein 43 kDa proteins seen in FTD has also been described in NPC [87, 88]. Psychotic symptoms have been reported in both FTD [89] and NPC [3], and—as frequently seen in NPC—patients with FTD may be misdiagnosed with schizophrenia or bipolar disorder prior to the accurate diagnosis of dementia [90]. However, neurological symptoms such as ataxia and gaze palsy would be highly atypical in FTD, as would hepatosplenomegaly. However, to date no study has compared the executive function findings in FTD with NPC to determine if the profile and/or severity of executive impairment differs across the two disorders.
Differentiating between the cognitive impairments of NPC and a primary psychiatric psychotic disorder such as schizophrenia is more challenging. Indeed, cognitive dysfunction is a core feature of schizophrenia and commonly includes similar deficits to those seen in NPC, predominantly in the domains of attention, working memory, verbal learning and memory, and executive functions [91]. However, the impairments seen in schizophrenia are posited to exist within a more widespread reduction in general cognitive functioning [92]. Additionally, the cognitive impairment of NPC typically progresses at a more rapid rate than that of schizophrenia, where cognitive impairment tends to be stable throughout the course of the illness in most patients [93]. Patients with NPC may be resistant to treatment with antipsychotic medications and can also show paradoxical worsening as a response [94]. The frank neurological symptoms and motor manifestations of NPC (including ataxia, vertical supranuclear gaze palsy, dystonia, rigidity, tremor and pyramidal signs) should also prompt a review of a primary psychiatric diagnosis.
5 The Origin of Psychiatric and Cognitive Symptoms in NPC
A number of LSDs present with neuropsychiatric symptoms at various stages in the disease process. Neurons, which are highly metabolically active cells, are very sensitive to derangements in metabolic processes—which may alter the synthesis of cellular components essential to neuronal function, or result in the accumulation of substances that are neurotoxic [95]. If these disruptions are severe, gross neurodevelopmental disturbance and encephalopathy occurs, but if they are mild and/or slowly progressive, their effects may be more subtle and/or delayed. Symptoms that occur as a result of derangements to neuronal development and metabolism appear to have a hierarchy of sorts, with more basic homeostatic brain functions requiring severe disruption to cause symptoms (such as coma and seizures), whereas mild disruption to CNS metabolic processes appears to cause more subtle alterations to cognition and emotion. The underlying maturational state of the CNS also plays a significant role, and metabolic disorders whose metabolic effect has a cumulative effect on neuronal function (such as LSDs) will often have their impact on later neurodevelopmental processes if mild, or very early neurodevelopmental processes if severe [96].
It is when the impingement of a progressive disorder upon CNS maturational trajectories is later in its onset that neuropsychiatric illness is likely to be the first presentation [71], and when a (mis)diagnosis of a primary rather than secondary psychiatric illness is likely to occur. Three-quarters of all psychiatric illness presents prior to the age of 24 years [97], and when disruption of neuronal function by a metabolic or storage disorder crosses the threshold that results in functional impairment of neuronal assemblies during the adolescent or early adult period, it is unsurprising that disorders that present in this period are more likely to be heralded by a psychiatric illness.
The key processes that can be disrupted during this period of neurodevelopment include a pruning of over-proliferated synapses in a posterior–anterior direction throughout the cortex and subcortical grey matter, which results in a reduction in grey matter volume [98]. Parallel to this is an expansion in white matter volume [99], largely due to increasing myelination of connections between prefrontal regions, and between dorsolateral prefrontal cortex (DLPFC) and temporoparieto-occipital association areas [100]. The disruption to frontotemporal connectivity in particular appears to be particularly psychotogenic [101], and disruption between DLPFC and association areas produces significant executive disturbance [71]. This is particularly relevant in NPC, where presentation in adolescence and early adulthood appears to result in a markedly elevated rate of psychotic illness [3]. A number of models have been proposed to link psychotic symptoms to neurobiological changes in both schizophrenia and NPC.
5.1 Dysconnectivity as a Substrate for Psychosis
Walterfang et al. [3] first proposed dysconnectivity as a model for underpinning psychosis in NPC, noting the significant evidence for disruptions to both micro- and macroconnectivity in patients and animal models. This model was drawn from the dysconnectivity hypothesis for schizophrenia first proposed by Friston and Frith [102] in 1995, which focused on altered functional, particularly frontotemporal, connectivity in the illness. It was Wernicke [103] who first posited that schizophrenia may result from anatomical disruption of association pathways, and Bleuler [104] in coining the term ‘schizophrenia’ described a functional “decoupling” of psychological processes underpinning the symptoms of the illness. Although the substrate for this dysconnectivity was not specified, many studies have confirmed that subtle changes in anatomical connectivity exist in schizophrenia, and that alterations to either anatomical micro-connectivity (at the synaptodendritic level) and macroconnectivity (at the level of axonal integrity and myelination) may produce psychotic symptoms [105], reflected in impaired functional coupling of the default mode network (DMN) [106]. As an example, Zalesky et al. [107] examined cortico–cortico connectivity via axonal fibre bundles, utilising DTI in 74 patients with schizophrenia, and found evidence of widespread dysconnectivity in white matter architecture compared with healthy controls, resulting in a cortex that was more sparse and less efficient in its connectivity, which further supports the idea of a ‘hub and spoke’ hypothesis contributing to the development of psychosis.
This model for psychosis in NPC is supported by the prevalence of psychosis in metachromatic leukodystrophy (MLD), a rare genetic neurometabolic disorder caused by deficiency of the enzyme arylsulfatase A. Psychosis has been found in up to 53% of adolescent and early adult-onset MLD patients [108]. The key neuropathology in MLD lesions is aberrant myelination, particularly affecting subcortical white matter and with a predilection for frontotemporal connecting fibres. This loss of cortico–cortical and cortico–subcortical connections is postulated to be critical for the development of psychosis in these patients, particularly when it occurs in the ‘psychotogenic window’ of adolescence and young adulthood [52, 108]. Black et al. [109] expand on this model, describing a model of psychosis in MLD involving widespread dysconnectivity and functional disruption, particularly in frontal–subcortical circuits.
Another genetic disorder in which schizophrenia-like psychosis is over-represented (occurring in 30% of young adult patients) is the 22q11.2 deletion syndrome (22Q11DS), often referred to as velocardiofacial syndrome (VCFS) [110]. White matter studies in VCFS patients using DTI have consistently found reduced white matter integrity, particularly in frontotemporal and frontolimbic tracts, including in the long tracts of superior longitudinal fasciculus and uncinate fasciculus [111].
In NPC, widespread and fulminant white matter changes in adults with the disease was first demonstrated by Walterfang et al. [47], and has been subsequently replicated by a number of other groups [20, 48, 49, 53], supporting this model of impaired macroconnectivity as underpinning psychotic illness in NPC patients. However, impairments at the level of macroconnectivity, microconnectivity, or connecting ‘hubs’ may all be, in part, responsible for psychiatric symptoms, particularly psychosis, in NPC.
5.2 Impaired Macroconnectivity and the Corollary Discharge System
If anatomical dysconnectivity at the level of myelinated axons is implicated in the pathophysiology of psychosis in both schizophrenia and NPC, how does this dysconnectivity produce symptoms? One way is through the disruption of the corollary discharge system. Corollary discharges refer to an efferent, or internal, ‘copy’ of an afferent motor signal. The terms corollary discharge and efferent (or efference) copy are often used interchangeably. These internal copies are thought to be transmitted to areas of the brain associated with sensory input as ‘anticipated sensation’ and collated with actual sensory input. When these two inputs are closely correlated this is thought to suppress the sensation associated with self-initiated movement [112]. First Feinberg [113] and later Frith [114] developed the idea that some of the symptoms of schizophrenia, particularly passivity phenomena and auditory hallucinations, might be explained by abnormalities in corollary discharges disrupting a person’s ability to distinguish between events initiated by the self and by the other; for example, internal thoughts are thus ‘mislabelled’ as external, or ‘non-self’, and experienced as external auditory hallucinations.
This theory was expanded upon by Whitford et al. [115], who suggested that structural abnormalities in fasciculi connecting motor-initiation areas of the frontal lobe with temporal and parietal cortices contribute to conduction delays in the efference signal. These delays then contribute to delays in the corollary discharge signals in sensory regions, meaning they occur after the actual sensory input has arrived and do not adequately suppress the sensation generated by self-initiated movement. Studies of auditory function in patients with schizophrenia have found a lack of attenuated response in the auditory cortex, which may account for auditory hallucinatory phenomena [116]. Similarly, this has been demonstrated in studies of patients with auditory hallucinations, passivity phenomena and high levels of schizotypy, who are more likely to experience a ‘ticklish’ sensation from self-applied tactile stimulation [117]. The plausibility for this hypothesis contributing to psychosis in NPC is based on two observations. Firstly, from extrapolating evidence of disrupted white matter integrity in NPC, and how in patients with schizophrenia this results in delayed corollary discharges and an inability to suppress consequences of willed actions [115]; and, secondly, from the recognised impairments in saccadic eye movements in NPC, which rely on efference copies for normal function [118, 119]. Given that white matter disruptions are widespread throughout the brain in both schizophrenia [120] and NPC [47], this makes anatomical dysconnectivity due to impairments to axons and their myelination an attractive substrate for psychosis-causing dysconnectivity in both illnesses.
5.3 Impaired Microconnectivity and Altered Synaptic Functioning
The current understanding of Friston et al.’s [121] ‘disconnection hypothesis’, however, focuses on impairments at the level of the synapse, particularly in disruptions to modulation of NMDA (N-methyl-d-aspartate)-mediated synaptic plasticity and efficacy. The allied ‘glutamatergic hypothesis’, which has usurped the older dopaminergic hypothesis of psychotic symptoms in schizophrenia, posits that glutamatergic over-activity in the prefrontal cortex causes widespread downstream dopaminergic dysfunction in cortical and subcortical regions [122]. There is abundant evidence that synaptic disruption occurs in NPC; NPC1 and NPC2 are richly located in synapses [123, 124] and neurodegeneration tends to occur in nerve terminals and then distal axons before affecting the neuronal soma [42]. Alterations in the glycosphingolipid:cholesterol ratio affect lipid rafts, which alter transductive signalling processes, particularly glutamatergic transmission [125]. Depletions of cholesterol are known to impact synaptic plasticity and transmission in the CA1 region of the hippocampus [126]. In NPC mice, impairments in long-term potentiation (LTP) in the hippocampus have been demonstrated, with a consequent reduction in synaptic plasticity and enhanced glutamatergic transmission [127, 128]. This may in part be due to altered synaptic vesicle turnover in the setting of NPC1 deficit, affecting inhibitory more than excitatory synapses [129], but also impaired lipid domain organisation affecting the NMDA-R (NMDA receptor) transduction pathway, thus affecting LTP induction via defective regulation of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid) receptor trafficking [128]. Notably, these changes have been shown to be at least partially reversible in the mouse model with the imino sugar miglustat [128]. NPC1 deficit appears to particularly impair presynaptic input to Purkinje cells in the cerebellum, one of the key regions of neuronal loss in NPC [130].
How might this synaptic impairment lead to psychosis? The dysconnectivity hypothesis posits that psychotic symptoms such as hallucinations, delusions and passivity phenomena result from the brain generating faulty ‘inferences’ to process and understand sensory phenomena [121]. Impaired modulation of synaptic efficacy, or postsynaptic gain, results in an inability to either augment or ignore the encoded precision (or uncertainty) of sensory evidence to allow for the selection of salient information that contextualises and updates our experiences [131]. Thus, the failure to precisely contextualise sensory evidence results in internal experiences being experienced as external, or beliefs or experiences being ascribed an abnormal (faulty) salience that results in ideas of reference or persecutory delusions [132]. These symptoms are seen in patients with schizophrenia, in addition to NPC patients with psychosis.
5.4 Impairment to Key Regional ‘Hubs’ Disrupting Network Activity
The large-scale networks that facilitate connectivity in the brain between disparate brain regions are composed of more than the axons that form the ‘wiring’ of these pathways: a number of richly connected network ‘hubs’ have a key role in the global topology of brain networks [133]. Crossley et al. [134] used DTI analysis in healthy volunteers to show that the most connected nodes were located in posterior cingulate cortex, thalamus, basal ganglia and hippocampus. They also showed computationally that when these nodes were affected, they disproportionately affected (and functionally degraded) these complex networks, and found that these nodes, in addition to insula, anterior cingulate, dorsolateral prefrontal and superior parietal cortex, tended to be preferentially affected in brain disorders such as schizophrenia and AD. The reason that these nodes may be more vulnerable to illness may be as they are more biologically ‘expensive’, with higher blood flow, glucose metabolism and longer connection distance [135]; thus, they may be preferentially affected in brain-wide disorders of metabolism, as they are the most metabolically active. Similarly, a disease that disrupts long-distance axonal projections or transport mechanisms may similarly disproportionately affect these hubs. It is known that this is characterised by a selective dysconnectivity amongst these central hub regions of the brain [120, 136], and in NPC these regions appear to be those most affected by altered cholesterol metabolism: axonal spheroids occurring in hippocampus, basal ganglia and cortex [41]; excess GM2/3 ganglioside storage occurring in thalamus, hippocampus, brainstem and cerebellum [42, 137, 138]; and NFTs in basal ganglia, hippocampus and cortex [23, 24]. These are also the regions of greatest grey matter tissue loss seen on MRI scans in NPC patients [47], and that neuropathological changes are greatest in these ‘hub’ regions may reflect their vulnerability to the metabolic and cellular derangements that occur with impaired intracellular cholesterol trafficking.
The cerebellum is the region where neuronal loss in NPC is the most fulminant [139], and this is reflected in the marked ataxia and ocular–motor impairments seen in adult and paediatric patients with the disease [6]. Cerebellar changes may at least partially underpin the executive changes seen in NPC patients [30]. The cerebellum is linked to the cerebrum through the cerebral peduncles and, through its rich connectivity to cortical and subcortical structures, has distinct contributions to a number of intrinsic connectivity networks including bilateral executive control networks, the salience network and the DMN [140]. Impairments in cerebellar functional connectivity, particularly to the thalamus and frontal cortex, have been demonstrated in schizophrenia [141], and suggests that the significant cerebellar neuronal loss that occurs in NPC patients may disrupt DMN connectivity and contribute to the genesis of psychotic symptoms.
Similarly, limbic structures in the medial temporal lobe have also been intimately implicated in the pathophysiology of schizophrenia, and have been seen as central to the neuropathology of the illness—particularly the hippocampus [142]. The hippocampus plays a central role in the formation of new declarative memories, and is particularly specialised for conjunctive memory formation (where inputs from multiple cortical regions are bound into an integrated memory trace) and pattern completion (where an extended representation is retrieved from a partial input). It is known to be particularly affected in schizophrenia at a neuronal [143], macroscopic [144] and functional [145] level. Current hypotheses focus on dysregulation of glutamatergic transmission affecting the CA1 region of the hippocampus (a subfield which has a high density of NMDA and AMPA receptors) which induces attenuated psychotic symptoms through CA1 hyperactivity, and progresses to psychosis as this dysregulation expands to projection fields within the hippocampus and also the frontal cortex, with reduced hippocampus and insula neuropil and interneurons [146]. Additionally, overactivity in the CA3 region of the hippocampus in schizophrenia, which situates objects within space and time, may lead to inappropriate binding of weakly related sensory representations, thus leading to hallucinations [147]. The dysregulation across these regions may produce pathological pattern completion, and its converse, pattern separation, resulting in false associations, referential thinking and delusional ideas [142]. The hippocampus is a convergent site of cholesterol accumulation, particularly in the CA3 region [148], as it is for ganglioside excess, NFT formation and neuronal loss in NPC, and volumetric reductions of the hippocampus have been shown in adult NPC patients [47, 51]. Enhanced glutamatergic neurotransmission has been shown in NPC mouse models [149], and the resultant hyperexcitable hippocampal neuronal populations may underpin seizure activity in the disorder, but could equally underpin psychotic symptoms. Additionally, hippocampal subfields in adult NPC patients have been examined, demonstrating prominent reductions in volume in CA1 and CA3, but not CA2, regions over time (Walterfang et al., unpublished data). Other medial temporal lobe structures may play an additional role. The insula plays a key role in processing of sensory inputs and with visual–tactile and auditory–visual integration [150] and shows reduced surface area in schizophrenia [151] with abnormal activation apparent on fMRI (functional MRI) in auditory hallucinations [152]. Additionally, this is an area of significant NFT accumulation [153] and regional volume loss in adult NPC patients. Abnormal activation of the insula results in unbalanced sensory–memory integration in hallucinations, and may contribute to the deficits in discriminating ‘self’ from ‘other’ in schizophrenia [150]. Other subcortical ‘hub’ regions that have been shown to be sites of significant neuronal loss and NFT formation in NPC such as the thalamus and basal ganglia [23, 24, 45, 47, 51] have similarly been demonstrated as key regions of pathology in, and implicit in psychiatric and cognitive symptoms of, schizophrenia [154, 155].
5.5 The Origins of Cognitive Impairment
The currently available, albeit limited, literature suggests that NPC results in a progressive dementia, with the predominant cognitive profile being an early and prominent impairment in executive and attention domains that progresses to involve other cognitive domains (including memory, visuospatial skills, language and psychomotor speed). The loss of cortico–cortical and cortico–subcortical connections involving the fronto-striatal circuits has been postulated to be the key reason for cognitive dysfunction in these patients, with thalamic and hippocampal reductions underpinning memory deficits, and cerebellar and striatal pathology, in combination with frontal cortical changes, interacting synergistically to lead to the prominent and early executive deficits seen in adult NPC [30, 52].
Given that the cerebellum is the site of most fulminant neuronal loss in NPC [43, 44, 137], despite the absence of NFTs in this brain structure (possibly due to lower levels of tau in Purkinje neurons [31]), it is likely that its contribution to cognitive impairment in NPC is significant. It has long been known that the cerebellum contributes input to the prefrontal cortex in the same way as it does the motor cortex [156], although the polysynaptic circuitry (entering cerebellum via a relay station in the pons, and exiting via relay stations in deep cerebellar nuclei and the thalamus) [157] connecting it to frontal cortex meant that antero/retrograde tracing techniques—which were predicated on monosynaptic connections—prevented its deep connectivity to frontal cortical regions being recognised for many years [158]. As noted by Schmahmann [159] almost three decades ago, in outlining the concept of cognitive dysmetria, “in the same way as the cerebellum regulates the rate, force, rhythm, and accuracy of movements, so may it regulate the speed, capacity, consistency, and appropriateness of mental or cognitive processes”. Thus, the striking loss of cerebellar neurons that occurs in NPC causes not only ataxia but dysmetria of thought, including altered executive control, working memory and language function [160]. The ‘cerebellar cognitive affective syndrome’ was described in patients with cerebellar damage, and consists of impairments in planning, flexibility, abstract reasoning, verbal fluency and working memory, particularly when the posterior cerebellum was most affected [161]. Notably, these corticocerebellar loops also pass through the thalamus, perhaps the next most significant site of neuronal loss in NPC [41, 43, 44], suggesting that these loops are further compromised by thalamic pathology in the disease. The thalamus itself is also intimately involved in cognitive control. Once thought to passively relay information to the cortex, thalamic neurons are known to modulate neural processing along pathways according to behavioural context. As such, they contribute to attentional control and new learning [162], and lesions to higher-order thalamic nuclei are known to produce severe attentional and memory deficits [163], in addition to impaired cognitive flexibility [164]. In NPC, this could be seen to compound the memory and executive deficits secondary to cerebellar pathology. Other areas of significant neuronal loss in NPC, including the hippocampus and basal ganglia, would be expected to result in declarative memory disturbance due to the former and executive dyscontrol and slowed information processing from the latter. When dysfunction in these key relay and modulation ‘hubs’ is considered, the pattern of cognitive impairment that could be expected—and which has largely been demonstrated in the small number of studies reviewed here—reveals itself as leading with executive disturbance, attentional impairment and cognitive slowing before more frank memory impairment emerges [30].
However, one apparent paradox to these models is that, whilst all patients with NPC will develop significant motor and cognitive impairment as the illness progresses, psychiatric illness is not integral: that is, it does not present in all patients who develop the disease. Psychosis may present in up to half of adolescent or young adult patients (markedly and significantly higher than the rate of schizophrenia in the non-NPC population—an 0.5% lifetime prevalence [165]), but psychotic symptoms rarely present in younger patients or those patients older than 40 years of age [3] (Table 1). One possible explanation for this may be drawn from other neurometabolic disorders that present with markedly higher rates of schizophrenia-like psychosis. MLD, a predominant disorder of anterior myelination, presents with a markedly elevated rate of psychosis in young adult patients [108]. Similarly, GM2 gangliosidosis (Tay Sach’s disease [TSD]) is a disorder of hexosaminidase A metabolism that leads to the neurotoxic accumulation of GM2 gangliosides, one of the gangliosides stored to excess in NPC, and where cerebellar and thalamic neurons are particularly affected and rates of psychosis, which often precedes motor symptoms by some years, in young adult patients approach 30–50% [166]. In both TSD and MLD, as in NPC, pre-adolescent patients show developmental delay and seizures [167, 168], but rarely psychosis; older patients also tend to present with cognitive and motor impairment, but again psychosis is rare [71]. These two disorders, with white matter and cerebellar/thalamic pathology, respectively, have significant neuropathological overlap with NPC, and present with similarly elevated rates of schizophrenia-like psychosis that frequently precedes cognitive and/or motor decline—but generally only when onset occurs between 15 and 30 years of age, described as the ‘psychotogenic window’ for neurometabolic disease [71]. Although the pathology of NPC, TSD and MLD differs significantly at a cellular level, the strikingly homologous elevations in psychosis in these disorders suggests that the commonality may be the late neurodevelopmental processes that are disrupted, rather than the metabolic processes themselves, which are ‘psychotogenic’. Alterations to frontotemporal and fronto-subcortical myelination, prefrontal dopaminergic fibre and transporter density increases and pruning, and frontotemporal pruning of NMDA receptors all occur during this phase; thus, the onset of significant pathology during this window could underpin the propensity towards psychosis seen in this period, but not before or after it. However, even when onset occurs during this period, psychosis is not inevitable; it is feasible that some individuals may have protective factors, which may include genetic modifiers at a cellular level or developmental or acquired ‘redundancies’ in disrupted networks that render them more robust to developmental disruptions.
An integrative model that connects changes in NPC patients at a cellular level through to altered micro- and macroconnectivity at the level of ‘hubs’ (subcortical grey matter regions) and ‘spokes’ (interconnecting white matter fibres) and their associated networks, resulting in cognitive and psychiatric symptoms, is outlined in Fig. 1.
6 Management of Psychiatric and Cognitive Symptoms in NPC
6.1 Symptomatic Treatments
Treatment of the most common neuropsychiatric presentation of NPC—a schizophrenia-like psychosis—has been described in numerous published case reports. High doses of antipsychotics are often required to achieve remission, which is often partial at best [1, 3, 62, 169]. Typical antipsychotics, in particular, are more likely to cause EPSE, particularly dystonia, which is generally a progressive and emergent neurological feature of the disease itself [62, 169]. Depression, when present, has been reported to respond to traditional antidepressant therapy, such as selective serotonin reuptake inhibitors [60], and hypomania has been shown to respond to sodium valproate [3, 72]. However, the limited data available limits the generalisability of these findings, and the effectiveness of symptomatic treatments for psychiatric illness in NPC remains largely unknown [69]. Although treatment with donepezil, a cholinesterase inhibitor that can improve cognitive impairment in AD, has been shown to enhance Purkinje cell survival in a murine NPC model, to date this agent has not been formally trialled as a symptomatic treatment for cognitive impairment in NPC [170].
6.2 Illness-Modifying Treatments
Unlike many neurodegenerative disorders, but like a number of other neurometabolic disorders, some illness-modifying treatments are available for NPC. Miglustat, an imino sugar that inhibits glucosylceramide synthase and thus reduces the toxic accumulation of GM2 and GM3 gangliosides, has been shown to improve or stabilise several neurological illness markers in NPC and was first granted regulatory approval in 2009 [13, 18,19,20, 171]. It has been shown to stabilise or slow cerebellar and thalamic volume loss on neuroimaging measures [18] and slow the loss of white matter micro-integrity in adult patients [20]. An orally available medication, it is usually administered up to 200 mg three times daily, with the chief adverse effect being diarrhoea in the setting of disaccharide intake (such as sucrose) as miglustat inhibits disaccharidases in the gut. Psychosis has been shown to respond to miglustat treatment alone in two patients. In the first reported patient, a florid psychosis presented at the age of 20 years and did not respond to treatment with olanzapine 20 mg/day; haloperidol and aripiprazole both caused significant dystonia and were withdrawn. Miglustat was then started, and after 3 months this patient was psychosis-free [65]. One further 27-year-old patient, originally diagnosed with schizophrenia, has been described as presenting with treatment-refractory psychosis and dystonia attributed to antipsychotic medication; however, treatment with miglustat resolved psychotic symptoms and improved neurological symptoms, and allowed antipsychotics to be ceased [63]. One paediatric patient who presented with depression alongside typical neurological features showed resolution of mood symptoms after 12 months’ treatment with miglustat [172]. This suggests that inasmuch as the psychotic symptoms of NPC are secondary to the underlying progressive neurobiology of the disease, addressing this pathology directly can result in a secondary improvement in psychotic symptoms, and that NPC should be considered in treatment-resistant psychotic illness with atypical features [63, 70].
The effect of miglustat on cognition has also been examined in a number of studies. Heitz et al. [82] did not find any significant improvement, or decline, in cognitive measures in 12 patients receiving up to 24 months of treatment in a retrospective case series. Patterson et al. [13] showed some improvement in MMSE scores over the course of 12 months of treatment in 20 patients over the age of 12 years treated with miglustat compared with nine patients receiving standard care. Two further studies showed stabilisation or improvement in most patients under treatment, although without control arms [173, 174].
Hydroxypropyl-β-cyclodextrin (HPCD), originally serendipitously discovered in animal trials for pregnanalone in preclinical models, has been shown to markedly improve neuronal cholesterol homeostasis and decrease neuronal pathology with intra-thecal administration [175, 176], and shows promise in slowing disease progression in humans [177]. Although not yet approved, a current phase IIb/III international trial is currently underway in adult and paediatric patients with NPC [180]. This compound shows great promise in treating the range of CNS manifestations of NPC, but as yet there have been no reports of psychiatric symptomatology in the disease responding to HPCD. However, the phase I–II trial data showed significantly decreased progression of cognitive impairment in HPCD-treated patients, suggesting that other disease-related manifestations, such as psychiatric symptoms, may also respond to treatment. A series of three patients treated with HPCD for 2–3 years showed stable or improved scores in all three patients using standardised IQ measures and computerised cognitive tasks [178]. Finally, a review of 16 early published cases of HPCD-treated NPC patients suggested that cognition stabilises or improves in most patients [179]. This suggests that, as in miglustat treatment, disease-modifying treatments that target the metabolic aspects of disease can improve the secondary (cognitive and/or psychiatric) symptoms of NPC disease.
7 Conclusions
NPC is a prototypical neurometabolic disorder that presents across the lifespan and, although metabolic in its origins, is fundamentally neurodegenerative in nature. Like other metabolic disorders that affect neurodevelopment, its protean age of onset means that some patients’ pathology affects neuronal function during a key window in late adolescent and early adulthood neurodevelopment that appears critical to its propensity to result in elevated rates of schizophrenia-like psychosis, with lesser rates of other psychiatric presentations; however, cognitive impairment is an inevitable and progressive symptom of the illness. There appear to be many pathways to psychosis in NPC, including alterations to connectivity at the level of the synapse but also myelinated axons, and also disrupted function of key grey matter ‘hubs’ in more distributed cortico-subcortical networks. Recognition of NPC as a possible cause of schizophrenia-like psychosis is important, as this and other metabolic disorders have specific treatments that address the metabolic defect or cellular consequences of this defect. Whilst symptomatic treatment can be partially effective, it may be that new illness-modifying treatments may also be crucial to treatment of psychiatric symptoms in addition to slowing disease progression. Clinicians’ awareness of NPC and related disorders that present with elevated rates of psychosis is critical to ensuring that presentations of ‘secondary’ psychiatric illness are appropriately diagnosed and treated.
References
Sévin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, et al. The adult form of Niemann-Pick disease type C. Brain. 2007;130:120–33.
Wassif CA, Cross JL, Iben J, Sanchez-pulido L, Platt FM, Ory DS, et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1 predicted by analysis of massively parallel sequencing data sets. Genet Med. 2016;18:41–8.
Walterfang M, Fietz M, Fahey M, Sullivan D, Leane P, Lubman DI, et al. The neuropsychiatry of Niemann-Pick type C disease in adulthood. J Neuropsychiatry Clin Neurosci. 2006;18:158–70. https://doi.org/10.1176/jnp.2006.18.2.158.
Vanier MT. Complex lipid trafficking in Niemann-Pick disease type C. J Inherit Metab Dis. 2014;38:187–99.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106:330–44.
Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16.
Bonnot O, Klünemann H-H, Velten C, Torres Martin JV, Walterfang M. Systematic review of psychiatric signs in Niemann-Pick disease type C. World J Biol Psychiatry. 2018. https://doi.org/10.1080/15622975.2018.1441548.
Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8:1–10.
Imrie J, Vijayaraghaven S, Whitehouse C, Harris S, Heptinstall L, Church H, et al. Niemann-Pick disease type C in adults. J Inherit Metab Dis. 2002;25:491–500.
Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol. 2015;15:257.
Jahnova H, Dvorakova L, Vlaskova H, Hulkova H, Poupetova H, Hrebicek M, et al. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis. 2014;9:140.
Sedel F, Baumann N, Turpin JC, Lyon-Caen O, Saudubray JM, Cohen D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J Inherit Metab Dis. 2007;30:631–41.
Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6:765–72.
Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018;13:140.
Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, et al. Development of a Suspicion Index to aid diagnosis of Niemann-Pick disease type C. Neurology. 2012;78:1560–7.
Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): a critical review. Mol Genet Metab. 2016;118:244–54. https://doi.org/10.1016/j.ymgme.2016.06.004.
Bonnot O, Klünemann HH, Sedel F, Tordjman S, Cohen D, Walterfang M. Diagnostic and treatment implications of psychosis secondary to treatable metabolic disorders in adults: a systematic review. Orphanet J Rare Dis. 2014;9:65.
Bowman EA, Walterfang M, Abel L, Desmond P, Fahey M, Velakoulis D. Longitudinal changes in cerebellar and subcortical volumes in adult-onset Niemann-Pick disease type C patients treated with miglustat. J Neurol. 2015;262:2106–14.
Abel LA, Walterfang M, Stainer MJ, Bowman EA, Velakoulis D. Longitudinal assessment of reflexive and volitional saccades in Niemann-Pick type C disease during treatment with miglustat. Orphanet J Rare Dis. 2015;10:1–8. https://doi.org/10.1186/s13023-015-0377-8.
Bowman EA, Velakoulis D, Desmond P, Walterfang M. Longitudinal changes in white matter fractional anisotropy in adult-onset Niemann-Pick disease type C patients treated with miglustat. JIMD Rep. 2018;39:39–43. https://doi.org/10.1007/8904_2017_42.
Arenas F, Garcia-Ruiz C, Fernandez-Checa JC. Intracellular cholesterol trafficking and impact in neurodegeneration. Front Mol Neurosci. 2017;10:382.
Benussi A, Cotelli MS, Padovani A, Borroni B. Recent neuroimaging, neurophysiological, and neuropathological advances for the understanding of NPC. F1000Res. 2018;7:194.
Love S, Bridges LR, Case CP. Neurofibrillary tangles in Niemann-Pick disease type C. Brain. 1995;118:119–29.
Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D’Agostino AN, et al. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995;89:227–38.
Hung YH, Faux NG, Killilea DW, Yanjanin N, Firnkes S, Volitakis I, et al. Altered transition metal homeostasis in Niemann-Pick disease, type C1. Metallomics. 2014;6:542–53.
Fu R, Yanjanin NM, Elrick MJ, Ware C, Lieberman AP, Porter FD. Apolipoprotein E genotype and neurological disease onset in Niemann-Pick disease, type C1. Am J Med Genet Part A. 2012;158A:2775–80.
Liu C-C, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–18.
Malnar M, Hecimovic S, Mattsson N, Zetterberg H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol Dis. 2014;72:37–47.
Auer IA, Schmidt ML, Lee VM-Y, Curry B, Suzuki K, Shin R-W, et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol. 1995;90:547–51.
Bergeron D, Poulin S, Laforce R. Cognition and anatomy of adult Niemann-Pick disease type C: insights for the Alzheimer field. Cogn Neuropsychol. 2018;35(3–4):209–22.
Bu B, Klunemann H, Suzuki K, Li J, Bird T, Jin LW, et al. Niemann-Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis. 2002;11(2):285–97.
Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903.
German DC, Liang C-L, Song T, Yazdani U, Xie C, Dietschy JM. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109:437–50.
Williams IM, Wallom KL, Smith DA, Al Eisa N, Smith C, Platt FM. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol Dis. 2014;67:9–17. https://doi.org/10.1016/j.nbd.2014.03.001.
Platt N, Speak AO, Colaco A, Gray J, Smith DA, Williams IM, et al. Immune dysfunction in Niemann-Pick disease type C. J Neurochem. 2016;136:74–80.
Yu T, Lieberman AP. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013;9:e1003462.
Takikita S, Fukuda T, Mohri I, Yagi T, Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J Neuropathol Exp Neurol. 2004;63:660–73.
Goodrum JF, Pentchev PG. Cholesterol reutilization during myelination of regenerating PNS axons is impaired in Niemann-Pick disease type C mice. J Neurosci Res. 1998;49:389–92.
Yan X, Yadav R, Gao M, Weng H-R. Interleukin-1 beta enhances endocytosis of glial glutamate transporters in the spinal dorsal horn through activating protein kinase C. Glia. 2014;62:1093–109.
Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet. 2007;16:1495–503.
Walkley SU, Baker HJ, Rattazzi MC, Haskins ME, Wu J-Y. Neuroaxonal dystrophy in neuronal storage disorders: evidence for major GABAergic neuron involvement. J Neurol Sci. 1991;104:1–8.
Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64.
Yamada A, Saji M, Ukita Y, Shinoda Y, Taniguchi M, Higaki K, et al. Progressive neuronal loss in the ventral posterior lateral and medial nuclei of thalamus in Niemann-Pick disease type C mouse brain. Brain Dev. 2001;23:288–97.
Lopez ME, Klein AD, Dimbil UJ, Scott MP. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J Neurosci. 2011;31:4367–78.
Pressey SNR, Smith DA, Wong AMS, Platt FM, Cooper JD. Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol Dis. 2012;45:1086–100.
Trouard TP, Heidenreich RA, Seeger JF, Erickson RP. Diffusion tensor imaging in Niemann-Pick Type C disease. Pediatr Neurol. 2005;33(5):325–30.
Walterfang M, Fahey M, Desmond P, Wood A, Seal ML, Steward C, et al. White and gray matter alterations in adults with Niemann-Pick disease type C: a cross-sectional study. Neurology. 2010;75:49–56.
Lee R, Apkarian K, Jung ES, Yanjanin N, Yoshida S, Mori S, et al. Corpus callosum diffusion tensor imaging and volume measures are associated with disease severity in pediatric Niemann-Pick disease type C1. Pediatr Neurol. 2014;51(669–674):e5.
Masingue M, Adanyeguh I, Nadjar Y, Sedel F, Galanaud D, Mochel F. Evolution of structural neuroimaging biomarkers in a series of adult patients with Niemann-Pick type C under treatment. Orphanet J Rare Dis. 2017;12:22.
Walterfang M, Macfarlane MD, Looi JCL, Abel L, Bowman E, Fahey MC, et al. Pontine-to-midbrain ratio indexes ocular-motor function and illness stage in adult Niemann-Pick disease type C. Eur J Neurol. 2012;19:462–7.
Walterfang M, Patenaude B, Abel LA, Kluenemann H, Bowman EA, Fahey MC, et al. Subcortical volumetric reductions in adult niemann-pick disease type C: a cross-sectional study. Am J Neuroradiol. 2013;34:1334–40.
Walterfang M, Bonnot O, Mocellin R, Velakoulis D. The neuropsychiatry of inborn errors of metabolism. J Inherit Metab Dis. 2013;36(4):687–702.
Lau MW, Lee RW, Miyamoto R, Jung ES, Yanjanin Farhat N, Yoshida S, et al. Role of diffusion tensor imaging in prognostication and treatment monitoring in Niemann-Pick disease type C1. Diseases. 2016;4(3):29.
Walterfang M, Abel LA, Desmond P, Fahey MC, Bowman EA, Velakoulis D. Cerebellar volume correlates with saccadic gain and ataxia in adult Niemann-Pick type C. Mol Genet Metab. 2013;108:85–9.
Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009;98:152–65.
Shulman LM, David NJ, Weiner WJ. Psychosis as the initial manifestation of adult-onset Niernann-Pick disease type C. Neurology. 1996;45:1739–43.
Campo JV, Robert S, Greg S, Debra B, Barbara G. Psychosis as a presentation of physical disease in adolescence: a case of Niemann-Pick disease, type C. Dev Med Child Neurol. 2008;40:126–9.
Josephs KA, Van Gerpen MW, Van Gerpen JA. Adult onset Niemann-Pick disease type C presenting with psychosis. J Neurol Neurosurg Psychiatry. 2003;74:528–9.
Sandu S, Jackowski-Dohrmann S, Ladner A, Haberhausen M, Bachmann C. Niemann-Pick disease type C1 presenting with psychosis in an adolescent male. Eur Child Adolesc Psychiatry. 2009;18:583–5.
Walterfang M, Fietz M, Abel L, Bowman E, Mocellin R, Velakoulis D. Gender dimorphism in siblings with schizophrenia-like psychosis due to Niemann-Pick disease type C. J Inherit Metab Dis. 2009;32:221–6.
Walterfang M, Kornberg A, Adams S, Fietz M, Velakoulis D. Post-ictal psychosis in adolescent Niemann-Pick disease type C. J Inherit Metab Dis. 2010;33:63–5.
Wouters S, De Meirleir L, Campforts E, Lampo A. Psychosis in an adolescent girl: a common manifestation in Niemann-Pick type C disease. Child Adolesc Psychiatry Ment Health. 2014;8:20.
Maubert A, Hanon C, Metton J. Niemann-Pick type C disease and psychosis: two siblings. Encephale. 2015;41:238–43.
Rebelo J, Oliveira M, Nunes P. Psychiatric manifestations of Niemann-Pick type C disease—two case reports. Eur Psychiatry. 2016;33:S466.
Szakszon K, Szegedi I, Magyar Á, Oláh É, Andrejkovics M, Balla P, et al. Complete recovery from psychosis upon miglustat treatment in a juvenile Niemann-Pick C patient. Eur J Paediatr Neurol. 2014;18:75–8.
Koens LH, Kuiper A, Coenen MA, Elting JWJ, de Vries JJ, Engelen M, et al. Ataxia, dystonia and myoclonus in adult patients with Niemann-Pick type C. Orphanet J Rare Dis. 2016;11:121.
Kawazoe T, Yamamoto T, Narita A, Ohno K, Adachi K, Nanba E, et al. Phenotypic variability of Niemann-Pick disease type C including a case with clinically pure schizophrenia: a case report. BMC Neurol. 2018;18:117.
Baumgartner A, Nia S, Geiblinger S, Baumgartner C. Psychiatric manifestation of adult form of Niemann-Pick type C. A case report. J Neurol Sci. 2013;333:e705.
Maubert A, Hanon C, Sedel F. Psychiatric disorders in adult form of Niemann-Pick disease type C. Encephale. 2016;42:208–13.
Bonnot O, Gama CS, Mengel E, Pineda M, Vanier MT, Watson L, et al. Psychiatric and neurological symptoms in patients with Niemann-Pick disease type C (NP-C): findings from the International NPC Registry. World J Biol Psychiatry. 2017;9:1–10.
Walterfang M, Bonnot O, Mocellin R, Velakoulis D. The neuropsychiatry of inborn errors of metabolism. J Inherit Metab Dis. 2013;36:687–702.
Sullivan D, Walterfang M, Velakoulis D. Bipolar disorder and Niemann-Pick disease type C. Am J Psychiatry. 2005;162:1021–2.
Bonnot O, Herrera P, Tordjman S, Walterfang M. Secondary psychosis induced by metabolic disorders. Front Neurosci. 2015;9:177.
Lossos A, Schlesinger I, Okon E, Al E. Adult-onset niemann-pick type c disease: clinical, biochemical, and genetic study. Arch Neurol. 1997;54:1536–41.
Abel LA, Walterfang M, Fietz M, Bowman EA, Velakoulis D. Saccades in adult Niemann-Pick disease type C reflect frontal, brainstem, and biochemical deficits. Neurology. 2009;72:1083–6.
Chien Y-H, Lee N-C, Tsai L-K, Huang A-C, Peng S-F, Chen S-J, et al. Treatment of Niemann-Pick disease type C in two children with miglustat: Initial responses and maintenance of effects over 1 year. J Inherit Metab Dis. 2007;30:826.
Chien YH, Peng SF, Yang CC, Lee NC, Tsai LK, Huang AC, et al. Long-term efficacy of miglustat in paediatric patients with Niemann-Pick disease type C. J Inherit Metab Dis. 2013;36:129–37.
Johnen A, Pawlowski M, Duning T. Distinguishing neurocognitive deficits in adult patients with NP-C from early onset Alzheimer’s dementia. Orphanet J Rare Dis. 2018;13(1):91.
Walterfang M, Siu R, Velakoulis D. The NUCOG: validity and reliability of a brief cognitive screening tool in neuropsychiatric patients. Aust N Z J Psychiatry. 2006;40:995–1002.
Hinton V, Vecchio D, Prady H, Wraith E, Patterson M, York N, et al. The cognitive phenotype of Niemann-Pick type C (NPC): neuropsychological characteristics of patients at baseline in a clinical trial with oral miglustat [abstract no. OPL120]. J Neurol Sci. 2005;238:S77.
Klarner B, Klünemann HH, Lürding R, Aslanidis C, Rupprecht R. Neuropsychological profile of adult patients with Niemann-Pick C1 (NPC1) mutations. J Inherit Metab Dis. 2007;30:60–7.
Heitz C, Epelbaum S, Nadjar Y. Cognitive impairment profile in adult patients with Niemann pick type C disease. Orphanet J Rare Dis. 2017;12(1):166.
Hendriksz CJ, Anheim M, Bauer P, Bonnot O, Chakrapani A, Corvol JC, et al. The hidden Niemann-Pick type C patient: clinical niches for a rare inherited metabolic disease. Curr Med Res Opin. 2017;33:877–90.
Schiffmann R. Niemann-Pick disease type C: from bench to bedside. JAMA. 1996;276:561–4.
Shulman LM, Lang AE, Jankovic J, David NJ, Weiner WJ. What is it? Case 1, 1995: psychosis, dementia, chorea, ataxia, and supranuclear gaze dysfunction. Mov Disord. 1995;10:257–62.
Salmon DP. Disorders of memory in Alzheimer’s disease. In: Cermak LS, editor. Handbook of neuropsychology: memory and its disorders, vol. 2. 2nd ed. Amsterdam: Elsevier Science Publishers B.V.; 2000. p. 155–95.
Dardis A, Zampieri S, Canterini S, Newell KL, Stuani C, Murrell JR, et al. Altered localization and functionality of TAR DNA Binding Protein 43 (TDP-43) in Niemann-Pick disease type C. Acta Neuropathol Commun. 2016;4:52.
Zhang M, Hallows JL, Wang X, Bu B, Wang W, Vincent I. Mitogen-activated protein kinase activity may not be necessary for the neuropathology of Niemann-Pick type C mice. J Neurochem. 2008;107:814–22.
Velakoulis D, Walterfang M, Mocellin R, Pantelis C, McLean C. Frontotemporal dementia presenting as schizophrenia-like psychosis in young people: clinicopathological series and review of cases. Br J Psychiatry. 2009;194:298–305.
Cooper JJ, Ovsiew F. The relationship between schizophrenia and frontotemporal dementia. J Geriatr Psychiatry Neurol. 2013;26:131–7.
Harvey PD. Cognitive impairment in schizophrenia: characteristics, assessment and treatment. Cambridge: Cambridge University Press; 2013.
O’Carroll R. Cognitive impairment in schizophrenia. Adv Psychiatr Treat. 2000;6:161–8.
Bowie CR, Harvey PD. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr Dis Treat. 2006;2:531–6.
Evans WRH, Hendriksz CJ. Niemann-Pick type C disease – the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment. BJPsych Bull. 2017;41:109–14.
Saudubray J-M. Neurometabolic disorders. J Inherit Metab Dis. 2009;32:595.
Walterfang M, Mocellin R, Velakoulis D. The neuropsychiatry of neuroendocrine and neurometabolic disorders. In: Sadock BJ, Sadock VA, Ruiz P, editors. Kaplan and Sadock’s comprehensive psychiatry. 11th ed. Philadelphia: Lippincott; 2009.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005;62:593–602.
Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaituzis AC, et al. Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA. 2004;101:8174–9.
Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human life span. Nat Neurosci. 2003;6:309–15.
Paus T, Zijdenbos A, Worsley K, Collins DL, Blumenthal J, Giedd JN, et al. Structural maturation of neural pathways in children and adolescents: in vivo study. Science. 1999;283:1908–11.
Walterfang M, Wood SJ, Velakoulis D, Copolov D, Pantelis C. Diseases of white matter and schizophrenia-like psychosis. Aust N Z J Psychiatry. 2005;39:746–56.
Friston K, Frith C. Schizophrenia: a disconnection syndrome? Clin Neurosci. 1995;3(2):89–97.
Wernicke C (1906) Grundrisse der Psychiatrie. Leipzig: Thieme.
Bleuler E, [Transl. Zinkin J]. Dementia praecox, or the group of schiozophrenias. New York: International Universities Press; 1950.
Walterfang M, Wood SJ, Velakoulis D, Pantelis C. Neuropathological, neurogenetic and neuroimaging evidence for white matter pathology in schizophrenia. Neurosci Biobehav Rev. 2006;30:918–48.
Broyd SJ, Demanuele C, Debener S, Helps SK, James CJ, Sonuga-Barke EJS. Default-mode brain dysfunction in mental disorders: a systematic review. Neurosci Biobehav Rev. 2009;33:279–96.
Zalesky A, Fornito A, Seal ML, Cocchi L, Westin CF, Bullmore ET, et al. Disrupted axonal fiber connectivity in schizophrenia. Biol Psychiatry. 2011;69:80–9.
Hyde TM, Ziegler JC, Weinberger DR. Psychiatric disturbances in metachromatic leukodystrophy: insights into the neurobiology of psychosis. Arch Neurol. 1992;49:401–6.
Black DN, Taber KH, Hurley RA. Metachromatic leukodystrophy: a model for the study of psychosis. J Neuropsychiatry Clin Neurosci. 2003;15:289–93.
Kikinis Z, Asami T, Bouix S, Finn CT, Ballinger T, Tworog-Dube E, et al. Reduced fractional anisotropy and axial diffusivity in white matter in 22q11.2 deletion syndrome: a pilot study. Schizophr Res. 2012;141:35–9.
Jalbrzikowski M, Villalon-Reina JE, Karlsgodt KH, Senturk D, Chow C, Thompson PM, et al. Altered white matter microstructure is associated with social cognition and psychotic symptoms in 22q11.2 microdeletion syndrome. Front Behav Neurosci. 2014;8:1–18.
Crapse TB, Sommer MA. Corollary discharge across the animal kingdom. Nat Rev Neurosci. 2008;9:587–600.
Feinberg I. Efference copy and corollary discharge: implications for thinking and its disorders. Schizophr Bull. 1978;4:636–40.
Frith CD (2014) The cognitive neuropsychology of schizophrenia. London: Psychology Press (First published 1992).
Whitford TJ, Ford JM, Mathalon DH, Kubicki M, Shenton ME. Schizophrenia, myelination, and delayed corollary discharges: a hypothesis. Schizophr Bull. 2012;38:486–94.
Blakemore S, Wolpert D, Frith C. Abnormalities in the awareness and control of action. Philos Trans R Soc B Biol Sci. 2000;355:1771–88.
Lemaitre AL, Luyat M, Lafargue G. Individuals with pronounced schizotypal traits are particularly successful in tickling themselves. Conscious Cogn. 2016;41:64–71.
Rottach KG, Von Maydell RD, Das VE, Zivotofsky AZ, Discenna AO, Gordon JL, et al. Evidence for independent feedback control of horizontal and vertical saccades from Niemann-Pick type C disease. Vis Res. 1997;37:3627–38.
Leigh RJ, Kennard C. Using saccades as a research tool in the clinical neurosciences. Brain. 2004;127:460–77.
Klauser P, Baker ST, Cropley VL, Bousman C, Fornito A, Cocchi L, et al. White matter disruptions in schizophrenia are spatially widespread and topologically converge on brain network hubs. Schizophr Bull. 2017;43:425–35.
Friston K, Brown HR, Siemerkus J, Stephan KE. The dysconnection hypothesis (2016). Schizophr Res. 2016;176:83–94.
Stahl SM. Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: dopamine, serotonin, and glutamate. CNS Spectr. 2018;23:187–91.
Hu CY, Ong WY, Patel SC. Regional distribution of NPC1 protein in monkey brain. J Neurocytol. 2000;29:765–73.
Xu S, Chen X, Wei X, Liu G, Wang Q. Presynaptic impairment in Niemann-Pick C1-deficient neurons: not dependent on presence of glial cells. Neurosci Lett. 2011;496:54–9.
Brachet A, Norwood S, Brouwers JF, Palomer E, Helms JB, Dotti CG, et al. LTP-triggered cholesterol redistribution activates Cdc42 and drives AMPA receptor synaptic delivery. J Cell Biol. 2015;208(6):791–806.
Frank C, Rufini S, Tancredi V, Forcina R, Grossi D, D’Arcangelo G. Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp Neurol. 2008;212:407–14.
Zhou S, Xu S, Yan Y, Yu H, Ling S, Luo J. Decreased purinergic inhibition of synaptic activity in a mouse model of Niemann-Pick disease type C. Hippocampus. 2011;21:212–9.
D’Arcangelo G, Grossi D, Racaniello M, Cardinale A, Zaratti A, Rufini S, et al. Miglustat reverts the impairment of synaptic plasticity in a mouse model of NPC disease. Neural Plast. 2016;2016:3830424.
Xu S, Zhou S, Xia D, Xia J, Chen G, Duan S, et al. Defects of synaptic vesicle turnover at excitatory and inhibitory synapses in Niemann-Pick C1-deficient neurons. Neuroscience. 2010;167:608–20.
Buard I, Pfrieger FW. Relevance of neuronal and glial NPC1 for synaptic input to cerebellar Purkinje cells. Mol Cell Neurosci. 2014;61:65–71.
Joyce DW, Averbeck BB, Frith CD, Shergill SS. Examining belief and confidence in schizophrenia. Psychol Med. 2013;43:2327–38.
Fletcher PC, Frith CD. Perceiving is believing: a Bayesian approach to explaining the positive symptoms of schizophrenia. Nat Rev Neurosci. 2008;10:48–58.
Sporns O, Honey CJ, Kötter R. Identification and classification of hubs in brain networks. PLoS ONE. 2007;2:e1049.
Crossley NA, Mechelli A, Scott J, Carletti F, Fox PT, McGuire P, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain. 2014;137:2382–95.
Tomasi D, Wang G-J, Volkow ND. Energetic cost of brain functional connectivity. Proc Natl Acad Sci USA. 2013;110:13642–7.
van den Heuvel MP, Sporns O, Collin G, Albert E. Abnormal rich club organization and functional brain dynamics in schizophrenia. JAMA Psychiatry. 2013;70:783–92.
German DC, Quintero EM, Liang C-L, Ng B, Punia S, Xie C, et al. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J Comp Neurol. 2001;433:415–25.
Taniguchi M, Shinoda Y, Ninomiya H, Vanier MT, Ohno K. Sites and temporal changes of gangliosides GM1/GM2 storage in the Niemann-Pick disease type C mouse brain. Brain Dev. 2001;23:414–21.
Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993;85:175–84.
Habas C, Kamdar N, Nguyen D, Keller K, Beckmann CF, Menon V, et al. Distinct cerebellar contributions to intrinsic connectivity networks. J Neurosci. 2009;29:8586–94.
Wang L, Zou F, Shao Y, Ye E, Jin X, Tan S, et al. Disruptive changes of cerebellar functional connectivity with the default mode network in schizophrenia. Schizophr Res. 2014;160:67–72.
Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167:1178–93.
Heckers S, Konradi C. Hippocampal neurons in schizophrenia. J Neural Transm. 2002;109:891–905.
Velakoulis D, Wood SJ, Wong MH, Albert E. Hippocampal and amygdala volumes according to psychosis stage and diagnosis: a magnetic resonance imaging study of chronic schizophrenia, first-episode psychosis, and ultra–high-risk individuals. Arch Gen Psychiatry. 2006;63:139–49.
Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, et al. Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry. 2014;171:549–56.
Lieberman JA, Girgis RR, Brucato G, Moore H, Provenzano F, Kegeles L, et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018;23(8):1764–72.
Behrendt R-P. Contribution of hippocampal region CA3 to consciousness and schizophrenic hallucinations. Neurosci Biobehav Rev. 2010;34:1121–36.
Treiber-Held S, Distl R, Meske V, Albert F, Ohm TG. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer’s disease. J Pathol. 2003;200:95–103.
D’Arcangelo G, Grossi D, De Chiara G, De Stefano MC, Cortese G, Citro G, et al. Glutamatergic neurotransmission in a mouse model of Niemann-Pick type C disease. Brain Res. 2011;1396:11–9.
Nagai M, Kishi K, Kato S. Insular cortex and neuropsychiatric disorders: a review of recent literature. Eur Psychiatry. 2007;22(6):387–94.
Crespo-Facorro B, Kim J-J, Andreasen NC, O ’leary DS, Bockholt HJ, Magnotta V. Insular cortex abnormalities in schizophrenia: a structural magnetic resonance imaging study of first-episode patients. Schizophr Res. 2000;46:35–43.
Shergill SS, Brammer MJ, Williams SC, Murray RM, Mcguire PK. Mapping auditory hallucinations in schizophrenia using functional magnetic resonance imaging. Arch Gen Psychiatry. 2000;57:1033–8.
Dougherty M, Lazar J, Klein JC, Diaz K, Gobillot T, Grunblatt E, et al. Genome sequencing in a case of Niemann-Pick type C. Cold Spring Harb Mol Case Stud. 2016;2:a001222.
Pergola G, Selvaggi P, Trizio S, Bertolino A, Blasi G. The role of the thalamus in schizophrenia from a neuroimaging perspective. Neurosci Biobehav Rev. 2015;54:57–75.
Simpson EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron. 2010;65:585–96.
Ramnani N. The primate cortico-cerebellar system: anatomy and function. Nat Rev Neurosci. 2006;7:511–22.
Schmahmann J, Pandya D. The cerebrocerebellar system. Int Rev Neurobiol. 1997;41:31–60.
Buckner RL. The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron. 2013;80:807–15.
Schmahmann JD. An emerging concept: the cerebellar contribution to higher function. Arch Neurol. 1991;48:1178–87.
Bellebaum C, Daum I. Cerebellar involvement in executive control. Cerebellum. 2007;6:184–92.
Schmahmann J, Sherman J. The cerebellar cognitive affective syndrome. Brain. 1998;121:561–79.
Halassa MM, Kastner S. Thalamic functions in distributed cognitive control. Nat Neurosci. 2017;20:1669–79.
Saalmann YB, Kastner S. The cognitive thalamus. Front Syst Neurosci. 2015;9:39.
Parnaudeau S, Bolkan SS, Kellendonk C. The mediodorsal thalamus: an essential partner of the prefrontal cortex for cognition. Biol Psychiatry. 2018;83:648–56.
Simeone JC, Ward AJ, Rotella P, Collins J, Windisch R. An evaluation of variation in published estimates of schizophrenia prevalence from 1990─2013: a systematic literature review. BMC Psychiatry. 2015;15:193.
MacQueen GM, Rosebush PI, Mazurek MF. Neuropsychiatric aspects of the adult variant of Tay-Sachs disease. J Neuropsychiatry Clin Neurosci. 1998;10:10–9.
Karimzadeh P, Jafari N, Nejad Biglari H, Jabbehdari S, Khayat Zadeh S, Ahmad Abadi F, et al. Neurometabolic diagnosis in children who referred as neurodevelopmental delay (a practical criteria, in Iranian pediatric patients). Iran J Child Neurol. 2016;10:73–81.
Kohlschütter A, Eichler F. Childhood leukodystrophies: a clinical perspective. Expert Rev Neurother. 2011;11:1485–96.
Maubert A, Hanon C, Metton J. Adult onset Niemann-Pick type C disease and psychosis: literature review. Encephale. 2013;39:315–9.
Seo Y, Shin Y, Kim H-S, Kang I, Hong I-S, Choi SW, et al. Donepezil enhances Purkinje cell survival and alleviates motor dysfunction by inhibiting cholesterol synthesis in a murine model of Niemann pick disease type C. J Neuropathol Exp Neurol. 2014;73:234–43.
Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M, et al. Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab. 2009;98:243–9. https://doi.org/10.1016/j.ymgme.2009.07.003.
Santos MLF, Raskin S, Telles DS, Löhr Junior A, Liberalesso PBN, Vieira SC, et al. Treatment of a child diagnosed with Niemann-Pick disease type C with miglustat: a case report in Brazil. J Inherit Metab Dis. 2008;31:357–61.
Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab. 2010;99:351–7.
Fecarotta S, Romano A, Della Casa R, Del Giudice E, Bruschini D, Mansi G, et al. Long term follow-up to evaluate the efficacy of miglustat treatment in Italian patients with Niemann-Pick disease type C. Orphanet J Rare Dis. 2015;10:22.
Liu B. Therapeutic potential of cyclodextrins in the treatment of Niemann-Pick type C disease. Clin Lipidol. 2012;7:289–301.
Vite CH, Bagel JH, Swain GP, Prociuk M, Sikora TU, Stein VM, et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med. 2015;7:276ra26.
Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet. 2017;390:1758–68. https://doi.org/10.1016/S0140-6736(17)31465-4.
Berry-Kravis E, Chin J, Hoffmann A, Winston A, Stoner R, LaGorio L, et al. Long-term treatment of Niemann-Pick type C1 disease with intrathecal 2-hydroxypropyl-β-cyclodextrin. Pediatr Neurol. 2018;80:24–34.
Megías-Vericat JE, García-Robles A, Company-Albir MJ, Fernández-Megía MJ, Pérez-Miralles FC, López-Briz E, et al. Early experience with compassionate use of 2 hydroxypropyl-beta-cyclodextrin for Niemann-Pick type C disease: review of initial published cases. Neurol Sci. 2017;38:727–43.
Vtesse Inc, a Mallinckrodt Pharmaceuticals Company. Study of VTS-270 (2-hydroxypropyl-β-cyclodextrin) to treat Niemann-Pick type C1 (NPC1) disease [ClinicalTrials.gov identifier NCT02534844]. National Institutes of Health, ClinicalTrials.gov. 2018. https://clinicaltrials.gov/ct2/show/NCT02534844. Accessed 11 Dec 2018.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding was received for medical illustrations for this article from an unrestricted educational grant provided by the Royal Melbourne Hospital.
Conflict of interest
Thomas Rego, Sarah Farrand, Anita M.Y. Goh, Dhamidhu Eratne, Wendy Kelso, Simone Mangelsdorf, Dennis Velakoulis and Mark Walterfang have no conflicts of interest to declare.
Rights and permissions
About this article

Cite this article
Rego, T., Farrand, S., Goh, A.M.Y. et al. Psychiatric and Cognitive Symptoms Associated with Niemann-Pick Type C Disease: Neurobiology and Management. CNS Drugs 33, 125–142 (2019). https://doi.org/10.1007/s40263-018-0599-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-018-0599-0